ClO radicals
R. S. Zhu and M. C. Lin
Citation: The Journal of Chemical Physics 118, 4094 (2003); doi: 10.1063/1.1540623 View online: http://dx.doi.org/10.1063/1.1540623
View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/118/9?ver=pdfcov
Published by the AIP Publishing
Articles you may be interested in
Ab initio studies of ClO x reactions. IX. Combination and disproportionation reactions of ClO and s- ClO 3 radicals
J. Chem. Phys. 119, 8897 (2003); 10.1063/1.1613632
Ab initio studies of ClO x reactions. VIII. Isomerization and decomposition of ClO 2 radicals and related bimolecular processes
J. Chem. Phys. 119, 2075 (2003); 10.1063/1.1585027
Ab initio studies of ClO x reactions. I. Kinetics and mechanism for the OH + ClO reaction
J. Chem. Phys. 116, 7452 (2002); 10.1063/1.1467057
Ab initio study of the CH 3 + O 2 reaction: Kinetics, mechanism and product branching probabilities
J. Chem. Phys. 115, 195 (2001); 10.1063/1.1376128
Temperature and pressure dependence of the addition reactions of HO to NO and to NO 2 . IV. Saturated laser-induced fluorescence measurements up to 1400 bar
J. Chem. Phys. 108, 5391 (1998); 10.1063/1.475971
Ab initio
studies of ClO
xreactions. IV. Kinetics and mechanism
for the self-reaction of ClO radicals
R. S. Zhu and M. C. Lina)
Department of Chemistry, Emory University, Atlanta, Georgia 30322
共Received 17 June 2002; accepted 4 December 2002兲
The self-reaction of ClO radicals has been investigated by ab initio molecular orbital and variational transition state theory calculations. Both singlet and triplet potential energy surfaces were predicted by the modified Gaussian-2 method. The reaction was shown to take place mainly over the singlet surface by forming ClOOCl (k1) and ClOClO (k1⬘). These association processes were found to be strongly pressure dependent and the predicted total rate constants are in good agreement with the experimental data. The predicted second- and third-order rate constants for formation of ClOOCl and ClOClO can be expressed, respectively, in units of cm3molecule⫺1s⫺1and cm6molecule⫺2s⫺1 by k1⬁⫽1.6⫻10⫺9T⫺0.67exp(⫺64/T), k⬁1⬘⫽6.4⫻10⫺9T⫺0.78exp(⫺76/T), and k10⫽8.31⫻10⫺20
T⫺4.96exp(⫺336/T), k10⬘⫽1.72⫻10⫺14T⫺6.99exp(⫺926/T) in the temperature range 180–500 K for N2as the third body. The observed T, P-dependent data could be best accounted for with the heat of formation of ClOOCl, ⌬fH0
o
(ClOOCl)⫽29.4⫾1 kcal/mol. The formation of Cl2⫹O2 共2兲, Cl
⫹ClOO 共3兲, and Cl⫹OClO 共4兲 products have been confirmed, with the predicted
pressure-independent rate constants: k2⫽1.09⫻10⫺13T0.66exp(⫺1892/T); k3⫽1.36⫻10⫺13T0.77 exp(⫺2168/T); k4⫽6.26⫻10⫺11T0.005exp(⫺2896/T), respectively, in units of cm
3
molecule⫺1s⫺1, covering the temperature range 200–1500 K. These results are also in reasonable agreement with existing experimental kinetic data. © 2003 American Institute of Physics.
关DOI: 10.1063/1.1540623兴
I. INTRODUCTION
The recombination and disproportionation reactions of ClO radicals have been investigated extensively on account of their relevance to the O3-destruction chemistry in the stratosphere.1–15 In principle, the reactions may take place via at least two long-lived intermediates; viz.
ClO⫹ClO→ClOOCl*→Cl2⫹O2 共a兲
→Cl⫹ClOO 共b兲
ClO⫹ClO→ClOClO*→Cl⫹OClO, 共c兲
where *denotes internal excitation. Potentially, a third inter-mediate, ClClO2, may be formed in the reverse Cl⫹OClO reaction, analogous to the association of OH with OClO, in which chloric acid (HOClO2) may be a major product, de-pending on the pressure of the system.16
Under higher pressure conditions, particularly at low temperatures, collisional deactivation of excited association products producing ClOOCl and ClOClO may be important kinetically. In fact, for the most stable intermediate, ClOOCl, its formation is the dominant process under the stratospheric condition.4,5,9,11,13,15 On the other hand, the disproportion-ation processes producing ClOO and OClO by 共b兲 and 共c兲, respectively, are endothermic by 3– 4 kcal/mol 共vide infra兲; accordingly, they cannot compete effectively with the recom-bination reaction in the stratosphere 共with low temperature
and medium pressure兲. These endothermic reactions may, however, become dominant processes in the combustion of ammonium perchlorate 共AP兲 propellant.
The effects of temperature and pressure on the total rate constant and product branching probabilities over a wide range of practical T, P-conditions (200 K⬍T⬍3000 K, 0
⬍P⬍200 atm) cannot be realistically studied in any
labora-tory experiments. In this series of theoretical studies,17–19we have employed high-level ab initio molecular orbital meth-ods in conjunction with statistical theory calculations to map out the detailed potential energy surfaces共PES兲 involved and calculate the associated rate constants and product branching ratios for applications over a wide range of conditions rel-evant to specific practical requirements. For the three dis-tinctly different systems, OH⫹ClO3,17 OH⫹ClO,18 and O
⫹OClO,19 which we have investigated recently, the results have been very encouraging. We could not only quantita-tively account for the experimentally determined total rate constants for these reactions, but also reliably predict the reported product branching ratios, typically measured under experimentally more readily accessible conditions.
Comparison between theory and experiment for a certain experimental condition does provide a much needed calibra-tion of the computed energies and the overall mechanism involved for a more reliable extrapolation to more extreme practical conditions 共e.g., at 1000–2500 K and 1–200 atm for AP combustion兲. In the present study, we have examined the effects of temperature and pressure on the recombination and disproportionation of ClO radicals combining ab initio MO and VRRKM calculations with a special emphasis on a兲National Science Council Distinguished Visiting Professor at Chiaotung
University, Hsinchu, Taiwan. Electronic mail: [email protected]
4094
0021-9606/2003/118(9)/4094/13/$20.00 © 2003 American Institute of Physics
the variation of product formation channels with tempera-ture, from 200 K of interest to stratosphere to 3000 K of importance to AP combustion.
II. COMPUTATIONAL METHODS
The geometry of the reactants, products, intermediates, and transition states of the title reaction have been fully op-timized by using the hybrid density functional B3LYP method 共Becke’s three-parameter nonlocal exchange functional20,21with the correlation functional of Lee, Yang, and Parr22兲 with the 6-311⫹G(3d f ) basis set. Vibrational frequencies employed to characterize stationary points, zero-point energy 共ZPE兲 corrections have also been calculated at this level of theory, and have been used for the rate constant calculations. All the stationary points have been positively identified for local minima 共with the number of imaginary frequencies NIMAG⫽0) and transition states 共with NIMAG⫽1). Intrinsic reaction coordinate 共IRC兲 calculations23 have been performed to confirm the connec-tion of each transiconnec-tion state with designated intermediate.
