Triplet exciton energy transfer in polyfluorene doped with heavy metal complexes studied using
photoluminescence and photoinduced absorption
H. H. Liao,1,2H. F. Meng,1,*S. F. Horng,2W. S. Lee,1J. M. Yang,2C. C. Liu,2J. T. Shy,3F. C. Chen,4and C. S. Hsu5
1Institute of Physics, National Chiao Tung University, Hsinchu 300, Taiwan, Republic of China 2Department of Electric Engineering, National Tsing Hua University, Hsinchu 300, Taiwan, Republic of China
3Department of Physics, National Tsing Hua University, Hsinchu 300, Taiwan, Republic of China 4Institute of Display, National Chiao Tung University, Hsinchu 300, Taiwan, Republic of China 5Department of Applied Chemistry, National Chiao Tung University, Hsinchu 300, Taiwan, Republic of China
共Received 12 April 2006; revised manuscript received 24 October 2006; published 15 December 2006兲
Modulated photoinduced absorption and photoluminescence are used to study triplet-to-triplet
Dexter energy transfer in Ir-complexes/polyfluorene blend systems. There is no Dexter energy transfer for red iridium 共III兲bis关2-共9,9-dibutylfluorenyl兲-1-isoquinoline共acetylacetonate兲兴 共DFIr兲 and red iridium 共III兲 bis共关2-共2-benzo-thienyl兲pyridinatoN,C3兴 acetyl-acetonate兲 共BtpIr兲 dopants. Although green iridium共III兲tris关2-共4-tolyl兲pyridinato-N,C2兴 关Ir共mppy兲3兴 has no triplet confinement in polyfluorene host, it has clear evidence for Dexter energy transfer. Aggregation and dopant lifetime are shown to significantly affect the energy transfer. The presence of Dexter transfer implies the possibility to harvest triplet excitons of polyfluo-rene in polymer light-emitting diodes even without carrier trapping and triplet exciton confinement.
DOI:10.1103/PhysRevB.74.245211 PACS number共s兲: 71.35.⫺y
I. INTRODUCTION
The formation and decay of spin-triplet excitons in poly-mer light-emitting diodes共PLED’s兲 are important and hotly debated subjects, partially because they determine the theo-retical limit of the quantum efficiency for both fluorescent and phosphorescent PLED’s.1 Among all the processes in-volved in luminescence, triplet-to-triplet energy transfer is probably the most elusive and least understood. Because only spin-singlet excitons of conjugated polymers emit light, the triplet-to-singlet branching ratio ␥ determines the quan-tum efficiency of fluorescent PLED’s.1–3 In our previous
study using infrared-induced absorption4it is found that␥is
a constant independent of electric field and temperature, sug-gesting that the exciton formation process is spin indepen-dent. This is consistent with a recent magnetic resonance experiment.5It is therefore expected that the branching ratio
for the nonemissive triplet excitons in PLED is no different from small-molecule light-emitting diodes 共OLED’s兲—i.e.,
␥= 3 for both cases.6These results imply that to harvest the
75% nonemissive triplet excitons in PLED’s is essential for achieving highly efficient PLED’s, just like the well-established case of OLED’s. A full understanding of the trip-let exciton in polymer is therefore highly desired.
The most promising way to harvest the triplet energy is to take the same strategy as OLED’s and introduce heavy-metal triplet emitters into the polymer to make phosphorescent PLED’s共PhPLED’s兲.7–9Indeed by employing triplet emitters
the efficiency of PLED’s is significantly improved10–12but is
still far below OLED’s. So far the reason for this difference is not clear and the basic physics of PhPLED’s is unknown. In the early period of phosphorescent small-molecule OLED’s共PhOLED’s兲, triplet emitters are considered to har-vest triplet excitons of the host by triplet-to-triplet Dexter energy transfer.7,8,13,14 Afterward, however, the high
effi-ciency of PhOLED’s is reported to be caused by carrier trapping9,10,15,16which is a sequential process of electron and
hole capture by the triplet emitters followed by direct recom-bination. It turns out that Dexter energy transfer and trapping mechanisms dominate in different host/guest PhOLED systems.17 As for PhPLED’s, previous studies show that
there is no Dexter energy transfer in main-chain conjugated polymers doped by a phosphor emitter,18–20 in particular the
common conjugated polymer host polyfluorene doped by Ir-complex phosphors. Dexter energy transfer is found only in side-chain conjugation polymers poly共vinylcarbazole兲 共PVK兲 at extremely low temperature. It is therefore an outstanding question whether the triplet excitons of polyfluorene can be harvested by triplet emitters through Dexter energy transfer to achieve highly efficient PhPLED’s or high efficiency can only be realized through trapping mechanisms by triplet emitters like the situation of PhOLED’s. In order to answer this question, in this work we use two polyfluorenes and three Ir complexes to study Dexter energy transfer in phosphor-doped conjugated polymer systems. Using our infrared-induced absorption setup,4 we are able to monitor
the behaviors of the three primary photoexcitations in the polymer/phosphor system, i.e., singlet exciton of the host polymer, triplet exciton of the host polymer, and triplet ex-citon of the phosphor. By comparing the decay dynamics of the polymer triplet exciton and phosphor triplet exciton, we observed Dexter energy transfer for a green Ir complex but not red complexes despite the lack of triplet exciton confine-ment in the green complex. The film morphology study shows that the blend with green Ir complex is more uniform than others. The triplet exciton lifetime is also shown to be critical to decide whether the transfer occurs or not.
