On: 27 April 2014, At: 23:33 Publisher: Taylor & Francis
Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
Liquid Crystals
Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/tlct20
Synthesis and characterization of
halogen-containing ferroelectric liquid crystals and side
chain liquid crystalline polymers
Ging-Ho Hsiue a , Yi-An Sha a , Shih-Jung Hsieh ac , Ru-Jong Jeng b & Wen-Jang Kuo a
a
Department of Chemical Engineering, National Tsing Hua University, Hsinchu, Taiwan 300, ROC
b
Institute of Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan 300, ROC
c
Department of Chemical Engineering, National Chung Hsing University, Taichung, Taiwan 402, ROC
Published online: 06 Aug 2010.
To cite this article: Ging-Ho Hsiue , Yi-An Sha , Shih-Jung Hsieh , Ru-Jong Jeng & Wen-Jang Kuo (2001) Synthesis and characterization of halogen-containing ferroelectric liquid crystals and side chain liquid crystalline polymers, Liquid Crystals, 28:3, 365-374, DOI: 10.1080/02678290010015324
To link to this article: http://dx.doi.org/10.1080/02678290010015324
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) contained in the publications on our platform. However, Taylor & Francis, our agents, and our licensors make no representations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of the Content. Any opinions and views expressed in this publication are the opinions and views of the authors, and are not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon and should be independently verified with primary sources of information. Taylor and Francis shall not be liable for any losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoever or howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use of the Content.
This article may be used for research, teaching, and private study purposes. Any substantial or
systematic reproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in any form to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http://www.tandfonline.com/page/terms-and-conditions
Liquid Crystals, 2001, Vol. 28, No. 3, 365± 374
Synthesis and characterization of halogen-containing ferroelectric
liquid crystals and side chain liquid crystalline polymers
GING-HO HSIUE*, YI-AN SHA, SHIH-JUNG HSIEH†, RU-JONG JENG‡ and WEN-JANG KUO
Department of Chemical Engineering, National Tsing Hua University, Hsinchu, Taiwan 300, ROC
† Institute of Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan 300, ROC
‡Department of Chemical Engineering, National Chung Hsing University, Taichung, Taiwan 402, ROC
(Received 6 June 2000; accepted 30 August 2000)
A new series of ferroelectric liquid crystals and side chain liquid crystalline polymers based on halogen-containing chiral centres has been synthesized. Chemical structures were analysed by NMR. Liquid crystal phases were characterized by diŒerential scanning calorimetry, optical polarizing microscopy, and X-ray diŒractometry. The behaviour of the liquid crystalline phases was investigated as a function of spacer units and diŒering terminal asymmetric moieties. It was found that phase transition temperatures decreased with increasing length of the oligooxyethylene spacer unit. DiŒering terminal asymmetric moieties led to diŒering mesophase phenomena. Furthermore, a wide temperature range (including room temperature) of a chiral smectic C phase was achieved.
1. Introduction unit favours a reduction in the phase transition temper-atures. As the number of oxyethylene units increases, During the past decade, ferroelectric liquid crystals
the transition temperature decreases [11]. The chiral (FLCs) have been extensively studied because of their
centre is usually the major part of the terminal chain fast response time and memory eŒect toward an applied
because the Psvalue is aŒected by the asymmetric atom electric eld [1–6]. These characteristics make them
and its position, and the length of the terminal chain suitable for electro-optical applications. Prompted by
unit. In addition, incorporation of a polar group (F, Cl, the development of surface stabilized ferroelectric liquid
Br, CN, CF3, etc.) onto the chiral centre close to the crystal (SSFLC) display technology [7], FLC materials
core can maximize the Psvalue [12, 13]. and SSFLC cells have been vigorously investigated [8].
In our previous work, a series of chlorine-containing For display use, FLC materials have mostly been
FLCs was synthesized and characterized [14]. These designed to provide a wide temperature range of the
monomers have a wide temperature range of the chiral FLC phase, including room temperature [9], and a
smectic C phase—including room temperature; their large value of the spontaneous polarization (Ps). These
chemical structures are shown below: properties are in uenced by the design of the molecular
structures of FLC mesogens. The fundamental molecular structure of an FLC includes a mesogenic group, a spacer chain unit, and a chiral centre unit (terminal chain
In this paper, a new series of ferroelectric liquid units). The mesogenic group often consists of at least
crystals and side chain liquid crystalline polymers is two linked rigid groups with lateral substituents [10].
reported. These liquid crystalline materials consist of The length and chemical structure of the spacer unit
various halogen-containin g chiral moieties (F, Cl, and appear to be important factors in LFC phase formation
Br), oligooxyethylen e spacers, and ester core units. The and temperature range. The oxyethylene group as spacer in uences of the varied halogen-containin g chiral tails and spacer units on mesophase formation are also discussed.