The total G2M energy with zero-point energy共ZPE兲 cor-rection is calculated as follows:24
E关共G2M共CC2兲兴⫽Ebas⫹⌬E共⫹兲⫹⌬E共2d f 兲
⫹⌬E共CC兲⫹⌬
⬘⫹⌬E共HLC,CC2兲
⫹ZPE共3d f 兲, Ebas⫽E关PMP4/6-311G共d,p兲兴,
⌬E共⫹兲⫽E关PMP4/6-311⫹G共d,p兲兴⫺Ebas,
⌬E共2d f 兲⫽E关PMP4/6-311G共2d f ,p兲兴⫺Ebas,
⌬E共CC兲⫽E关CCSD共T兲/6-311G共d,p兲兴⫺Ebas, ⌬⬘⫽E关UMP2/6-311⫹G共3d f ,2p兲兴 ⫺E关UMP2/6-311G共2d f ,p兲兴 ⫺E关UMP2/6-311⫹G共d,p兲兴 ⫹E关UMP2/6-311G共d,p兲兴, ⌬E共HLC,CC2兲⫽⫺5.78n⫺0.19n␣ in units of mhartree,
where n␣ and n are the numbers of valence electrons, n␣
⭓n. All calculations were carried out withGAUSSIAN 98.25 The rate constants were computed with a microcanonical variational RRKM共VARIFLEX26兲 code, which solves the
mas-ter equation27,28involving multistep vibrational energy trans-fers for the excited intermediate ClOOCl† or ClOClO†. The energies for the intermediates and transition states calculated at the G2M共CC2兲 level were used in the calculation.
For the barrierless transition states, the Morse potential
E共R兲⫽De关1⫺e⫺共R⫺Re兲兴2,
was used to represent the potential energy along the mini-mum energy path of each individual reaction coordinate. In the above equation, R is the reaction coordinate 共i.e., the distance between the two bonding atoms, O–O or O–Cl in this work兲, De is the bond energy excluding zero-point
en-ergy, and Re is the equilibrium value of R. For the tight transition states, the numbers of states were evaluated ac-cording to the rigid-rotor harmonic-oscillator approximation. For the formation of higher barrier disproportionate products in reactions共a兲 to 共c兲, we have employed the CHEM-RATE code of Mokrushin et al.29 to couple all low-lying re-action channels including isomerization processes by solving the T, P-dependent master equation.
III. RESULTS AND DISCUSSION
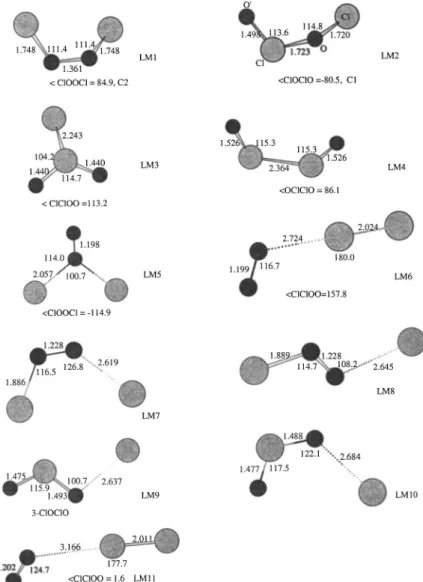
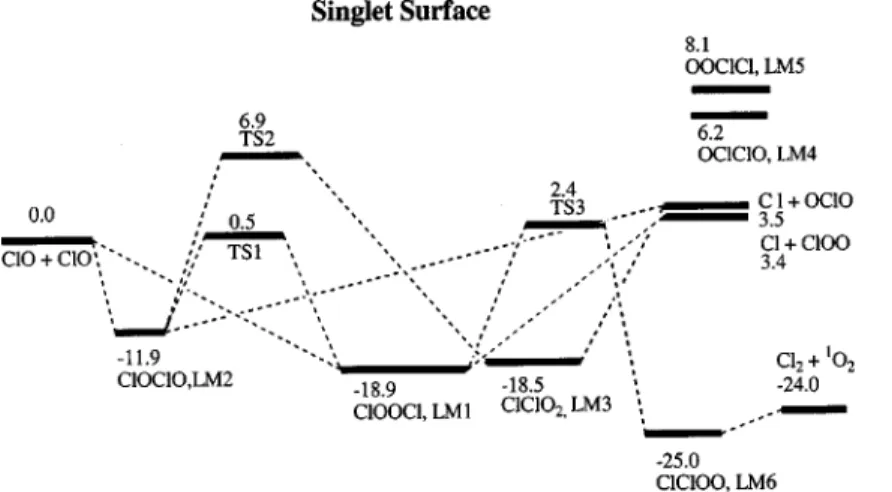
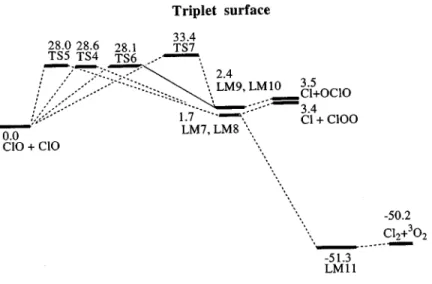
A. Potential energy surface and reaction mechanism The optimized geometries of the intermediates and tran-sition states are shown in Figs. 1 and 2, respectively. The singlet and triplet potential energy diagrams obtained at the G2M//B3LYP/6-311⫹G(3d f ) level are presented in Figs. 3 and 4. The predicted vibrational frequencies and rotational constants are summarized in Table I to compare with the available experimental vibrational frequencies of ClOOCl and its isomers.
1. Cl2O2isomers
a. Equilibrium geometries and frequencies.
Theoreti-cally, most investigators30–38 focused their studies on the three low-lying isomers, ClOOCl, ClOClO, and ClClO2. Lee
et al.37have investigated these isomers using different meth-ods and basis sets关including CCSD共T兲 with large ANO basis sets for single-point energies calculation兴; they indicated that
f-type basis functions are necessary to obtain accurate
equi-librium geometries and frequencies. Christen et al.38showed that density functional theory 共DFT兲 calculations using hy-brid functionals with large basis sets reproduced the experi-mental data well.
In this section, the structures and frequencies of five iso-mers, ClOOCl, ClClO2, ClOClO, OClClO, and OOClCl, la-beled as LM1–LM5 in Fig. 1, calculated at the B3LYP/6-311⫹G(3d f ), are compared with previously pre-dicted values and available experimental data. The first iso-mer, ClOOCl, has C2 symmetry; the predicted Cl–O bond length at the B3LYP/6-311⫹G(3d f ) level, 1.748 Å, can be compared with the calculated values, 1.731,37 1.753,37 1.711,37 1.706,38 1.731,38 1.746 Å 共Ref. 38兲 obtained at the MP2/TZ2P, CCSD共T兲/TZ2P, MP2/TZ2Pf, MP2/cc-PVQZ, B3PW91/6-311⫹G(3d2 f ), B3LYP/aug-cc-PVTZ⫹d lev-els, respectively. Apparently, the Cl–O bond lengths pre-dicted by DFT and CCSD共T兲 methods appear to be too long even with larger basis sets, but the value predicted by the MP2 method with big basis sets seems to be closer to the experimental value, 1.704 Å.39 The O–O bond length ob-tained at the B3LYP/6-311⫹G(3d f ) level, 1.361 Å, is 0.065 Å shorter than the experimental value39 and can be compared with 1.421, 1.411, 1.407, 1.409, 1.354, and 1.371 Å using the above six methods, respectively. These data show that the predicted Cl–O and O–O bond lengths depend strongly on the size of the basis set and the method. All of the calculated ClOO bond angles and ClOOCl dihedral angles deviate from experimental values, 110.1° and 81.0°,39 by approximately ⫾1–2° and ⫾1–3°, respectively. Calcula-tions in Ref. 38 indicated that the HF method totally fails in
predicting the structure of ClOOCl. For the ClClO2 isomer with Cssymmetry, the Cl–Cl and Cl–O bond lengths, 2.243
and 1.440 Å obtained at the B3LYP/6-311⫹G(3d f ) level are in good agreement with experimental values, 2.198共Ref. 40兲 and 2.22,41 and 1.436 共Ref. 40兲 and 1.440 Å,41 respec-tively.