II. EXPERIMENT
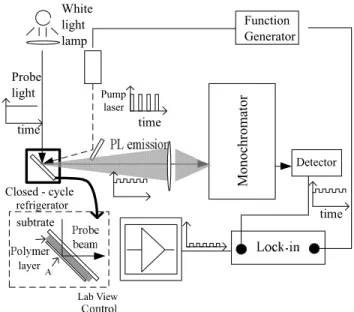
The triplet exciton is detected by photoinduced absorption 共PA兲.4 Figure 1 shows the experimental setup for the PA
spectroscopy. A 20-mW, 405-nm pump laser 共Power Tech-nology兲 is modulated by a function generator 共HP 33120A兲
with a square-wave form, and the laser beam is focused on the sample and generates the modulated singlet exciton den-sity. The sample is placed in a low-temperature closed-cycle refrigerator共Janis CCS-150, 10–300 K兲. Some singlet exci-tons decay to the ground state, and others become triplet excitons through intersystem crossing 共ISC兲 process. The triplet exciton generation is also modulated due to the modu-lation of singlet exciton density. Although it is not possible to detect the triplet exciton by its radiative decay due to spin selection rule, we can detect them by its spin-allowed higher-level absorption. A continued-wave共CW兲 white light probe beam generated by a 250-W quartz-tungsten-halogen lamp is focused on the same spot of the sample as the probe. The probe beam goes through the glass substrate, then the poly-mer film, and is reflected by a Al layer coated on the polypoly-mer film. The optical path of the probe beam is shown in the inset of Fig. 1. The reflected probe beam contains two parts in-cluding a modulated component caused by the triplet-exciton higher-level absorption and a remnant CW component. The reflected beam is collected into a monochromator 共CVI DK240, with a 700-nm high-pass filter兲 and detected by a Si detector 共New Focus 2001, with 103 gain兲, which is con-nected to a lock-in amplifier 共SRS 830兲 whose reference channel is connected to the synchronous port of the function generator. The detector receives a small modulated ac and a dc signal. The dc part is removed by the dc filter of the lock-in amplifier, and only the ac signal with the same fre-quency of the function generator will be extracted and dis-played in the in-phase and quadrature-phase channels. All the instruments and the data are controlled and recorded by the Labview program through IEEE-488 interface. When the pump modulation frequency is lower than 1 /共2T兲, where
Tis host triplet exciton lifetime, the signal will only appear
in the in-phase channel of the lock-in amplifier. When the modulation frequency is higher than 1 /共2T兲, the signal in
the in-phase channel starts to decay and the signal starts to appear in the quadrature-phase channel of the amplifier.
When the modulation is even faster so that the triplet exci-tons do not respond to the modulation, the quadrature-phase channel will decay eventually.21Analyzing the phase relation at different frequencies enables one to distinguish a slow signal共with long lifetime, in the quadrature-phase channel兲 superimposed on a fast signal 共with short lifetime, in the in-phase channel兲.22,24Theoretically frequency domain
mea-surement should give equivalent information as the time do-main measurement. Since the relevant time scales in this problem are the triplet exciton lifetime and the Dexter trans-fer over milliseconds,26it is more convenient to work in the
frequency domain up to only kHz. It is also easy to separate the fast Ir intrinsic emission from the slow emission from Dexter transfer in the polymer/phosphor blended system by the different frequency response. Two polyfluorenes are used: poly共9,9-dioctylfluorenyl-2,7-diyl兲 end capped with dimethylphenyl共PFO-DMP兲 and poly关9,9-di-共2-ethylhexyl兲-fluorenyl-2, 7-diyl兴 end capped with N,N-bis共4-methylphenyl兲-4-aniline 共PF-MPA兲. Three Ir complexes are used: iridium共III兲tris关2-共4-tolyl兲pyridinato-N, C2兴 关Ir共mppy兲3兴, iridium 共III兲 bis关2-共9,9-dibutylfluorenyl兲-1-isoquinoline共acetylacetonate兲兴 共DFIr兲, and iridium 共III兲 bis共关2-共2-benzo-thienyl兲pyridinatoN, C3兴共acetyl-acetonate兲 共BtpIr兲. These materials are all purchased from American Dye Source.
III. MODULATION SPECTRA
Figure2shows the relevant transitions. The Ir triplet ex-citon can be formed by either the Förster or Dexter energy transfer route. The former has a fast response to modulation because of the sub-nanosecond lifetime of host singlet exciton. The latter has long transient because the host triplet exciton lifetimeTis in the ms range at low
tempera-ture, which is far longer than temperature-independent iridium lifetime Ir in the s range. Therefore, if both Förster energy transfer and Dexter energy transfer occur, the emission of Ir will have a fast component in the s 共Ir兲 time scale and a delayed component in the ms 共T兲 time
scale. We use a lock-in amplifier to monitor the delayed pho-toluminescence共PL兲 at the modulation frequency f such that 1 / 2T⬍ f ⬍1/2Ir, The fast Ir emission signals from Förster energy transfer 共f ⬍1/2T兲 will always appear in
the in-phase channel of the amplifier and the delayed Ir emis-sion from Dexter energy transfer共f ⬍1/2Ir兲兲 will appear in the quadrature-phase channel. By comparing the dynamics of the host triplet exciton detected by PA and guest triplet exciton detected by PL, we are able to unambiguously decide if Dexter energy transfer happens in Ir-complex-doped poly-fluorene.