*Author for correspondence; e-mail: [email protected]
L iquid Crystals ISSN 0267-829 2 print/ISSN 1366-585 5 online © 2001 Taylor & Francis Ltd
http://www.tandf.co.uk/journals DOI: 10.1080/02678290010015324
2. Experimental heating and second cooling scans. A Nikon Micro-photo-Fx polarizing optical microscope (POM) equipped 2.1. Materials
Allyl bromide, 2-chloroethanol , 2-(2-chloroethoxy) - with a Mettler FP82 hot stage was used to observe the thermal transitions and anisotropic textures. X-ray ethanol, 2-[2-(2-chloroethoxy )ethoxy]ethanol, hydrogen
uoride–pyridine, tri uoromethanesulfonic anhydride, diŒraction (XRD) measurements were performed with a Rigaku R-axis IIC powder diŒractometer. A mono-tetrabutylammoniu m uoride hydrate, and hydrogen
hexachloroplatinat e(IV) hydrate were purchased from chromatized X-ray beam from nickel- ltered CuK a radiation with a wavelength of 0.15406 nm was used. A Adrich Chemical Co. 4,4¾ -Dihydroxybiphenyl, N,N¾
-dicyclohexylcarbodiimide (DCC), 4,4¾ -biphenol, l-iso- thermal controller was added to the X-ray system for thermal measurement with a tolerance of Ô 0.5ßC. leucine, sodium nitrite, and pyridine were purchased from
Tokyo Chemical Industry Co. Ltd.
4-Dimethylamino-pyridine (DMAP) and benzophenone were purchased 2.3. Synthesis of monomers
The syntheses of the liquid crystalline monomers are from Lancaster Chemicals Ltd;
poly(methylhydrogen-siloxane) ([g]5 30) was purchased from United Chemical outlined in schemes 1–3. Technologies, Inc; hexane, ethyl acetate, dichloromethane ,
acetonitrile, and methanol were purchased from TEDIA. 2.3.1. 4-(2-Allyloxyethox y)benzoic acid (S-1);
4-[2-(2-ally loxyethoxy)ethoxy]benzoi c acid (S-2);
All these chemicals were used as received.
Tetrahydro-furan and toluene purchased from TEDIA were distilled 4-{2-[2-(2-allyloxyethox y)ethoxy]ethox y}benzoic acid (S-3)
over sodium using benzophenone as indicator under a
nitrogen atmosphere. The syntheses of these compounds via esteri cation
and etheri cation reactions have been reported previously [14].
2.2. Characterization methods
1H NMR and 19F NMR spectra were obtained on
Bruker AM-400 NMR or Jeol JNM-FX100 spectro- 2.3.2. (2S,3S)-2-Fluoro-3-methylpentanoi c acid (Fs-1)
In a 500 ml round-bottom ask, hydrogen uoride-meters. Transition temperatures were determined as the
maxima of endothermic or exothermic peaks using a pyridine (100 g) was added dropwise to pyridine (60 ml ) under N2 at 0ßC; after 10 min, l-isoleucine (7.87 g, Seiko SSC 5200 diŒerential scanning calorimeter (DSC);
heating and cooling rates were 10ßC minÕ 1. The transition 60 mmol ) was added to the reaction mixture. Sodium nitrite powder (6.21 g, 90 mmol) was then added under temperatures were speci cally obtained from the rst
Scheme 1. Synthesis of monomers
MDn12Fs (n5 1, 2, 3).
367
Halogen-containin g FL Cs
Scheme 2. Synthesis of monomer MD312Fr.
N2 at 0ß C. After stirring the reaction mixture for 5 h, after evaporating the solvent, a dark yellow oily pro-cooling was removed, and the mixture held at room duct was obtained; yield 80%. 1H NMR (DMSO-d
6): temperature for 24 h. The reaction mixture was then d5 0.84 (t, 3H, CH2 CH3), 0.87 (d, 3H, CH(CH3) ), added to 500 ml of water and the mixture was extracted 1.02–1.29 and 1.29–1.53 (m, 2H, CH2CH3), 1.53–1.57 with ethyl ether. The organic phase solvent was evaporate d (m, 1H, CHCH2), 3.79 (m, 1H, CH(OH) ).
and a colourless oily product was nally obtained after distillation (78–80ßC, 4 mm Hg); yield 27.6%.1H NMR 2.3.4. Ethyl (2S,3S)-2-hydroxy-3-methylpentanoat e (DMSO-d6): d5 0.97 (t, 3H, CH3 CH2 ), 1.12 (d, 3H, (Fr-2) CH(CH3) ), 1.44 and 1.63 (m, 2H, CH3CH2 ), 2.17 A mixture of 39.5 g (299 mmol ) of Fr-1, 7.5 ml of (m, 1H, CH(CH3) ), 4.93 (d, 1H, CH(F) ).19F NMR
H2SO4 and 300 ml of anhydrous ethanol was stirred (DMSO-d6): d5 Õ 243.42 (s, CH(F)).
under re ux for 24 h; ethanol was then distilled oŒ. The reaction mixture was extracted with ethyl ether after 2.3.3. (2S,3S)-2-Hydroxy-3-methylpentanoi c acid (Fr-1)
saturated with an aqueous solution of NaHCO3. The l-isoleucine (49.5 g, 377 mmol ) was added to 2.67M
organic phase solvent was evaporated oŒ, and a colour-H2SO4 aqueous solution (586 ml) at 0ßC under N2.
less oily product obtained by distillation (62–65ß C, Sodium nitrite (39.4 g, 570 mmol) dissolved in water
6 mm Hg). 1H NMR (DMSO-d
6): d5 0.91 (t, 3H, (150 ml ) was added dropwise under N2 at 0ß C and the
CH2CH3), 0.99 (d, 3H, CHCH3 C2H5), 1.13–1.53 mixture stirred at room temperature overnight. The
reaction mixture was then extracted with ethyl ether; (m, 2H, CH2CH3), 1.31 (t, 3H, COOCH2CH3),
Scheme 3. Synthesis of monomers
MDn12Bs (n5 1, 2, 3).