Our predicted Cl–Cl and Cl–O bond lengths are close to those values, 2.279, 1.468 Å; 1.202, 1.434 Å; 2.199, 1.429 Å obtained at the CCSD共T兲/TZ2P,37 B3PW91/6-311
⫹G(3d2 f ),38
and B3PW91/cc-pVQZ38 levels. For this structure, basis set and electron correlation effects are more important than that of ClOOCl.37For the ClOClO isomer, no experimental structure parameters are available. Calculations by either the DFT or the coupled-cluster method show that basis set has a strong effect on the middle ClO bond length. For instance, at the B3LYP/6-311G(d) level it is 1.837 Å; with larger basis sets, it decreases to 1.737 Å共Ref. 38兲 and 1.723 Å 共this work兲 at the B3LYP/cc-pVQZ and B3LYP/6-311(3d f ) level, respectively. From CCSD共T兲/ DZP to CCSD共T兲/TZ2P, the decrease is even larger, 0.126 Å.37 This bond is also more sensitive to the correlation ef-fect; for example, at the CCSD/TZ2P and CCSD共T兲/TZ2P levels, the predicted values are 1.739 and 1.814 Å.37Again, this structure appears to be method dependent. The other two
isomers, OClClO and OOClCl, were previously optimized by Jensen and Oddershede42at the HF/3-21G*level, with no structure parameters available in their paper. Figure 1 shows the structural parameters of these two molecules with C2and
Cs symmetry at the B3LYP/6-311⫹G(3d f ) level.
The frequencies of ClOOCl, ClClO2, and ClOClO are shown in Table I. Our results at the B3LYP/6-311
⫹G(3d f ) level are similar to those obtained at the
B3PW91/6-311⫹G(3d2 f )38 and B3LYP/aug-cc-pVTZ
⫹d.38From Table I one can see that the frequencies calcu-lated at the B3LYP/6-311⫹G(3d f ) level agree well with the experimental values of Mu¨ller et al.,41 Chen and Lee,43 and Jacobs and co-workers.44 Many other complexes with loose structures, LM6 –LM11, have also been located; they will be discussed in the following related parts.
b. Relative energies. Here, we mainly compare the
sta-bility of LM1 to LM5. The calculated G2M energies relative to ClO⫹ClO are presented in Fig. 3. The results show that the relative stability of the Cl2O2 isomers is approximately: ClOOCl⬇ClClO2⬎ClOClO⬎OClClO⬎OOClCl. At the G2M//B3LYP/6-311⫹G(3d f ) level, ClOOCl lies 18.9 kcal/mol below the reactants, the dissociation energy (D0) is the same as that predicted by Stanton et al.33 using many-body perturbation theory 共MBPT兲 and the infinite-order
FIG. 1. The optimized geometry of intermediates com-puted at the B3LYP/6-311⫹G(3d f ) level for the ClO⫹ClO reaction.
coupled-cluster 共CC兲 approximation. The predicted value of
D0 共18.9 kcal/mol兲 is also close to the experimentally esti-mated enthalpy changes, 17.3,9 19.5,13 and 18.1 共Ref. 13兲 kcal/mol at 200–300 K, corresponding to the values of 16.5, 18.6, and 17.2 reported at 0 K, but is different from the results computed at the G2 and CCSD共T兲/关65321/54321兴// CCSD/TZ2P levels, 22.1 kcal/mol45 and 14.2 kcal/mol,37 re-spectively. Further discussion on this important quantity will continue in the following section. For ClClO2, which lies above ClOOCl by only 0.4 kcal/mol, the result is close to those relative to ClOOCl obtained by McGrath et al.31 and Lee et al.,37 1.0 kcal/mol and 0.9⫾2.0 kcal/mol, respec-tively, but, according to Stanton et al.33 and Li et al.,45 ClOOCl is more stable than ClClO2by 6.7 and 7.7 kcal/mol, respectively. The third isomer, ClOClO, is predicted to be 7.0 kcal/mol less stable than ClOOCl; this energy difference is
the same as that reported by McGrath et al.31 and also close to the best estimated result of Lee et al.,37 10.1
⫾4 kcal/mol. At the G2 level, the energy difference was
pre-dicted to be 11.7 kcal/mol.45 Finally, the other two isomers, OClClO and OOClCl, were predicted to lie 6.1 and 8.1 kcal/mol, respectively, above the reactants, as shown in Fig. 3. From the above comparison one can see that al-though uncertainties exist among the values computed by different methods, the relative stability of these isomers re-mains the same, i.e., ClOOCl⬇ClClO2⬎ClOClO⬎OClClO
⬇OOClCl.
c. Isomerization. As OClClO and OOClCl have higher
energies, only the isomerization processes involving the three low-lying isomers are investigated. TS1 and TS2 共see Figs. 2 and 3兲 correspond to the isomerization from ClOClO to ClOOCl and ClClO2; they lie above the reactants by 0.5
FIG. 2. The geometry of the transition states involved in the ClO⫹ClO reaction at the B3LYP/6-311
⫹G(3d f ) level.
FIG. 3. The schematic diagram of the singlet potential energy surface for the ClO–ClO system computed at the G2M level.
and 6.9 kcal/mol, respectively. The potential effect of the former isomerization process, which has a much smaller bar-rier, will be discussed later.
2. Formation of ClO¿ClO\Cl2¿1,3O2
As discussed in the above section, many authors have theoretically investigated the structures and stabilities of the Cl2O2 isomers; however, the dissociation mechanisms of these isomers have not been reported before. In principle, Cl2⫹1O2 and3O2 can be produced via the singlet and triplet potential energy surfaces, respectively. Over the singlet sur-face, shown in Fig. 3, the ClOOCl intermediate can dissoci-ate to give Cl2⫹1O
2 via a four-center transition state TS3 in which the O–O bond length, 1.209 Å, is much close to that of 1O2, 1.203 Å, obtained at the same level of theory. The breaking two Cl–O bonds lengthen unequally by 0.135 and 0.956 Å, compared with those in ClOOCl. TS3 lies 3.6 kcal/ mol above the reactants; this value is close to the experimen-tal activation energy, 2.7⫾0.8, 2.3⫾1.0, and 3.2
⫾0.2 kcal/mol reported by Hayman et al.,7 Clyne et al.,3共b兲 and Nickolaisen et al.,13 respectively. IRC analysis23results
show that the forward side of TS3 connects to a loose com-plex, LM6, which is 1.0 kcal/mol lower than the Cl2⫹1O2 products.