Ir共mppy兲3 and DFIr are dissolved in xylene and BtpIr in tetrahydrofuran 共THF兲. We try four doping concentrations 共0%, 5%, 10%, 20%兲 and compared these solutions. The highest concentrations in which Ir complexes can be well dissolved without any precipitation are 20%, 10%, and 5% for Ir共mppy兲3, DFIr, and BtpIr, respectively. The solution is then spin-coated to form a 100-nm-thick film on a precleaned glass substrate. A 1000-Å Al layer is evaporated on the poly-mer film as the reflection coating. The PA frequency
depen-time time time Monochromator White light lamp Function Generator Probe light Closed - cycle refrigerator Pump laser layer Detector subtrate Lab View
FIG. 1. The setup of PA and quadrature-phase channel PL ex-periment. The inset is the sample structure and the optical path.
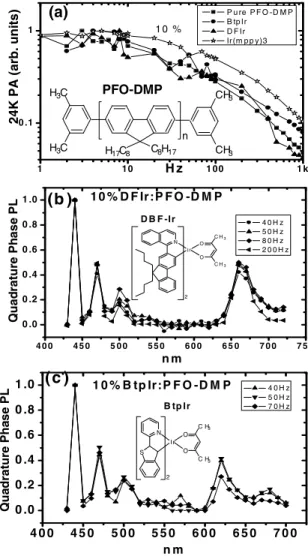
dence of the red DFIr- or red BtpIr-doped and intrinsic PFO-DMP films at 24 K are shown in Fig.3共a兲. All Ir dopants are compared at the same concentration of 10% despite some precipitation for BtpIr. The effect of concentration will be discussed below. There is no difference between two red Ir-doped and intrinsic PFO-DMP triplet excitons, indicating that the host triplet exciton lifetime Tis not influenced by
DFIr and BtpIr. From Fig.3共a兲and the relation= 1 /共2f兲 the PFO-DMP triplet exciton lifetime is approximately 15 ms共at f =10 Hz兲.21Figures3共b兲 and3共c兲are the normal-ized quadrature-phase channel PL spectra of PFO-DMP doped with BtpIr and DFIr, respectively. As described above, if the modulation frequency exceeds 1 /共2T兲, the delayed Ir
triplet excitons from Dexter energy transfer would appear in the quadrature-phase channel. Although the delayed red PL signals do appear in the quadrature-phase channel as shown in Figs.3共b兲and3共c兲, it is not due to Dexter energy transfer because the delayed signal appears in the blue host singlet exciton simultaneously. The spectra at different modulation frequencies are all the same after normalization, implying that the red and blue peaks are delayed by the same mecha-nism. We attribute this to the weak delayed fluorescence caused by the triplet-triplet annihilation.17 Through Förster
energy transfer the delayed singlet excitons are transferred to Ir to form the delayed Ir emissions. There is no Dexter en-ergy transfer in PFO-DMP doped by either BtpIr or DFIr.
The PA frequency dependence of green Ir共mppy兲3 doped PFO-DMP is also shown in Fig.3共a兲. The striking difference from the red Ir complexes BtpIr and DFIr is that Ir共mppy兲3
significantly affects the modulation response of the host trip-let exciton and the host triptrip-let lifetime is decreased by 2 times in magnitude to 8 ms 共⬃20 Hz兲. This indicates that there is a new channel for the decay of the host triplet exci-ton. In order to decide if this additional channel is Dexter energy transfer, the host triplet and Ir triplet excitons are monitored by PA and quadrature-phase channel PL measure-ments, respectively. It is seen in Fig. 4共a兲 that the PA in-phase channel signal starts to decrease and the PA quadrature-phase channel begins to rise as the frequency is larger than the host triplet exciton lifetimeT.
The key feature is that the delayed PA signal directly translates to the quadrature-phase channel as a slow compo-nent of green Ir-complex PL. In fact the quadrature-phase channel of Ir PL monitored at its emission peak at 535 nm has an identical frequency response to the PA quadrature-phase channel signals. This correlation between PA and PL suggests that the delayed emission of Ir共mppy兲3is due to the Dexter route from the host triplet excitons. The normalized quadrature-phase channel PL spectra at different frequencies
(a)
(b)
2 5 0 3 00 3 50 40 0 45 0 5 0 0 5 5 0 60 0 6 5 0 7 0 0 7 50 8 0 0 0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2 D F Ir Ir(m p p y )3 B tp Ir P F O -D M P PL (ar b. uni ts ) n m Ir(m p p y)3 A b s o rp tio n P F -M P AS
T
ττττ T ττττ S Förster Dexter R GIr
ττττ IrFIG. 2.共a兲 shows the relative exciton energies of the materials. SandTare host singlet and triplet exciton lifetimes, respectively.