1.73–1.93 (m, 1H, CH(CH3) ), 2.78 (br, s, 1H, graphy on silica gel with ethyl acetate and hexane as eluant. A colourless product was obtained by distil-CH(OH) ), 4.08 (d, 1H, CH(OH) ), 4.26 (q, 2H,
COOCH2CH3). lation (59–63ßC, 9 mm Hg); yield 81.9%. 1H NMR
(DMSO-d6): d5 0.95 (t, 3H, CH2CH3), 0.96 (d, 3H, CHCH3 C2H5), 1.19–1.65 (m, 2H, CH2CH3), 1.32 2.3.5. Ethyl (2S,3S)-2-(tri
uoromethyl)sulfonyloxy-3-methylpentanoat e (Fr-3) (t, 3H, COOCH2CH3), 1.75–2.12 (m, 1H, CHCH3 ),
4.28 (q, 2H, COOCH2CH3), 5.01 (d, 1H, CHF ). In a 500 ml polyproplene reactor, 34.5 g (325 mmol)
of Fr-2, 200 ml of anhydrous dichloromethane and 19F NMR (DMSO-d
6): d5 Õ 251.26 ( CHF ). 18.6 ml of anhydrous pyridine was stirred under N2 at
0ß C. Tri uoromethylsulfonic anhydride (50 g, 177 mmol)
2.3.7. (2R,3S)-2-Fluoro-3-methylpentanoi c acid (Fr-5)
was added dropwise. After 2 h the reaction mixture was
A mixture of 16.7 g (103 mmol ) of Fr-4, 6 ml of 50 wt % added to 500 ml of water and the mixture extracted
aqueous sodium hydroxide, 20 ml of ethanol, and 200 ml twice with dichloromethane. The organic phase was
of water was stirred under re ux for 24 h; ethanol was extracted with a 10 wt % aqueous solution of HCl, and
then distilled oŒ. After cooling at 0ßC, the reaction the solvent evaporated to yield a colourless product
mixture was acidi ed with 5N hydrochloric acid and after distillation (55–61ßC, 0.4 mm Hg); yield 85.4%.
extracted with ethyl acetate. The organic phase solvent 1H NMR (DMSO-d6): d5 0.95 (t, 3H, CH2CH3), 1.07
was evaporated oŒ, and a colourless oily product (d, 3H, CHCH3 C2H5), 1.19–1.64 (m, 2H, CH2CH3),
obtained by distillation (78–80ßC, 4 mm Hg); yield 76%. 1.33 (t, 3H, COOCH2CH3), 2.05–2.27 (m, 1H, 1 H NMR (DMSO-d6): d5 0.89 (t, 3H, CH3CH2 ), CHCH3 ), 4.31 (q, 2H, COOCH2CH3), 5.01 (d, 1H, 0.97 (d, 3H, CH(CH3) ), 1.06–1.32, 1.32–1.56 (m, 2H, CH(OSO2CF3) ). CH3CH2 ), 1.71–2.13 (m, 1H, CH(CH3) ), 5.01 (d, 1H, CHF ). 19F NMR (DMSO-d 6): d5 Õ 251.26 2.3.6. Ethyl (2R,3S)-2- uoro-3-methylpentanoat e (Fr-4) ( CH(F) ). A solution of tetrabutylammoniu m uoride hydrate
(40 g) in 150 ml acetonitrile was added dropwise to a
solution of Fr-3 (36.7 g, 133 mmol) in 150 ml aceto- 2.3.8. (2S,3S)-2-Bromo-3-methylpentanoi c acid (Bs-1)
l-isoleucine (7.9 g, 60 mmol ) and 75 ml 6N HBr were nitrile at 80ßC under N2. After stirring the reaction
mixture under re ux for 3 h, the acetonitrile was distilled placed in a 500 ml round-bottom ask equipped with a magnetic stirrer at 0ßC. Sodium nitrite (6.21 g, 90 mmol ) oŒ. The crude product was puri ed by ash
369
Halogen-containin g FL Cs
aqueous solution was added dropwise below 5ßC under (Bs-2) 75.5. (Fs-2) 1H NMR (CDCl3): d5 0.97 (t, 3H, CH3CH2 ), 1.12 (d, 3H, CH(CH3) ), 1.44 and 1.63 N2. After stirring the reaction mixture for 5 h, cooling
was removed, and the mixture held at room temperature (m, 2H, CH3CH2 ), 2.17 (m, 1H, CH(CH3) ), 4.93 (d, 1H, CHF ), 6.89, 7.15, 7.42 and 7.52 (4d, 8H, for 24 h; it was then extracted with ethyl ether. The
organic phase solvent was evaporated, and a red oily aromatic protons) ; 19F NMR (CDCl3): d5 Õ 243.42 ( CHF ). (Fr-6) 1H NMR (CDCl3): d5 0.97 (t, 3H, product obtained by distillation (140ßC, 20 mm Hg);
yield 73%. 1H NMR (DMSO-d6): d5 0.96 (t, 3H, CH3CH2 ), 1.12 (d, 3H, CH(CH3) ), 1.44 and 1.63 (m, 2H, CH3CH2 ), 2.17 (m, 1H, CH(CH3) ), 5.05 CH2CH3), 1.13 (d, 3H, CHCH3 ), 1.39 and 1.83 (m, 2H, CH2CH3), 2.18 (m, 1H, CHCH3), 4.09 (d, 1H, (d, 1H, CHF ), 6.87, 7.12, 7.39 and 7.52 (4d, 8H, aromatic protons) ; 19F NMR (CDCl3): d5 Õ 251.26 CHBr ), 11.8 ( br, COOH). ( CHF ). (Bs-2) 1H NMR (CDCl3): d5 0.96 (t, 3H, CH2CH3), 1.13 (d, 3H, CHCH3 ), 1.39 and 1.83 2.3.9. 4,4¾ -Dihydroxybipheny l (2S,3S)-2- uoro-3-methyl-pentanoate (Fs-2); 4,4¾ -dihydroxybiphenyl (m, 2H, CH2CH3), 2.18 (m, 1H, CHCH3), 4.09 (d, 1H, CHBr ), 6.89, 7.15, 7.42 and 7.52 (4d, 8H, aromatic (2R,3S)-2- uoro-3-methylpentanoat e (Fr-6);
4,4¾ -dihydroxybipheny l (2S,3S)-2-bromo- protons).