On the triplet surface, the most likely process is that ClO and ClO form3cis-ClOOCl共see Figs. 1 and 4兲 followed by
dissociation to produce Cl2⫹3O2. However, the calculated result indicates that the formation of 3cis-ClOOCl occurs
with a very high barrier, 28.6 kcal/mol. Formation of 3trans-ClOOCl 共LM8兲 also has a similar high barrier, 28.0 kcal/mol, as shown in Fig. 4. Evidently, formation of triplet ClOOCl is energetically unfavorable. Similarly, trans- and
cis-ClOClO共LM9, LM10兲 also have large entrance barriers,
28.1 and 33.4 kcal/mol, corresponding to TS6 and TS7, re-spectively. It should be mentioned that the high barriers for the entrance step on triplet potential energy surfaces were also found in the HO⫹ClO 共Ref. 18兲 and HO⫹OClO 共Ref. 16兲 reactions. For the HO⫹ClO→3HOOCl and HO
⫹OClO→3HOOClO reactions, their barriers were found to be 17.2 共Ref. 18兲 and 35.0 共Ref. 16兲 kcal/mol, respectively. The transition state between3cis-ClOOCl and Cl2⫹3O2was not located at the B3LYP/6-311⫹G(3d f ) level of theory;
FIG. 4. The schematic diagram of the triplet potential energy surface for the ClO–ClO system computed at the G2M level.
TABLE I. Vibrational frequencies and rotational constants for the reactants, the key intermediates, and transi-tion states of the ClO⫹ClO reaction computed at the B3LYP/6-311⫹G(3d f ) level of theory.
Species B共GHZ兲 Frequencies ClO 0.0, 18.5, 18.5 861.4 ClOOCla 13.5, 2.3, 2.1 127 (127⫾20), 326, 443 共418兲, 551 共543兲, 638 共647, 648兲, 844 共754, 752兲 ClOClOa 14.9, 2.4, 2.2 116, 258, 354 共338.4兲, 441 共440.4兲, 622 共695.9兲, 1015 共994.5兲 ClClO2b 9.4, 3.5, 2.7 243共251.4兲, 264 共271.4兲, 441 共440.5兲, 524 共522.7兲, 1061 共1041.5兲, 1229 共1218兲 OClClO 14.4, 2.2, 2.1 87, 159, 209, 248, 934, 974 OOClCl 6.4, 2.8, 2.0 163, 226, 324, 501, 538, 1427 TS1 19.6, 1.7, 1.6 159i, 125, 241, 444, 780, 926 TS2 10.6, 2.5, 2.2 475i, 130, 226, 479, 981, 1211 TS3 5.8, 3.2, 2.1 280i, 163, 249, 325, 557, 1386 a
Italic and regular fonts of the frequencies in the parentheses are from the experimental results of Refs. 43 and 44, respectively.
bThe frequencies in the parentheses are from the experimental results of Ref. 41.
thus, the formation of3O2 from3cis-ClOOCl may be a bar-rierless process.
3. Formation of ClOO¿Cl and OClO¿Cl
Besides the molecular elimination reaction producing Cl2⫹1O2, ClOOCl can also dissociate barrierlessly to give Cl⫹ClOO, as shown in Fig. 3. Similarly, triplet cis- and
trans-ClOOCl 共see Figs. 1 and 4兲 can dissociate to Cl ⫹ClOO also but, as mentioned before, their high entrance
barriers, 28.6 and 28.0 kcal/mol, corresponding to TS4 and TS5 共see Fig. 4兲, respectively, prevent the formation of Cl
⫹ClOO via these triplet states.
The analogous reactions producing Cl⫹OClO, as shown in Figs. 3 and 4, can occur by the decomposition of ClOClO, ClClO2, 3cis- and3trans-ClOClO共LM9 and LM10兲.
How-ever, again, because of the high barriers at TS2, TS6, and TS7共see Figs. 3 and 4兲, only the singlet ClOClO dissociation path will be discussed in the later rate constant calculation.
The endothermicities for the formation of Cl⫹ClOO and Cl⫹OClO are predicted to be 3.4 and 3.5 kcal/mol, respec-tively; these values can be compared with the activation en-ergies 4.9⫾0.7 and 2.7⫾0.3 kcal/mol determined by Nickolaisen et al.13The heats of formation for the three low-lying Cl2O2 and ClO2 isomers are discussed below.
4. Heats of formation of ClOOCl, ClClO2, ClOClO,
ClOO, and OClO
In order to establish the reliability of the rate constant calculations, besides the comparison of the structural param-eters, frequencies, and transition state barriers with available theoretical or experimental data given in the previous sec-tions, here we also compare the heats of formation for some species with available values. The heats of formation at 0 K for ClO and Cl are known to be 24.15⫾0.024 and 28.59
⫾0.0014, respectively.46Other heats of formation are calcu-lated based on these values using the computed⌬rH0o. Table II presents the values and other calculated or experimental data for comparison. It can be seen that for ClOOCl, our result 共29.4 kcal/mol兲 is close to those predicted by Stanton
et al.33 with the infinite-order coupled-cluster共CC兲 approxi-mation and Li et al.45with the G2 method. All are in reason-able agreement with the experimental values, 29.7共Ref. 13兲 and 32.1共Ref. 9兲 kcal/mol. The highest value, 34.2 kcal/mol obtained by Lee et al.,37 was based on the calculated en-thalpy change for the isodesmic reaction, ClOOCl⫹H2O
⫽HOOH⫹Cl2O, using the heat of formation of Cl2O, 21.4
⫾1.6 kcal/mol. As stated in their Note Added in Proof,37 if the recent value of⌬fH0
o
(Cl2O)⫽19.8⫾0.5 kcal/mol46was used, the same isodesmic reaction led to 32.6⫾1.0 kcal/mol for the heat of formation for ClOOCl, which is consistent with 32.6⫾3.2 kcal/mol30 predicted at the MP2/6-31⫹
⫹G(3d,2p) level based on the same isodesmic reaction.
This lower value essentially overlaps with our value within the combined uncertainty in theory and experiment. Because of the high sensitivity of the predicted ClO⫹ClO association rates to the D0 共ClO–OCl兲 value, we will compare the pre-dicted rate constant as a function of D0 in the next section. For ClClO2 and ClOClO, there are large uncertainties; the maximum deviation among the results predicted by different methods reaches as much as 8 kcal/mol. For OClO and ClOO, our results 23.2 and 23.1 kcal/mol, are in good agree-ment with experiagree-mental values, 23.7⫾1.9 共Ref. 46兲 and 23.8⫾0.7 共Ref. 46兲, respectively; they are consistent with our previously predicted values based on the reactions 3O
⫹OClO→ClO⫹3O 2
19 and HO⫹ClO→H⫹OClO,18 24.7 and 25.3 kcal/mol, respectively. It should be mentioned that the G2 method45 overestimated the heats of formation of OClO and ClOO by 4.5 and 6.4 kcal/mol, respectively, com-pared with experimental values. The deviation may arise from the unreliable structures optimized at the MP2/6-31G(d) level employed in the standard G2 method. B. Rate constant calculations
Variational TST and RRKM calculations have been car-ried out for the following reaction channels:
ClO⫹ClO→ClOOCl*→ k1 ClOOCl共⫹M兲 → k2 Cl2⫹O2 → k3 Cl⫹ClOO, ClO⫹ClO→ClOClO*→ k 1 ⬘ ClOClO共⫹M兲 → k4 Cl⫹OClO.