Förster energy transfer is from host singlet excitons to Ir singlet excitons and then to its triplet excitons. Dexter energy transfer is from host triplet excitons to Ir triplet excitons. The labels S and T are for first singlet excited state and first triplet excited state. The energy levels of Ir represent the energy levels of Ir-complex triplet excitons. 共b兲 shows the absorption and emission spectrum of the materials. 1 10 1 00 1 k 0 .1 1 H z 24K P A (ar b . u n its) P ure P F O -D M P B tpIr D F Ir Ir(m pp y)3 1 0 % 4 0 0 4 5 0 5 0 0 5 5 0 6 0 0 6 5 0 7 0 0 7 5 0 0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 Ir O O C H3 C H3 N 2 Q u ad ra tu re P h ase P L n m 4 0 H z 5 0 H z 8 0 H z 2 0 0 H z 1 0 % D F Ir:P F O -D M P
(b )
D B F -Ir 4 0 0 4 5 0 5 0 0 5 5 0 6 0 0 6 5 0 7 0 0 0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 Ir O O C H3 C H3 N S 2 4 0 H z 5 0 H z 7 0 H z Qu a d ra tu re P h a s e P L n m 1 0 % B tp Ir:P F O -D M P(c )
B tp Ir C8H17 n H17C8 CH3 CH3 H3C H3C PFO-DMP (a)FIG. 3. 共a兲 is the PA frequency dependence of pure PFO-DMP and Ir-complex-doped PFO-DMP films. The inset is the structure of PFO-DMP.共b兲 is the quadrature-phase channel PL spectrum of 10% DFIr:PFO-DMP film. The inset is the structure of DFIr.共c兲 is the quadrature-phase channel PL spectrum of 10% BtpIr:PFO-DMP film. The inset is the structure of BtpIr.
and the in-phase channel PL spectrum of the Ir共mppy兲3:PFO-DMP system are shown in Fig. 4共b兲. Al-though quadrature-phase channel PL spectra also show the delayed fluorescence coming from the triplet-triplet annihila-tion as in red Ir complexes, the magnitude of the green Ir共mppy兲3 emission is strongly frequency dependent, con-trary to the previous cases of BtpIr and DFIr. Indeed this green component is prominent only when the frequency is around 1 / 2T. This delayed extra Ir emission cannot come
from the delayed fluorescence through Förster energy trans-fer because of the apparent diftrans-ference in modulation re-sponses at a frequency far smaller than the inverse of both of their intrinsic lifetimes. Dexter energy transfer from the host triplet to guest triplet is the only possibility to explain these results.
Further proof comes from the temperature dependence. Since the delayed Ir emission comes from the host triplet
excitons, it must have a strong temperature dependence as the host triplet exciton lifetime decreases rapidly with rising temperature.4Figure4共c兲shows the delayed PL of Ir共mppy兲3
at three temperatures. It is obvious that when the temperature is increased the maximum is shifted to higher frequency, indicating that the delay is shortened at higher temperature. This provides strong evidence that the Ir emission is derived from the host triplet exciton. The uncertainty in the existence of triplet-to-triplet Dexter energy transfer in PLED doped with phosphor is therefore settled. Another host polyfluorene polymer PF-MPA is doped by Ir共mppy兲3. It is found that all general features are the same as PFO-DMP doped by Ir共mppy兲3. The Dexter energy transfer to Ir共mppy兲3is there-fore expected to be a general behavior for polyfluorene.
IV. MORPHOLOGY AND CONCENTRATION EFFECTS
In Fig. 5共a兲, PFO-DMP doped by 5% Ir共mppy兲3 is also measured in order to study the concentration effects. The PFO-DMP triplet exciton lifetime is reduced from 1.8 ms to 0.5 ms by Ir共mppy兲3doping at 100 K. Figure5共b兲 shows PA in-phase, PA quadrature-phase, and Ir共mppy兲3PL