3-methylpentanoat e (Bs-2)
These compounds were synthesized by similar 2.3.10. Synthesis of FL C monomers MDn12Fs, MDn12Fr, and MDn12Bs
methods. As an example, the synthesis of 4,4¾
-dihydroxy-biphenyl (2S,3S)-3- uoro-3-methylpentanoat e (Fs-2) is These products were synthesized, respectively, in the same manner as compounds Fs-2, Fr-6, or Bs-2;1H NMR described as follows. In a 100 ml round-bottom ask,
(2S,3S)-2- uoro-3-methyl pentanoic acid (3 g, 22.3 mmol), spectra are listed in table 1.
MDn12Fs stands for the following series: 4¾
-[(2S,3S)-4,4¾ -dihydroxybipheny l (11 g, 59 mmol ),
dimethylamino-pyridine (0.5 g, 4 mmol),N,N¾ -dicyclohexylcarbodiimid e 2- uoro-3-methylpe ntanoyloxy]-4-biphenyl 4-(2-allyloxy-ethoxy)benzoate (MD112Fs); 4¾ -[(2S,3S)-2-
uoro-3-(4.1 g, 20 mmol ), and dried THF (50 ml ) were stirred
under N2 at 0ß C overnight. The solution was ltered methylpentanoyl oxy]-4-biphenyl 4-[2-(2-allyl-oxyetho xy)-ethoxy]benzoate (MD212Fs); 4¾ - [( 2S,3S)-2-
uoro-and the ltrate evaporated. The product was puri ed
by chromatograph y on silica gel with ethyl acetate 3-methylpentanoylox y]-4-biphenyl 4-{2-[2-(2-allyloxy-ethoxy)ethoxy]ethoxy}benzoat e (MD312Fs).
and hexane as eluant. Yield (Fs-2) 70.5%, (Fr-6) 69.7,
Table 1. Chemical shift and optical rotation values of compounds MDn12Fs, MD312Fr and MDn12Bs.
Compound [a]25
d a NMR spectrab
MD112Fs Õ 9.789 1H NMR: 0.97 (t, 3H, CH
3CH2 ), 1.12 (d, 3H, CH(CH3) ), 1.44 and 1.63 (m, 2H, CH3CH2 ), 2.17 (m, 1H, CH(CH3) ), 4.93 (dd, 1H, CHF ), 3.81–4.20 (m, 6H, CH2 (OCH2CH2) ), 5.23 and 5.9 (m, 3H, CH2 CH ), 6.9–8.15 (6d, 12 aromatic protons).19F NMR:Õ 243.42 ( CHF ). MD212Fs Õ 8.366 1H NMR: 0.98 (t, 3H, CH
3CH2 ), 1.12 (d, 3H, CH(CH3) ), 1.44 and 1.65 (m, 2H, CH3CH2 ), 2.17 (m, 1H, CH(CH3) ), 4.93 (dd, 1H, CHF ), 3.61–4.22 (m, 10H, CH2 (OCH2CH2)2 ), 5.23 and 5.9 (m, 3H, CH2 CH ), 6.9–8.15 (6d, 12 aromatic protons).19F NMR:Õ 243.42 ( CHF ). MD312Fs Õ 8.647 1H NMR: 0.99 (t, 3H, CH
3CH2 ), 1.12 (d, 3H, CH(CH3) ), 1.44 and 1.67 (m, 2H, CH3CH2 ), 2.19 (m, 1H, CH(CH3) ), 4.93 (dd, 1H, CHF ), 3.58–4.22 (m, 14H, CH2 (OCH2CH2)3 ), 5.23 and 5.9 (m, 3H, CH2 CH ), 6.9–8.15 (6d, 12 aromatic protons).19F NMR:Õ 243.42 ( CHF ). MD312Fr Õ 6.17 1H NMR: 0.99 (t, 3H, CH
3CH2 ), 1.09 (d, 3H, CH(CH3) ), 1.44 and 1.63 (m, 2H, CH3CH2 ), 2.20 (m, 1H, CH(CH3) ), 5.05 (dd, 1H, CHF ), 3.58–4.22 (m, 14H, CH2 (OCH2CH2)3 ), 5.23 and 5.9 (m, 3H, CH2 CH ), 6.98–8.15 (6d, 12 aromatic protons).19F NMR:Õ 251.26 ( CHF ). MD112Bs Õ 10.042 1H NMR: 0.95 (t, 3H, CH
2CH3), 1.12 (d, 3H, CHCH3 ), 1.38 and 1.68 (m, 2H, CH2CH3), 2.18 (m, 1H, CHCH3), 4.21 (dd, 1H, CHBr ), 3.81–4.20 (m, 6H, CH2 (OCH2CH2) ), 5.23 and 5.9 (m, 3H, CH2 CH ), 6.9–8.16 (6d, 14 aromatic protons).