The energies used in the calculation are plotted in Fig. 3 and the vibrational frequencies and rotational constants are listed in Table I. The LJ parameters required for the RRKM calcu-lations for the quenching of ClOOCl*, ⫽533 K and
⫽4.1 Å, were derived from deconvoluting the LJ potential TABLE II. Comparison of the predicted heats of formation of different species共at 0 K, in kcal/mol兲 in this work
with available data.
Species Predicted Expt.
ClOOCl 29.4,a29.4共Ref. 33兲, 30.6 共Ref. 45兲, 32.6⫾3.2 共Ref. 30兲,
34.2共Ref. 37兲
29.7共Ref. 13兲, 32.1 共Ref. 9兲 ClClO2 29.8,a33.8共Ref. 30兲, 35.1 共Ref. 37兲, 37.1 共Ref. 33兲, 37.5
共Ref. 45兲
ClOClO 36.4,a42.4共Ref. 45兲, 44.3 共Ref. 37兲
OClO 23.2,a24.7共Ref. 19兲, 25.3 共Ref. 18兲, 28.2 共Ref. 45兲 23.7⫾1.9 共Ref. 46兲
ClOO 23.1,a23.5共Ref. 18兲, 30.2 共Ref. 45兲 23.8⫾0.7 共Ref. 46兲
a
This work.
of the He–ClOOCl system obtained by our ab initio calcu-lation at the B3LYP/6-311⫹G(3d f ) level. The and pa-rameters for the He–ClOOCl collision pair were determined to be 73 K and 3.3 Å by fitting the LJ function,47 V ⫽4关(/r)12⫺(/r)6兴. The LJ parameters for He,
⫽10 K and⫽2.55 Å, were taken from the literature.48 The rate constants for the dominant association reactions (k1 and k1⬘) were calculated with the VARIFLEX code,
26 whereas those for the disproportionation reactions, k2, k3, and k4, were computed with theCHEMRATEcode
29
coupling all intermediates involved in the forward and reverse reac-tions as shown below:
Scheme 1:
ClO⫹ClO↔ClOOCl*→ClOOCl共⫹M兲
→Cl2⫹O2 →Cl⫹ClOO ↔ClOClO*→ClOClO共⫹M兲 →Cl⫹OClO →ClO⫹ClO共loss兲. Scheme 2:
ClO⫹ClO↔ClOClO*→ClOClO共⫹M兲
→Cl⫹OClO
↔ClOOCl*→ClOOCl共⫹M兲
→Cl2⫹O2 →Cl⫹ClOO →ClO⫹ClO共loss兲. In theCHEMRATEcalculation, the transition state parameters for the barrierless association and decomposition processes were evaluated canonically for each temperature and critical separation, r#(T), based on the maximum Gibbs free-energy criterion as described previously for radical–radical reactions.49,50
1. Pressure and third-body effects on formation of ClOOCl and ClOClO
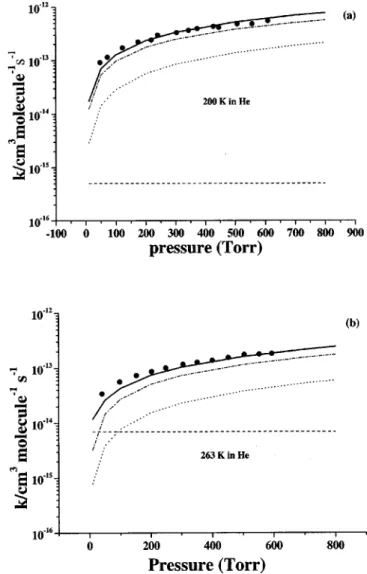
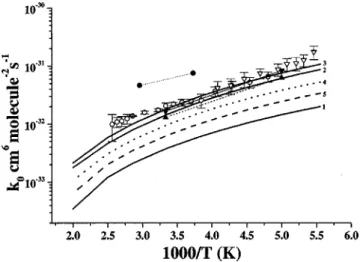
The association of ClO radicals was found to be strongly pressure dependent.11,13,15 The formation of both ClOOCl and ClOClO occurs barrierlessly; therefore, variational treat-ment was employed to obtain the dissociation potential en-ergy function by varying the respective breaking O–O and Cl–O bonds from 1.361 to 4.161 Å and 1.723 to 3.723 Å at an interval of 0.1 Å; other geometry parameters were fully optimized at the B3LYP/6-311⫹G(3d f ) level. The com-puted potential energies could be fitted to the Morse function with the parameters ⫽1.498 and 1.361 Å⫺1for the disso-ciation of ClO–OCl and ClO–ClO, respectively. Figures 5–7 show the predicted and experimental rate coefficients at dif-ferent temperatures as a function of He, O2, and N2pressure. In these figures, the solid, dash-dotted, dotted, and dashed curves correspond to the predicted values of the total rate constant kt, k1, k1⬘, and (k2⫹k3⫹k4), respectively. From these figures one can see most notably that k1⬘is not
negli-gible, for instance, accounting for about 18%–29% of the total rate from 10– 800 Torr in N2 at 200 K and that (k2
⫹k3⫹k4) is pressure independent and the association reac-tions forming ClOOCl and ClOClO are pressure dependent for all of the third bodies at all temperatures, with the former being dominant. The predicted total rate constants (kt) are in
excellent agreement with experimental data.11,13,15At higher temperatures 共e.g., 263 K兲 and low pressures 共⬍10 Torr兲, decomposition channels (k2⫹k3⫹k4) become competitive, especially in He.
The third-body efficiencies of the buffer gases in this system are similar to those in the HO–CO system.51 The predicted order of third-body efficiencies is SF6⬎N2⬇O2
⬎He with the values relative to N2: 1.51 : 1.00 : 0.86 : 0.40 under the 263 K and 50– 800 Torr conditions. The
具
⌬Edown典
used for fitting the experimental results in different third body is displayed in Table III, together with the modeled k0 and k⬁ in comparison with experimental results at 300 K for M⫽He, Ar, N2, O2, CF4, and SF6. It can be seen that the predicted k0, the sum of k10 and k1⬘
0
, is in good agreement with those experimental values10,11,13for different third
bod-FIG. 5. Comparison of the predicted rate constants with the experimental values at 200 K共a兲 and 263 K 共b兲 in He. Dashed, dotted, dash-dotted, and solid lines represent the predicted (k2⫹k3⫹k4), k1⬘, k1, and kt, respec-tively.䊉, Ref. 11.
ies; k⬁(k1⬁⫹k1⬁⬘) is, however, about 1 order of magnitude larger than the experimentally extrapolated values,11,13 sug-gesting that the extrapolation is likely insufficient due to the lack of data in the higher pressure regime. A similar obser-vation was made in our study of the O⫹OClO reaction.19
It is clear that in order to well present the experimental results, the ClOClO formation channel should be taken into account 共see Figs. 5–7兲 and that single Troe fits to these fall-off data cannot be quantitatively employed for interpola-tion or extrapolainterpola-tion of P-dependent data. In addiinterpola-tion, the high-pressure limits obtained from these data in the rather narrow P range are not reliable as mentioned above.