1 1 0 1 0 0 1 k 1 0 k 0 . 0 1 0 . 1 1 N N H3C C H3 H3C I r N H z I r ( m p p y )3 P L q u a d r a t u r e p h a s e P A i n - p h a s e P A q u a d r a t u r e p h a s e I r ( m p p y )3 4 0 0 4 2 0 4 4 0 4 6 0 4 8 0 5 0 0 5 2 0 5 4 0 5 6 0 5 8 0 6 0 0 0 . 0 0 . 1 0 . 2 0 . 3 0 . 4 0 . 5 0 . 6 0 . 7 0 . 8 0 . 9 1 . 0 1 . 1 10 0K q ua dr at ur e-ph as e P L n m 2 0 0 H z 5 0 0 H z 1 k H z I n - p h a s e P L 1 0 % I r ( m p p y )3: P F O - D M P 1 0 1 0 0 1 k 1 0 k 0 . 0 5 . 0 x 1 0 - 7 1 . 0 x 1 0 - 6 1 . 5 x 1 0 - 6 2 . 0 x 1 0 - 6 2 . 5 x 1 0 - 6 3 . 0 x 1 0 - 6 3 . 5 x 1 0 - 6 4 . 0 x 1 0 - 6 1 0 % I r ( m p p y )3: P F O - D M P 2 4 K 1 0 0 K 53 5n m q ua dr at ur e-ph as e PL H z 2 4 K 1 0 0 K 1 5 0 K 1 5 0 K ( a ) ( b ) ( c ) 1 1 0 1 0 0 1 k 1 0 k 1 0 0 k 0 . 2 0 . 3 0 . 4 0 . 5 0 . 6 0 . 7 0 . 8 0 . 91 1 . 1 24 K P L (a rb . u ni t) H z I r ( m p p y )3 D B F I r B t p I r 100K (arb. units)
FIG. 4. 共a兲 shows the in-phase channel PA, quadrature-phase
channel PA and quadrature-phase channel PL
frequency-dependences of 10% Ir共mppy兲3:PFO-DMP films at 100 K. The inset is the structure of Ir共mppy兲3.共b兲 is the quadrature-phase channel PL spectra normalized at 439 nm at different modulation frequency and an in-phase PL spectrum. 共c兲 is the Ir共mppy兲3 emission peak quadrature-phase PL frequency dependences at three temperatures. The inset is the in-phase PL frequency dependences of three Ir complexes. 10m100m 1 10 100 1k 10k 100k 10-3 10-2 10-1 100 100K P A (ar b . u n it s ) Hz
5% Ir(mppy)3doped PFO-DMP
Pure PFO-DMP (a) 1 10 100 1k 10k 10-2 10-1 100 100K (ar b . u n it s ) Hz PA in-phase PA quadrature phase PFO PL quadrature phase Ir(mppy)3PL quadrature phase
(b) 400 420 440 460 480 500 520 540 560 580 600 0 1 2 3 4 100K q u ad ratu re-p h ase PL nm In-phase PL 200Hz 500Hz 700Hz 1kHz 5% 5%
FIG. 5. 共a兲 is the normalized PA at 100 K for 5%
Ir共mppy兲3-doped PFO-DMP and pure PFO-DMP. Inset is a 5%
Ir共mppy兲3-doped PFO-DMP quadrature-phase channel PL spectrum at different frequency and in-phase channel PL spectrum.共b兲 shows the Ir共mppy兲3 in-phase channel PA, Ir共mppy兲3 quadrature-phase channel PA, PFO-DMP PL quadrature-phase channel PL, and Ir共mppy兲3 quadrature-phase channel PL frequency dependences. The PFO-DMP PL quadrature-phase channel PL is almost fre-quency independent.
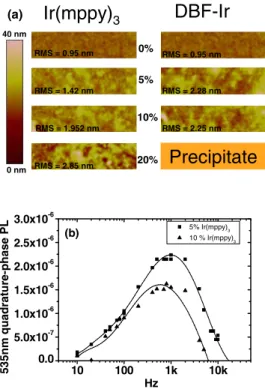
quadrature-phase frequency modulations, which are all simi-lar to 10% results and indicate that Dexter energy transfer exists for 5% as well. Atomic-force microscope 共AFM兲 is used to characterize the concentration dependence of the morphology. AFM images of two dopants with four different doping concentrations are shown in Fig.6共a兲. BtpIr results are not shown here because they have precipitations when the concentration is over 5% and the optical microscope im-age shows the film is very rough, indicating that the aggre-gation of BtpIr in PFO-DMP is so serious that the host/guest average distance is not short enough to make Dexter energy transfer happen. For Ir共mppy兲3, the root-mean-square 共rms兲 roughness increases with doping concentration and does not exceed 2 nm before 10%. When 5% DFIr is doped into PFO-DMP, the roughness immediately exceeds 2 nm, but the roughness is not changed when the concentration is 10%. This means that the DFIr morphology is less uniform than Ir共mppy兲3, and it might be the reason why Dexter energy transfer cannot be observed in DFIr doped PFO-DMP sys-tems. Twenty percent of the DFIr-doped PFO-DMP solution has precipitation, so the AFM image is not shown. Figure 6共b兲 shows the un-normalized Ir共mppy兲3 quadrature-phase PL intensity for 5% and 10% doping concentrations. Inter-estingly 5% has higher quadrature-phase PL intensity than 10%, implying that Dexter energy transfer from the PFO-DMP triplet state to Ir共mppy兲3 is more efficient at 5% dop-ing. Since the morphology of 5% is more uniform than 10% doping as shown in Fig. 6共a兲, Dexter energy transfer effi-ciency is significantly correlated to the film morphology.