MD212Bs Õ 15.187 1H NMR: 0.95 (t, 3H, CH2CH3), 1.12 (d, 3H, CHCH3 ), 1.38 and 1.68 (m, 2H, CH2CH3), 2.18 (m, 1H, CHCH3), 4.21 (dd, 1H, CHBr ), 3.81–4.32 (m, 10H, CH2 (OCH2CH2)2 ), 5.23 and 5.9 (m, 3H, CH2 CH ), 6.9–8.16 (6d, 12 aromatic protons).
MD312Bs Õ 7.338 1H NMR: 0.95 (t, 3H, CH2CH3), 1.12 (d, 3H, CHCH3 ), 1.38 and 1.68 (m, 2H, CH2CH3), 2.18 (m, 1H, CHCH3), 4.21 (dd, 1H, CHBr ), 3.81–4.31 (m, 14H, CH2 (OCH2CH2)3 ), 5.23 and 5.9 (m, 3H, CH2 CH ), 6.9–8.16 (6d, 12 aromatic protons).
aThese values were measured in CHCl
3at 25ßC. bThese values were measured in CDCl
3, using 400 MHz NMR spectroscopy (internal standard tetramethylsilane).
MD312Fr stands for the following compound: chiral centre of MDn12Bs. The chemical structures of
compounds MDn12Fs and MD312Fr were also
identi-4¾ -[(2R,3S)-2- uoro-3-methylpentanoylox y]-4-biphenyl
4 - {2 - [2 - ( 2 - allyloxyethoxy)ethoxy]ethoxy}benzoate ed by 19F NMR. In the19F NMR spectra, the uorine chemical shifts appeared at Õ 243.49 and Õ 251.24 ppm (MD312Fr).
MDn12Bs stands for the following series: 4¾ -[(2S,3S)- for MDn12Fs and MD312Fr, respectively. Optical
rotations, [a]5
D, of these monomers are summarized 2-bromo-3-methylpentanoyloxy ]-4-biphenyl
4-(2-allyl-oxy-ethoxy)benzoate (MD112Bs); 4¾ -[(2S,3S)-2-bromo- in table 1.
The FLCPs were synthesized by hydrosilation reactions 3-methylpentanoylox y]-4-biphenyl 4-[2-(2- allyl-
oxy-ethoxy)ethoxy]benzoate (MD212Bs); 4¾ -[(2S,3S)-2-bromo- in which 10 mol % excess of the LFC monomers was employed to react with the Si–H groups on the poly-3-methylpentanoyloxy [-4-biphenyl
4-{2-[2-(2-allyl-oxy-ethoxy)ethoxy]ethoxy}benzoat e (MD312Bs). (methylhydrogens iloxane) backbone. Chemical structures of the FLCPs were characterized by 1H NMR spectra in which the Si–H peak (4.7 ppm) and vinyl protons
2.4. Synthesis of FL CPs, PS312Fr, PS312Fs, PS312C
and PS312Bs of the CH2 CH group appearing between 5.23 and
5.90 ppm vanished after reaction. This con rmed that The structures of the synthesized liquid crystalline
poly-siloxanes are shown in scheme 4. They were synthesized complete reaction between FLC monomers and Si–H groups had taken place.
by similar methods; as an example, the procedure for PS312Bs is described below.
FLC monomer, MD312Bs (0.5 g, 10 mol % excess 3.2. T hermal properties
Phase sequences and their corresponding transition versus the Si–H groups present in the polysiloxane), was
dissolved in 50 ml of freshly distilled toluene together temperatures for compounds MDn12Fs, MD312Fr,
MDn12C, and MDn12Bs are shown in table 2.
with the appropriate amount of poly(methylhydrogen-siloxane). This solution was heated to re ux under N2.
Hydrogen hexachloroplatinat e(IV) hydrate (100 mg) in 3.2.1. MDn12Fs series
There are three compounds in this series; they con-dry THF was then injected via a syringe, and the
solution heated at re ux under N2 for 24 h. After this sist of the (2S,3S)-2- uoro-3-methylpentanoylox y chiral
moiety and an oxyethylene spacer unit. MD112Fs and reaction time, 1H NMR analysis indicated that the
hydrosilation reaction was completed. The solution was MD212Fs exhibited similar liquid crystal behaviours with a cholesteric–chiral smectic C–chiral smectic F sequence. evaporated under reduced pressure to give crude yellow
powder; this product was further puri ed by precipitation No chiral smectic F phase was observed in MD312Fs. The phase assignment was made by POM and XRD. with methanol, and dried under vacuum.