Although the formation of bimolecular products (Cl
⫹OClO/ClOO and Cl2⫹O2) from the ClO⫹ClO reaction is insensitive to the value of the well depth, D0(ClO–OCl), the rate constant for the formation of ClOOCl is, however, very sensitive on account of the relatively shallow well. In view of the spread of the D0values, from 18.9 to 14.1 kcal/mol as alluded to above, we have compared the predicted bimolecu-lar rate constants for the ClO⫹ClO reaction with three dif-ferent dissociation energies at 200 and 263 K in N2as shown in Figs. 7共a兲 and 7共b兲. In these figures, lines 1, 2, and 3
rep-resent the kt predicted by D0(ClO–OCl)⫽18.9, 16.3, and 14.1 kcal/mol; lines 4 and 5 represent k1
⬘
and (k2⫹k3⫹k4), which are included in the total rate constant. Evi-dently, the lower values of D0 共14.1 and 16.3 kcal/mol兲 un-derpredict the bimolecular rates due to the faster back-dissociation of the excited ClOOCl*. For example, with
D0⫽16.3 and 14.1 kcal/mol at 263 K, the predicted total rate constants decrease by as much as 34% and 52% in the pres-sure range of 50– 800 Torr, respectively, comparing with the value calculated with D0⫽18.9 kcal/mol, which agrees closely with experimental data as illustrated above. An addi-tional comparison will be made below using five indepen-dent sets of experimental data over a wide range of tempera-tures.
For practical applications, we have analyzed our theoret-ical fall-off curves in terms of the following empirtheoret-ical equa-tions to obtain broadening parameters (Fc):
52,53
k⫽a关b/共1⫹b兲兴F,
log F⫽log Fc/关1⫹共log b兲2兴,
FIG. 6. Comparison of the predicted rate constants with the experimental values at 200 K共a兲 and 263 K 共b兲 in O2. Dashed, dotted, dash-dotted, and
solid lines represent the predicted (k2⫹k3⫹k4), k1⬘, k1, and kt, respec-tively.䊉, Ref. 11.
FIG. 7. The variation of the predicted rate constants with D0共ClO–OCl兲 at
200 K共a兲 and 263 K 共b兲 in N2. Solid, dashed, dotted lines 1, 2, 3 represent
the predicted ktusing D0⫽18.9, 16.3, and 14.1 kcal/mol for ClOOCl
asso-ciation process, respectively; lines 4 and 5 represent k1⬘and (k2⫹k3⫹k4);
symbols are the experimental values.䊉, Ref. 11; 䉭, Ref. 13 at 208 K; 䉭, Ref. 13 at 195 K.
using the predicted high- and low-pressure limits for N2 as the third body. For the ClOOCl association path, we obtained
Fc⫽0.24, 0.22, 0.22, and 0.27 at 200, 263, 298, and 400 K,
respectively. In the fitting, a⫽k⬁⫽3.40, 3.07, 2.90, and 2.51 in units of 10⫺11cm3molecule⫺1s⫺1, and b⫽k0关M兴/k⬁ with k0⫽6.19, 2.35, 1.47, and 0.464 in units of
10⫺32cm6molecule⫺2s⫺1, respectively. For the ClOClO as-sociation path, Fc⫽0.72, 0.74, 0.75, and 0.83 at 200, 263,
298, and 400 K with a⫽k⫺1⬁ ⫽6.97, 6.19, 5.80, and 4.90 in units of 10⫺11cm3molecule⫺1s⫺1 and k⫺10 ⫽14.5, 5.94, 3.83, and 1.23 in units of 10⫺33cm6molecule⫺2s⫺1, respec-tively.
2. Temperature effect
a. Association/decomposition processes. Under the
con-dition of interest to the stratosphere O3 chemistry, the ClO
⫹ClO reaction is dominated by the association and
decom-position processes. In Fig. 8, the predicted and experimental rate constants for the association processes in N2 as a
func-tion of temperature are plotted and compared. It can be seen that the total rate constants for the formation of ClOOCl and ClOClO computed with our predicted dissociation energies for both molecules are in reasonable agreement with experi-mental results given by Trolier et al.,11 Nickolaisen et al.,13 Bloss et al.,15and Atkinson et al.;54they are, however, about one order of magnitude lower than that reported by Hayman
et al.7in a similar temperature range. The predicted second-and third-order rate constants for formation of ClOOCl second-and ClOClO can be expressed, respectively, by
k1⬁⫽1.6⫻10⫺9T⫺0.67exp共⫺64/T兲cm3molecule⫺1s⫺1, k1⬁⬘⫽6.4⫻10⫺9T⫺0.78exp共⫺76/T兲cm3molecule⫺1s⫺1, k10⫽8.31⫻10⫺20T⫺4.96exp共⫺336/T兲cm6molecule⫺2s⫺1, k1 ⬘ 0 ⫽1.72⫻10⫺14 T⫺6.99exp共⫺926/T兲cm6molecule⫺2s⫺1, for the temperature range 180–500 K. Similarly, the unimo-lecular decomposition rate constants for the dissociation of ClOOCl and ClOClO can be given by the following expres-sions for the high- and low-pressure limits:
k⫺1⬁ ⫽6.3⫻1019T⫺1.32exp共⫺9999/T兲s⫺1, k1 ⬘ ⬁⫽5.99⫻1020T⫺1.63exp共⫺6474/T兲s⫺1, k10⫽4.64⫻108T⫺5.2exp共⫺10 159/T兲cm3molecule⫺1s⫺1, k⫺10 ⬘⫽6.81⫻106T⫺4.9exp共⫺6488/T兲cm3molecule⫺1s⫺1. The optical isomers of ClOOCl have been considered in the prediction of ClOOCl dissociation constant.
In Fig. 8, we have also compared the predicted total third-order rate constants 共dotted and dashed lines 4 and 5兲 with D0(ClO– OCl)⫽16.3 and 14.1 kcal/mol. These lower values of dissociation energy again underpredict the third-order rate constant noticeably; at 200 K, for example, the predicted values are a factor of 2 and 3, respectively, below experimental data, which agree quantitatively with the result calculated with the 18.9 kcal/mol dissociation energy evalu-ated by the G2M method.
b. Equilibrium constants. K1 p and K1⬘p. The predicted
equilibrium constants for reactions共1兲 and 共1⬘兲
FIG. 8. Comparison of the predicted third-order rate constants with the experimental values in N2. Lines 1, 2, and 3 represent the predicted
forma-tion rate constants of ClOClO, ClOOCl, and their sum, i.e., k1⬘, k1, and
k1⫹k1⬘; dotted and dashed lines 4, and 5 represent the predicted kt using
D0⫽16.3 and 14.1 kcal/mol for ClOOCl association process, respectively;
symbols are the experimental values䊊, Ref. 共13兲; ---䊉---, Ref. 7; ---䉱---, Ref. 11; --䉲--, Ref. 55; 䉮, Ref. 15.
TABLE III. Comparison of the predicted and experimental k⬁ and k0in different third bodies for ClO⫹ClO
→Cl2O2⫹M at 300 K.