V. ELECTROLUMINESCENCE
Figure7shows the device electroluminescence共EL兲 char-acteristics of 5% Ir共mppy兲3doped DMP and pure PFO-DMP. The device structure is ITO/PEDOT:PSS/polymer 共1000 Å兲/CsF共10 Å兲/Al 共1000 Å兲. PEDOT:PSS is the hole injection polymer poly共3,4-ethylenedioxythiophene兲 doped with polystyrene sulphonated acid. The device turn-on volt-age is increased enormously from 3 V to 10 V, apparently due to Ir共mppy兲3carrier trapping induced by Ir共mppy兲3. The device efficiency is very poor, indicating huge imbalance of electron and hole by the trapping. Although we prove that Dexter energy transfer exists in polyfluorene doped by Ir complex under optical excitation, this blend system is often dominated by the trapping mechanism in an organic phosphorescent EL device—for example, 4 , 4
⬘
-N , N⬘
-dicarbazole-biphenyl 共CBP兲 doped by fac tris共2-phenylpyridine兲iridium 关Ir共mppy兲3兴.6However, recently it isdemonstrated that the Ir共mppy兲3:CBP system can still use
10 100 1k 10k 0.0 5.0x10-7 1.0x10-6 1.5x10-6 2.0x10-6 2.5x10-6 3.0x10-6 5 35nm quadrat u re-phas e P L Hz 5% Ir(mppy)3 10 % Ir(mppy)3 (b)
Precipitate
Ir(mppy)
3DBF-Ir
RMS = 0.95 nm RMS = 1.42 nm RMS = 1.952 nm RMS = 2.85 nm RMS = 0.95 nm RMS = 2.28 nm RMS = 2.25 nm 0% 5% 10% 20% (a) 40 nm 0 nmFIG. 6. 共Color兲 共a兲 shows the AFM images for Ir共mppy兲3and DFIr with four different doping concentrations. 共b兲 is the un-normalized Ir共mppy兲3 quadrature-phase channel PL intensity at emission peak 535 nm. The 5% Ir共mppy兲3-doped PFO-DMP film has a better morphology than 10% Ir共mppy兲3, so that Dexter energy transfer is more efficient for 5% doping.
0 2 4 6 8 10 12 14 16 0 200 400 600 800 1000 1200 1400 1600 1800 2000 L u minescnece (C d /m 2) V 5 % Ir(m pp y)3 do ped PF O -D M P P ure P FO -D M P 5% P ure (b) 0 2 4 6 8 10 12 14 16 0 20 40 60 80 100 120 140 160 180 200 Cu rr en t d e n s it y ( m A/ cm 2 ) V 5 % Ir(m p py) 3 d o pe d P FO -DM P P u re PF O -D M P 5% P ure (a) 400 500 600 700 0.0 0.2 0.4 0.6 0.8 1.0 E L ( a rb . u n it ) nm Ir(mppy)3 (c)
FIG. 7. EL device characteristics for 5% Ir共mppy兲3-doped PFO-DMP and pure PFO-PFO-DMP.共a兲 is current density versus voltage. 共b兲 is luminescence versus voltage. 共c兲 is the 5% Ir共mppy兲3-doped PFO-DMP EL spectrum.
Dexter energy transfer to achieve high EL efficiency with proper multilayer structures.27 These works indicate that
Ir-doped polyfluorene also has the possibility to use Dexter energy transfer to achieve high EL efficiency if the morphol-ogy problems are overcome and a proper multilayer structure can be fabricated.28
VI. DISCUSSIONS AND RATE EQUATIONS The interesting remaining question is what causes the dif-ferent behaviors of green Ir共mppy兲3and the two red Ir com-plexes. The triplet exciton levels of the red-emitting DFIr and BtpIr are smaller than the PFO-DMP triplet exciton level as shown in Fig. 2共a兲. Despite the good triplet exciton confinement,23there is surprisingly no Dexter energy transfer
in these two cases. On the other hand, there is no共or poor兲 triplet exciton confinement for Ir共mppy兲3. Nevertheless, Dexter energy transfer does exist. This illustrates that the triplet exciton confinement is not the main condition for Dexter energy transfer in Ir-doped polyfluorene systems as previously believed.23The main differences between two red
Ir complexes and Ir共mppy兲3 are the aggregation and decay lifetime. BtpIr has serious aggregation even in good solvent THF. Although DFIr has fluorene ligands and good solubility,15 the morphology is not as uniform as Ir共mppy兲3 in polyfluorene. So Dexter energy transfer can only be ob-served in Ir共mppy兲3-doped systems, with a uniform distribu-tion of dopants. The average distance between the host exci-ton and the dopant is therefore much smaller for a given doping concentration. The second important factor is the fast triplet exciton lifetime for Ir共mppy兲3. From in-phase PL shown in the inset of Fig. 4共c兲, the lifetimes of BtpIr and DFIr are 16s共at 10 kHz兲 and 5s共at 30 kHz兲,25
respec-tively. On the other hand, the PL in-phase channel signals do not have a significant drop in our frequency range 共from 1 Hz to 100 kHz兲, indicating that Ir共mppy兲3 triplet lifetime is far below 1s.14With fast decay at the dopant, the triplet exciton can hardly come back to the host once transferred to the dopant. The dopant therefore becomes an efficient drain of the triplet energy than the host itself.