An optical polarized micrograph reveals an oily streak texture at 80ßC which corresponds to a cholesteric
3. Results and discussion
3.1. Synthesis structure. Figure 1 presents the temperature-dependen t
XRD diagrams obtained from a powder sample of The synthetic routes for FLCs and FLCPs are
out-lined in schemes 1–4. Chemical structures of the com- MD312Fs at 30, 40, 50, 60, and 70ßC. Upon further cooling from the cholesteric phase, a sharp low angle pounds were identi ed by 1H NMR and 19F NMR. In
the 1H NMR spectra, the chemical shift appearing at re ection (associated with the smectic layer) and a broad wide angle re ection (associated with lateral packings) 4.9 ppm can be associated with the CHF proton of
the chiral centre of MDn12Fs. The peak appearing were observed. Curve A exhibits a diŒused re ection at 4.81 AÃ and a very weak re ection at 28.26 AÃ , which at 5.1 ppm can be assigned to the CHF proton of
the chiral centre of MD312Fr. The peak appearing at correspond to smectic layers. Moreover, the d-spacing
of the rst order re ection reduces from 28.36 to 27.26 AÃ 4.21 ppm can be assigned to the CHBr proton of the
Scheme 4. The polysiloxane series PS312Fr, PS312Fs, PS312C and PS312Bs.
371
Halogen-containin g FL Cs
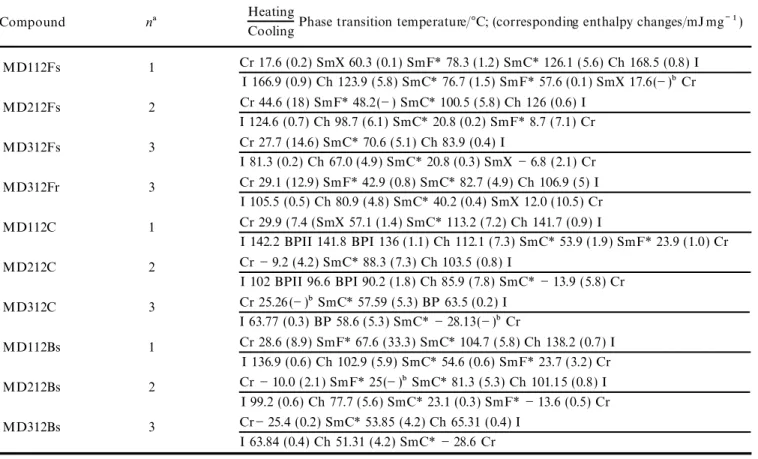
Table 2. Phase transitions and phase transition enthalpies for compounds MDn12Fs, MD312Fr, MDn12C and MDn12Bs:
Cr5 crystalline phase, SmX5 high order smectic phase, SmF*5 chiral smectic F phase, SmC*5 chiral smectic C phase, Ch5 cholesteric phase, I5 isotropic phase.
Heating
CoolingPhase transition temperature/ßC; (corresponding enthalpy changes/mJ mgÕ 1)
Compound na MD112Fs 1 Cr 17.6 (0.2) SmX 60.3 (0.1) SmF* 78.3 (1.2) SmC* 126.1 (5.6) Ch 168.5 (0.8) I I 166.9 (0.9) Ch 123.9 (5.8) SmC* 76.7 (1.5) SmF* 57.6 (0.1) SmX 17.6(Õ )bCr MD212Fs 2 Cr 44.6 (18) SmF* 48.2(Õ ) SmC* 100.5 (5.8) Ch 126 (0.6) I I 124.6 (0.7) Ch 98.7 (6.1) SmC* 20.8 (0.2) SmF* 8.7 (7.1) Cr MD312Fs 3 Cr 27.7 (14.6) SmC* 70.6 (5.1) Ch 83.9 (0.4) I I 81.3 (0.2) Ch 67.0 (4.9) SmC* 20.8 (0.3) SmXÕ 6.8 (2.1) Cr MD312Fr 3 Cr 29.1 (12.9) SmF* 42.9 (0.8) SmC* 82.7 (4.9) Ch 106.9 (5) I I 105.5 (0.5) Ch 80.9 (4.8) SmC* 40.2 (0.4) SmX 12.0 (10.5) Cr MD112C 1 Cr 29.9 (7.4 (SmX 57.1 (1.4) SmC* 113.2 (7.2) Ch 141.7 (0.9) I I 142.2 BPII 141.8 BPI 136 (1.1) Ch 112.1 (7.3) SmC* 53.9 (1.9) SmF* 23.9 (1.0) Cr MD212C 2 CrÕ 9.2 (4.2) SmC* 88.3 (7.3) Ch 103.5 (0.8) I I 102 BPII 96.6 BPI 90.2 (1.8) Ch 85.9 (7.8) SmC*Õ 13.9 (5.8) Cr MD312C 3 Cr 25.26 (Õ )bSmC* 57.59 (5.3) BP 63.5 (0.2) I I 63.77 (0.3) BP 58.6 (5.3) SmC*Õ 28.13(Õ )bCr MD112Bs 1 Cr 28.6 (8.9) SmF* 67.6 (33.3) SmC* 104.7 ( 5.8) Ch 138.2 (0.7) I I 136.9 (0.6) Ch 102.9 (5.9) SmC* 54.6 (0.6) SmF* 23.7 (3.2) Cr MD212Bs 2 CrÕ 10.0 (2.1) SmF* 25(Õ )bSmC* 81.3 (5.3) Ch 101.15 (0.8) I I 99.2 (0.6) Ch 77.7 (5.6) SmC* 23.1 (0.3) SmF*Õ 13.6 (0.5) Cr MD312Bs 3 CrÕ 25.4 (0.2) SmC* 53.85 (4.2) Ch 65.31 ( 0.4) I I 63.84 (0.4) Ch 51.31 (4.2) SmC*Õ 28.6 Cr
an corresponds to spacer length.
bEnthalpies were too small to be evaluated.