M P/Torr ka 具⌬Ecmdown⫺1典
He 0 0.74,b0.99⫾(0.05) 共Ref. 13兲, 0.46⫾0.04 共Ref. 11兲 120 Ar 0 1.34,b1.71⫾0.06 共Ref. 13兲 400 N2 ⬁ 87, b (6⫾2) 共Ref. 13兲, 8.0 共Ref. 11兲 N2 0 1.85, b 1.96⫾0.24 共Ref. 13兲, 1.8⫾0.5 共Ref. 10兲, 1.34 ⫾0.09 共Ref. 11兲 400 O2 0 1.54, b 1.24⫾0.09 共Ref. 13兲, 1.10⫾0.08 共Ref. 11兲, 1.7 ⫾0.5 共Ref. 10兲 350 CF4 0 2.1,b2.6⫾0.17 共Ref. 13兲 500 SF6 0 3.37,b3.15⫾0.14 共Ref. 13兲, 2.22⫾0.14 共Ref. 11兲 1000 ak, the sum of k
1and k1⬘ ; is given in units of 10⫺32cm6molecule⫺2s⫺1for k0and 10⫺12cm3molecule⫺1s⫺1
for k⬁.
bThis work.
ClO⫹ClO⫽ClOOCl, 共1兲
ClO⫹ClO⫽ClOClO, 共1⬘兲
are shown in Fig. 9 by solid lines 1 and 2, respectively, for comparison with experimental values assuming the presence of only reaction 共1兲. The predicted K1 p is apparently larger than experimental values9,13 by almost 2 orders of magni-tude, whereas K1⬘p is lower by 3–5 orders of magnitude in
the experimental temperature range based on the predicted enthalpy changes, ⫺18.9 and ⫺11.9 kcal/mol, respectively. The total Kp 共dash-dot-dotted line 3兲 predicted by Slanina
and Filip36using⌬1H0
o⬃⫺16.1 kcal/mol including the three stable isomers共ClOOCl, ClOClO, and ClClO2) is also com-pared in Fig. 9; their result is about one and half orders of magnitude lower than the experimental values. As our pre-dicted pressure dependence and the third-body effect on the association rate constants at different temperatures using the predicted enthalpy changes, ⌬1H0
o⫽⫺18.9 and ⌬ 1⬘H0
0
⫽⫺11.9 kcal/mol, are quite reasonable, the deviation results
most likely from the contributions of reaction 共1⬘兲 共which was not included in all previous kinetic data analyses兲 and radical reactions involving ClOOCl. Slanina and Filip36 as-sumed that ClClO2 could contribute to the total observed
Kp, but from the calculated PES we see that ClClO2 is not easily accessible from ClO⫹ClO due to the high isomeriza-tion barrier at TS2 共see Fig. 3兲; its contribution to the total
Kp may be neglected.
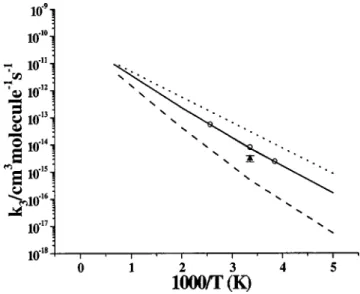
c. Cl2⫹O2formation. The reported rate constant for the
formation of Cl2⫹O2 exhibits an order of magnitude scatter at 298 K.5,6,12,13,54 –58As described in the ab initio calculation part, ClOOCl can dissociate to Cl2⫹O2 via a four-center transition state TS3 with a barrier of 3.6 kcal/mol above the reactants. With this barrier height, the result of our
CHEMRATE calculation based on Schemes 1 and 2 given
above, as shown in Fig. 10, is in reasonable agreement with the values reported by DeMore et al.55and Simon et al.12 at low temperature, but the model slightly overestimated at high temperatures compared with the most recent
experimen-tal values of Nickolaisen et al.13 However, at 1250 K, our predicted value is close to the one given by Park.56
The predicted k2 can be expressed by k2⫽1.09
⫻10⫺13T0.66exp(⫺1892/T) cm3molecule⫺1s⫺1. For com-parison, the contribution from Scheme 2 is also shown as a dotted line in Fig. 10. The result indicates that below 298 K, the indirect contribution from Scheme 2 to k2 via ClOClO* is less than 7%; however, at higher temperatures, the contri-bution increases and reaches around 40% at 1500 K.
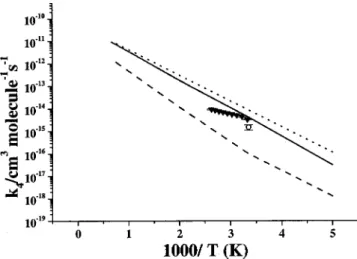
d. Cl⫹ClOO formation. ClOOCl can also dissociate
barrierlessly to produce Cl⫹ClOO without a well-defined transition state. The variational potential energies were ob-tained by varying the breaking O–Cl bond from 1.748 to 3.548 Å at an interval of 0.1 Å. The computed potential
FIG. 9. Comparison of the predicted temperature dependence for equilib-rium constants K1 pand K1⬘pwith experimental values. -䊉-, Ref. 13; -䊊-, Ref. 9. The meanings of lines 1–3 can be found in the related part in the text.
FIG. 10. Comparison of the predicted bimolecular reaction rate constant for ClO⫹ClO→Cl2⫹O2 with the experimental values. Solid line is the
pre-dicted total value coupling Schemes 1 and 2; dotted line is the contribution from Scheme 2. -䊉-, Ref. 55; -䉱-, Ref. 13;丣, Ref. 12;䉭, Ref. 6; 䊐, Ref. 5;⫻, Ref. 56; 䉮, Ref. 57; 䉲; Ref. 58.
FIG. 11. Comparison of the predicted bimolecular reaction rate constant for ClO⫹ClO→Cl⫹ClOO with the experimental values. Dotted and solid lines are calculated by using predicted 23.1 and experimental 23.8 kcal/mol for the heat of formation for ClOO, respectively. Dashed line is the contribution from Scheme 2 using the experimental heat of formation for ClOO. -䊊-, Ref. 13; -䉱-, Ref. 5.
energies could be fitted to the Morse function with the pa-rameter, ⫽3.343 Å⫺1. The predicted and experimental heats of formation for ClOO, 23.1 and 23.8 kcal/mol, were both used in the rate constant calculation. The results are given by dotted and solid curves, respectively, in Fig. 11. The latter, obtained with the experimental heat of formation for ClOO, is shown to give a much better agreement with ex-perimental data. The obvious deviation between the two the-oretical curves stemming from a rather small difference in the endothermicity employed, 0.7 kcal/mol, clearly illustrates the sensitivity of the computed rate constants to the predicted energetics. The result corresponding to the solid curve can be expressed by k3⫽1.36⫻10⫺13T0.77exp(⫺2168/T) cm3molecule⫺1s⫺1, covering the temperature range 200– 1500 K. The small contribution from Scheme 2 to k3, similar to that for Cl2⫹O2 formation, is shown as a dashed line in Fig. 11.
e. Cl⫹OClO formation. In principle, Cl⫹OClO can be
formed by the dissociation of ClOClO and ClClO2; how-ever, due to the higher isomerization barrier from ClOClO to ClClO2 共see Fig. 3兲, formation of Cl⫹OClO from ClClO2 can be neglected. For the decomposition of ClOClO, the variational potential energies were obtained by varying the breaking O–Cl bond from 1.720 to 5.12 Å at an interval of 0.1 Å, and⫽1.732 Å⫺1 is determined by fitting the calcu-lated potential to the Morse function. Figure 12 compares the predicted data with experimental values. The dotted and solid lines represent the results by using the predicted and experimental heats of formation for OClO, 23.2 and 23.7 kcal/mol, respectively. The latter result indicates that the predicted values given by k4⫽6.26⫻10⫺11T0.005
⫻exp(⫺2896/T) cm3molecule⫺1s⫺1 covering the tempera-ture range of 200 and 1500 K are in close agreement with the reported data by Cox and Derwent5and Nickolaisen et al.13 at lower temperatures, but deviate noticeably from experi-mental values at higher temperatures. The deviation may arise in part from the large uncertainty in the heat of
forma-tion of OClO, 23.7⫾1.9 kcal/mol46共see Table II兲. In Fig. 12, we also present the indirect contribution from Scheme 1 to
k4, shown as a dashed line. The result suggests that the isomerization between ClOOCl* and ClOClO* has a minor effect on k4, less than 5% below room temperature.