The general behaviors of Dexter energy transfer can be quantitatively modeled by the rate equations. Consider first the situation that the guest triplet exciton energy is higher than the host triplet exciton, presumably the case for the green Ir complex. Let the energy difference be⌬. Assume the host triplet exciton density is nTper host monomer and
the guest molecule density is ngper host monomer. nTand ng
are both dimensionless and ngis always smaller than 1. f is
the probability that the guest molecule is in its triplet excited state—i.e., that there is one triplet exciton on that molecule. Since there is no energy barrier for the guest triplet exciton to transfers back to the host, the number of triplet exciton transfer from guest to host per host monomer per unit time can be written as wgh= kDngf, where kDis the intrinsic Dexter
energy transfer rate. On the other hand, the Dexter energy transfer from the host to the guest has an energy barrier, so the transfer rate is whg= kDngnTe−⌬.is the inverse of
ther-mal energy kBT. T is temperature. In case of detailed balance
we have wgh= whg, which gives the Boltzmann distribution
f = nTe−⌬. Under a constant generation rate G of host triplet
exciton per host monomer due to intersystem crossing from the host singlet exciton, the rate equations are
n˙T= − kDe−⌬ngnT+ kDngf − kTnT+ G, 共1兲
f˙ng= kDe−⌬ngnT− kDngf − kgfng. 共2兲
Here kTand kgare the inverse lifetimes of the host and guest
triplet excitons, respectively. The nonlinear decay like triplet-triplet annihilation is not included here because our system is under weak CW pumping. Indeed, the PA signals are always proportional to the pumping intensity in all mea-surements. For any initially formed host triplet exciton, eventually there are two possibilities for its final decay: ei-ther through its own direct decay to the ground state or through the decay of the guest triplet state. By definition the latter belongs to the Dexter energy transfer of the blend sys-tem and it is the fraction of the decay which can be detected by the slow component in the PL which shows up in the quadrature phase. The Dexter fraction z is expressed as z =kgngf
kTnT. The steady-state solution of Eq. 共2兲 gives
z =kgng
kT
kDnge−⌬
kDng+ kgng
. 共3兲
In PFO/ Ir共mppy兲3 blend, the Dexter transfer rate kDng is at
most one one order of magnitude higher than the direct de-cay rate kTof the host triplet exciton as shown in Fig.3共a兲.
Since kTis about 10 s−1 at 100 K, kDng is at most 100 s−1.
On the other hand, the guest decay rate kg is as large as
106 s−1. Therefore, for n
g= 0.1 共10% doping兲, we always
have kgngⰇkDng in the denominator. As a result the Dexter
transfer fraction z is simplified to
z =kDng
kT
e−⌬. 共4兲
This form has transparent interpretations. The prefactor de-scribes the competition between direct decay共kT兲 and Dexter
energy transfer 共kDng兲. Because of the energy barrier for
Dexter transfer, the Boltzmann factor is also present. The result suggests that even without energy confinement some fraction of Dexter energy transfer is still possible if the host decay is very slow and the Dexter transfer is very fast. Our measurement can be used to give an estimate for the energy barrier⌬ as below. As discussed above kDng
kT is estimated to be about 10. The strength of the quadrature phase signal sug-gests that z is about 10−2 at 100 K. Equation 共4兲 then gives ⌬⯝0.07 eV. In other words, while triplet exciton confine-ment is not necessary for Dexter transfer, the triplet energy for the green Ir can only be slightly larger than the host triplet exciton. Since the Ir共mppy兲3 triplet exciton energy is 2.38 eV from its phosphorescence, the host triplet exciton energy is estimated to at least 2.3 eV. This implies that a previous extremely-low-temperature optical measurement of the PFO triplet exciton energy of 2.1 eV is about 0.2 eV too low.29 However, a room-temperature thermal measurement
gives a quite consistent value of 2.3 eV.30 Equation共4兲
tem-perature exponentially with a fixed ⌬. In Fig. 4共c兲 it does decrease as the temperature drops from 150 K to 24 K, sup-porting the assumption that energy is required in order to make the transfer. The decrease is, however, weaker than what Eq. 共4兲 would predict. The weak temperature depen-dence might result from the random distribution of both the host and guest triplet energies due to morphological disorder.31Because of the small barrier, it is likely that there
is some overlap between the host triplet and the low-energy tail of the guest triplet. Such an overlap will make the tem-perature dependence weaker. Moreover, the value of⌬ itself could be temperature dependent since the mean energies of the host and guest are expected to be different functions of temperature.
Let us then consider the situation that the guest triplet exciton energy is lower than the host by⌬, presumably for the case of the red Ir complexes. The rate equations become
n˙T= kDe−⌬ngf − kDngnT− kTnT+ G, 共5兲
f˙ng= − kDe−⌬ngf + kDngnT− kgfng. 共6兲
The Dexter transfer fraction z is defined as before. The steady-state solution for Eq.共6兲 gives
z =kDng kT 1 1 +kD kg e−⌬ . 共7兲
It now becomes clear that even with a favorable energy and high doping of ng⬃0.1, significant Dexter transfer
oc-curs only when the intrinsic Dexter transfer rate kD
over-whelms the direct decay of the host triplet exciton. The ab-sence of Dexter transfer for red Ir complexes suggests that kDng is smaller than 10−3 s−1 at low temperature. This
im-plies that Dexter transfer depends sensitively on the particu-lar molecuparticu-lar structures and aggregations. Even with the right energy, unfavorable structure or blend morphology could make the transition matrix element practically zero.
VII. CONCLUSION
In conclusion, PA and quadrature-phase channel PL have been employed to detect the Dexter energy transfer from polymer host to Ir guest in the polyfluorene blend systems. We observed Dexter energy transfer for green Ir共mppy兲3but not for red DFIr and red BtpIr guests. The film morphology is found to be important for the existence of Dexter energy transfer. A fast Ir共mppy兲3 triplet exciton lifetime is respon-sible for Dexter energy transfer without triplet confinement. Based on this result it is possibility to utilize Ir complexes to harvest triplet excitons even without carrier trapping or trip-let exciton confinement. In particular, a polymer host doped by blue phosphor with impossible triplet confinement could still be used for efficient blue PhPLEDX’s if the lifetime of phosphor is fast enough.