(curve A to curve E) as the temperature of measurement 3.2.2. MD312Fr
This compound contained the (2R,3S)-2- uoro-3-decreases from 70 to 30ßC. The temperature dependence
of the layer spacing for MDn12Fs is presented in gure 2. methylpentanoylox y chiral moiety and three oxyethylene spacer units. A cholesteric–chiral smectic C–chiral In one example, the optical polarizing micrograph of
MD312Fs reveals a striated fan texture ( gure 3 ) from smectic F liquid crystal sequence was found. Upon cooling from the cholesteric phase, the chiral smectic C 27.7 to 70.6ßC. These results imply the presence of a
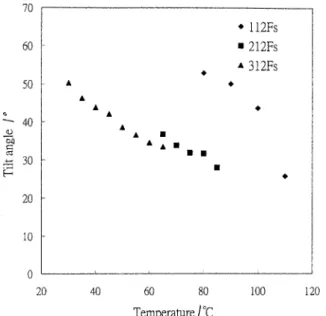
tilted SmC* phase. In gure 4, the tilt angle is plotted phase, and chiral smectic F with striated fan textures were formed. The (2R,3S)-2- uoro-3-methy lpentanoylox y
as a function of temperature in the chiral smectic C
phase; it decreased with increasing temperature. The tilt chiral moiety showed a poorer mesomorphic behaviour as compared with MD312Fs.
angles were calculated from equation (1);dSmC*(Max) was chosen to be the maximum layer spacing of the chiral
smectic C phase. 3.2.3. MDn12C series
The MDn12C series contained the
(2S,3S)-2-chloro-h5 cosÕ 1
A
dSmC*dSmC*(Max)
B
. (1 ) 3-methylpentanoylox y chiral moiety and oxyethylene spacer units (n5 1–3) [14]. The three compounds in this series are all mesomorphic; all three compounds The temperature angle of the chiral smectic phase
was about 50ßC. From gure 5, it can be seen that the display the chiral smectic C phase. The mesophase ranges of MDn12C and MDn12Fs are shown in table 2. increasing number of oxyethylene units signi cantly
depresses the phase transition temperature [15]. This A wider chiral smectic C phase and lower clearing temperature for MDn12C were observed. MD112Fs depression has been attributed to the increased exibility
of the C–O bonds. and MD212Fs are more inclined to form the ordered
Figure 1. X-ray diŒraction measurements for MD312Fs.
Figure 3. Optical polarizing micrograph of MD312Fs showing the chiral smectic C phase at 50ßC (400Ö ).
3.2.4. MDn12Bs series
The MDn12Bs series contained the
(2S,3S)-2-bromo-3-methylpentanoylox y chiral moiety and oxyethylene Figure 2. Layer spacing as a function of temperature in the spacer units (n5 1–3). MD112Bs and MD212Bs exhibited
chiral smectic C phase of compounds MDn12Fs.
similar liquid crystal behaviours with a cholesteric– chiral smectic C–chiral smectic F liquid crystal sequence. The chiral smectic F phase was not observed for the chiral smectic F phase as compared with MD112C
and MD212C. This implies that the decreasing dipole compound with longest spacer length, MD312Bs. XRD measurements and POM veri ed the assignment of moment and increasing atomic size of the halogen in
the chiral moiety can decrease the clearing temperature the mesophases for these compounds. For compounds MDn12Bs, a sharp low angle re ection and a broad wide
and widen the chiral smectic C phase.
373
Halogen-containin g FL Cs
Figure 6. Optical polarizing micrograph of MD112Bs showing the chiral smectic C phase at 75ßC (400Ö ).
Figure 4. Tilt angles as a function of temperature in the chiral smectic C phase of compounds MDn12Fs.
Figure 7. Plots of transition temperature versusn, the number of oxyethylene spacer units for compounds MDn12Bs. series is higher than for the MDn12Bs series. This may
result from the increase in dipole moment and decrease in molecular size of the asymmetric chiral center [16]. Figure 5. Plots of transition temperature versusn, the number
of oxyethylene spacer units for compounds MDn12Fs. 3.2.5. Polymer series PS312Fr, PS312Fs, PS312C and PS312Bs
The phase sequences and corresponding transition angle re ection angle were observed in XRD diagrams;
these were respectively assigned to lateral packing and temperatures for the (n5 3) members of these series are shown in table 3. Polymers PS312Fs, PS312Fr, and the smectic layer below the cholesteric point. Moreover,
the tilt angle decreases as the temperature increases. In PS312C exhibited similar liquid crystal behaviour, with cholesteric and chiral smectic C phases on the heating one example, the optical polarizing micrograph reveals
a striated fan texture ( gure 6) from 67.6 to 104.7ß C for and cooling scans. XRD measurement for PS312Fs and PS312Fr show a smectic diŒraction pattern (a sharp low MD112Bs. This indicates the formation of the tilted
chiral smectic C phase. The temperature range of the angle re ection and a broad wide angle re ection). Layer spacings were plotted as a function of temperature in chiral smectic phase was about 80ßC for MD312Bs. The
clearing temperature and phase transition temperature the chiral smectic C phase ( gure 8); the layer spacing increases as the temperature increases, con rming the decreased as the number of oxyethylene units increased
( gure 7) . Moreover, the clearing point of the MDn12Fs presence of the chiral smectic C phase. On the other
Table 3. Phase transitions and phase transition enthalpies more bulky molecular size of asymmetric chiral centre for polymers PS312Fs, PS312Fr, PS312C, and PS312Bs: [16], which reduces the phase stability of ferroelectric g5 glassy state, SmF*5 chiral smectic F phase, SmC*5 liquid crystal homopolymer.
chiral smectic C phase, Ch5 cholesteric phase, I5 isotropic phase.