ACKNOWLEDGMENTS
This work was supported by the National Science Council and the Excellence Project of the Ministry of Education of the Republic of China. The authors also thank RiTEK Cor-poration for supplying ITO substrates.
*Corresponding author. Electronic address: [email protected] 1J. S. Wilson, A. S. Dhoot, A. J. A. B. Seeley, M. S. Khan, A.
Köhler, and R. H. Friend, Nature共London兲 413, 828 共2001兲. 2M. Wohlgenannt, K. Tandon, S. Mazumdar, S. Ramasesha, and Z.
V. Vardeny, Nature共London兲 409, 494 共2001兲.
3L. C. Lin, H. F. Meng, J. T. Shy, S. F. Horng, L. S. Yu, C. H. Chen, H. H. Liaw, C. C. Huang, K. Y. Peng, and S. A. Chen, Phys. Rev. Lett. 90, 036601共2003兲.
4H. H. Liao, H. F. Meng, S. F. Horng, J. T. Shy, K. Chen, and C. S. Hsu, Phys. Rev. B 72, 113203共2005兲.
5M. K. Lee, M. Segal, Z. G. Soos, J. Shinar, and M. A. Baldo, Phys. Rev. Lett. 94, 137403共2005兲.
6M. A. Baldo, D. F. O’Brien, M. E. Thompson, and S. R. Forrest, Phys. Rev. B 60, 14422共1999兲.
7M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson, and S. R. Forrest, Nature 共London兲 395, 151 共1998兲.
8M. A. Baldo, M. E. Thompson, and S. R. Forrest, Nature 共Lon-don兲 403, 750 共2000兲.
9C. Adachi, M. A. Baldo, M. E. Thompson, and S. R. Forrest, J. Appl. Phys. 90, 5048共2001兲.
10X. Gong, J. C. Ostrowski, G. C. Bazan, D. Moses, and A. J. Heeger, Appl. Phys. Lett. 81, 3711共2002兲.
11X. H. Yang and D. Neher, Appl. Phys. Lett. 84, 2476共2004兲. 12S. A. Choulis, V. E. Choong, M. K. Mathai, and F. So, Appl.
Phys. Lett. 87, 113503共2005兲.
13V. Cleave, G. Yahioglu, P. Le Barny, R. H. Friend, and N. Tessler, Adv. Mater.共Weinheim, Ger.兲 11, 285 共1999兲.
14M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson, and S. R. Forrest, Appl. Phys. Lett. 75, 4共1999兲.
15X. Gong, J. C. Ostrowski, D. Moses, G. C. Bazan, and A. J. Heeger, Adv. Funct. Mater. 13, 439共2003兲.
16X. Gong, J. C. Ostrowski, D. Moses, G. C. Bazan, and A. J. Heeger, J. Polym. Sci., Part B: Polym. Phys. 41, 2691共2003兲. 17M. A. Baldo and S. R. Forrest, Phys. Rev. B 62, 10958共2000兲. 18I. H. Campbell, D. L. Smith, S. Tretiak, R. L. Martin, C. J. Neef,
and J. P. Ferraris, Phys. Rev. B 65, 085210共2002兲.
19P. A. Lane, L. C. Palilis, D. F. O’Brien, C. Giebeler, A. J. Cadby, D. G. Lidzey, A. J. Campbell, W. Blau, and D. D. C. Bradley, Phys. Rev. B 63, 235206共2001兲.
20Y. Y. Noh, C. L. Lee, J. J. Kim, and K. Yase, J. Chem. Phys. 118, 2853共2003兲.
21M. Wohlgenannt and Z. V. Vardeny, Synth. Met. 125, 55共2002兲. 22A. S. Dhoot and N. C. Greenham, Adv. Mater.共Weinheim, Ger.兲
14, 1834共2002兲.
23F. C. Chen, G. He, and Y. Yang, Appl. Phys. Lett. 82, 1006 共2003兲.
24X. Gong, W. Ma, J. C. Ostrowski, G. C. Bazan, D. Moses, and A. J. Heeger, Adv. Mater.共Weinheim, Ger.兲 16, 615 共2004兲. 25C. Adachi, M. A. Baldo, and S. R. Forrest, S. Lamansky, M. E.
Thompson, and R. C. Kwong, Appl. Phys. Lett. 78, 1622 共2001兲.
26C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson, and S. R. Forrest, Appl. Phys. Lett. 79, 2082共2001兲.
27Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson, and S. R. Forrest, Nature共London兲 440, 908 共2006兲.
28S. R. Tseng, S. C. Lin, H. F. Meng, H. H. Liao, C. H. Yeh, H. C.
Lai, S. F. Horng, and C. S. Hsu, Appl. Phys. Lett. 88, 163501 共2006兲.
29C. Rothe and A. P. Monkman, Phys. Rev. B 68, 075208共2003兲. 30A. P. Monkman, H. D. Burrows, L. J. Hartwell, L. E. Horsburgh, I. Hamblett, and S. Navaratnam, Phys. Rev. Lett. 86, 1358 共2001兲.
31V. I. Arkhipov, P. Heremans, E. V. Emelianova, and H. Bassler, Phys. Rev. B 71, 045214共2005兲.