4. Conclusion
A new series of ferroelectric liquid crystal monomers Heating
CoolingPhase transition temperature/ßC;
Polymer na and polymers consisting of a halogenated chiral centre,
(corresponding enthalpy changes/mJ mgÕ 1) oligooxyethylen e spacers, and an ester core unit con-taining three aromatic rings have been synthesized. All PS312Fr 3 g 25.9 SmC* 139.4 Ch 160.5 I of the compounds exhibit the chiral smectic C phase I 150.5 Ch 131.4 SmC* 24.4 g except PS312Bs. Wide SmC* temperature ranges were obtained in these monomers (~90ßC) and polymers PS312Fs 3 g 28.6 SmC* 143 Ch 163 I
I 153.34 Ch 137.4 SmC* 32.4 g (~100ßC). Several MDn12Fs, MDn12Fr, and MDn12Bs
compounds (n5 1, 2) exhibited chiral smectic F phases. PS312C 3 g 17 SmC* 131.9 Ch 149.8 I
I 145.5 Ch 128.1 SmC* 12 g When the number of oxyethylene spacer units increased, the clearing and phase transition temperatures decreased.
PS312Bs 3 —
I 105.4 (0.6) Ch 7.4 (0.7) g The lack of mesophases in PS312Bs is possibly due to the bulky substituted group at the chiral centre which
an corresponds to spacer length. disturbs the orientation of the side chain liquid crystal
polymer.
The authors thank the National Science Council of Republic of China for nancial support of this work (NSC 89-2216-E-007-00 6).
References
[1] Meyer, R. B., Liebert, L., Strzelecki, L., and Keller, P., 1975,J. Phys. Paris L ett., 36, L-69.
[2] Clark, N. A., and Lagerwall, S. T., 1980, Appl. Phys.
L ett., 36, 899.
[3] Zental, R., Reckert, G., and Reck, B., 1987, L iq.
Cryst., 2, 83.
[4] Parmar, D. S., Clark, N. A., Keller, P., Walba, D. M., and Wand, M. D., 1990,J. Phys. Paris.,
51, 355.
[5] Dumon, M., Nguyen, H. T., Mauzac, M., Destrade, C., and Gasparoux, H., 1991, L iq. Cryst.,
10, 475.
[6] Naciri, J., Pfeiffer, S., and Shashidhar, 1991, L iq.
Cryst., 10, 585.
[7] Lu, R., Xu, K., and Lu, Z., 1999, L iq. Cryst., 26, 553. [8] Keller, P., 1984, Mol. Cryst. liq. Cryst., 102, 295. [9] Adams, T. G., and Sinta, R., 1989, Mol. Cryst. liq. Figure 8. Layer spacing as a function of temperature in the Cryst., 177, 145.
chiral smectic C phase of polymers PS312Fs and PS312Fr. [10] Collings, P. J., and Hird, M., 1997, Introduction to
L iquid Crystals (London: Taylor & Francis Ltd), p. 51.
[11] Chen, J. H., Chang, R. C., Hsiue, G. H., Guu, F. W., and Wu, S. L., 1995,L iq. Cryst., 18, 291.
[12] Sierra, T., Serrano, J. L., Ros, M. B., Ezurra, A., and hand, PS312Fs, PS312Fr, and PS312C reveal wider
tem-Zubia, J., 1992,J. Am. chem. Soc., 114, 7645. perature ranges of the SmC* phase as compared with
[13] Collings, P. J., and Hird, M., 1997, Introduction to MD312Fr, MD312Fs, and MD312C. For PS312C, the L iquid Crystals (London: Taylor & Francis Ltd), p. 119. temperature range of the chiral smectic C phase was [14] Hsiue, G. H., Wu, J. L., and Chen, J. H., 1996, L iq. about 120ß C. A exible polysiloxane backbone enhances Cryst., 21, 449.
[15] Kitamura, T., Fujii, T., and Mukoh, A., 1984, Mol. the decoupling between side chain and main chain,
Cryst. liq. Cryst., 108, 333. tending to give rise to a higher thermal stability of the
[16] Collings, P. J., and Hird, M., 1997, Introduction to mesophases, including the chiral smectic C phase [17]. L iquid Crystals (London: Taylor & Francis Ltd), p. 120. Sample PS312Bs exhibited fewer mesomorphic phases [17] Hsiue, G. H., and Chen, J. H., 1995, Macromolecules, and no chiral smectic C phase. This may result from the 28, 4366.


![Table 3. Phase transitions and phase transition enthalpies more bulky molecular size of asymmetric chiral centre for polymers PS312Fs, PS312Fr, PS312C, and PS312Bs: [16], which reduces the phase stability of ferroelectric g 5 glassy state, SmF* 5 chiral sm](https://thumb-ap.123doks.com/thumbv2/9libinfo/7935727.157368/11.855.48.411.143.367/transitions-transition-enthalpies-molecular-asymmetric-polymers-stability-ferroelectric.webp)