3. Comparison of k2¿k3and k2Õk3
with experimental values
Experimentally, Nickolaisen et al.13 and Horowitz
et al.14reported the directly measured results for k2⫹k3 and
k2/k3in the temperature ranges of 270–390 K and 285–331 K, under the corresponding pressures of 1–36 and 503–576 Torr, respectively. Figures 13共a兲 and 13共b兲 compare the pre-dicted and experimental data. It can be seen from Fig. 13共a兲 that the predicted values for k2⫹k3 agree closely with the result of Nickolaisen et al.13 The predicted k2/k3 ratios lie between the values reported by Horowitz et al.14 and those calculated with the individual k2 and k3 obtained by Nickolaisen et al.13The large scatter in the experimental re-sults reflects the difficulty in determining the product branch-ing ratios experimentally.
FIG. 12. Comparison of the predicted bimolecular reaction rate constant for ClO⫹ClO→Cl⫹OClO with the experimental values. Dotted and solid lines are calculated by using predicted 23.2 and experimental 23.7 kcal/mol for the heat of formation for ClOO, respectively. Dashed line is the contribution from Scheme 1 using the experimental heat of formation for ClOO. -䊉-, Ref. 13; -䊊-, Ref. 5.
FIG. 13. Comparison of the predicted共a兲 k2⫹k3and共b兲 k2/k3with
experi-mental values. Solid line is the predicted result.䊊, Ref. 13; 䊉, Ref. 14.
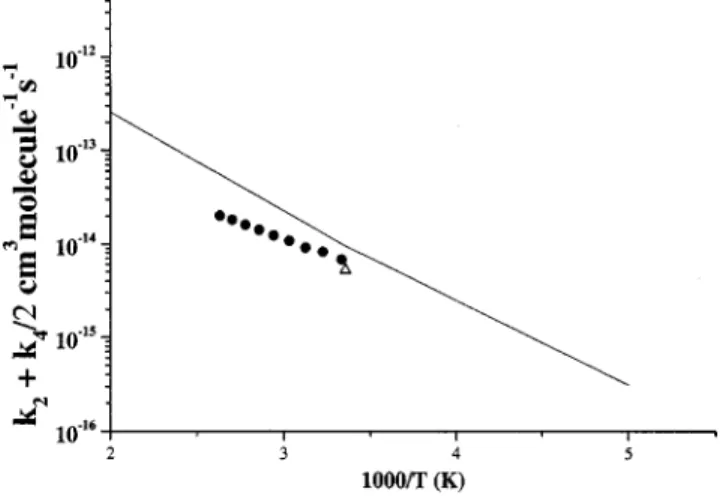
4. Comparison of k2¿k4Õ2 with experimental values
Figure 14 shows the comparison of the predicted k2
⫹k4/2 with experimental results of Nickolaisen et al.13 As already shown in Figs. 10 and 12, at high temperatures theory overestimated both k2 and k4; this is also reflected in the comparison. The differences between the predicted and experimental values of k2 and k4 could be attributed to the difficulty in differentiating experimentally the reaction prod-ucts, particularly Cl2⫹O2 versus Cl⫹ClOO 共whose reaction quickly generates the former product pair兲. Take the result at 298 K as an example; the product branching ratios for Cl2
⫹O2, Cl⫹ClOO, and Cl⫹OClO estimated by Cox and Derwent,5Simon et al.,12Nickolaisen et al.,13 and Horowitz
et al.,14 vary noticeably: (0.49⫾0.11,0.34⫾0.09,0.17
⫾0.05); (0.33⫾0.08,0.33⫾0.07,0.34⫾0.12); 共0.30, 0.49,
0.21兲; and 共0.39, 0.40, 0.20兲, respectively. The ratio predicted in this work 共0.39, 0.40, 0.21兲, is in reasonable agreement with those reported by Nickolaisen, Horowitz, and co-workers.
IV. CONCLUDING REMARKS
The kinetics and mechanism for the self-reaction of ClO radicals have been investigated at the G2M/B3LYP/6-311
⫹G(3d f ) level of theory in conjunction with statistical rate
constant calculations. Experimentally determined rate con-stants for the bimolecular association process producing Cl2O2 could be quantitatively accounted for only with the inclusion of both ClOOCl and ClOClO dimer products. The latter accounts for about 23% of the dimer yield at 200 K and 22% at room temperature at 100 Torr N2. The rate constants for the formation of Cl2⫹O2 (k2), Cl⫹ClOO (k3), and Cl
⫹OClO (k4), could be reasonably accounted for theoreti-cally, however, with noticeable overestimation in k2 and k4, compared with those reported recently by Sander and co-workers.13 The origins of the deviations, aside from the potential predictive error of the theory in energetics 共⫾1 kcal/mol based on the heats of formation obtained computa-tionally for OClO, ClOO, and ClOOCl兲, omissions of many important stable species in kinetic modeling of
experimen-tally measured data might also have contributed to errors in evaluating rate constants and product branching ratios. For example
Cl⫹OClO⫹M→ClClO2⫹M,
ClO⫹OClO⫹M→ClOClO2⫹M, etc.
The formation of these and other stable intermediates共such as ClOClO兲 and their reactions with Cl, ClO, and OClO may significantly affect the predicted concentrations of these re-active species, which were monitored experimentally in most kinetic studies. To reliably examine the effects of these and other ClOx reactions, efforts are under way to calculate the rate constants for the association of Cl and ClO with OClO and the reactions of Cl2O2 and Cl2O3 isomers with these radicals.
Finally, it should be mentioned that although all existing kinetic data for the recombination and disproportionation re-actions of ClO radicals could be quantitatively accounted for with our predicted energies within⫾1.0 kcal/mol, the obvi-ous discrepancy between the heat of formation of ClOOCl derived from the computed dimerization energy, ⌬fH0o (ClOOCl)⫽29.4 kcal/mol, and the value estimated by a pre-sumably more reliable isodesmic calculation based on the Cl2O⫹HOOH⫽ClOOCl⫹H2O reaction, 32.6⫾1.0 kcal/mol, deserves further investigation. The latter, larger heat of for-mation significantly underpredicts the independently deter-mined, more reliable recombination kinetic data by several research groups.
ACKNOWLEDGMENTS
This work is sponsored by the Office of Naval Research under Contract No. N00014-02-1-0133, Dr. J. Goldwasser, program manager. We appreciate the useful discussion with Dr. D. M. Golden on the kinetics for the recombination of ClO radicals.
1R. S. Stolarski and R. J. Cicerone, Can. J. Chem. 52, 1610共1974兲. 2F. S. Rowland and M. J. Molina, Rev. Geophys. Space Phys. 78, 5341
共1975兲; F. S. Rowland, Annu. Rev. Phys. Chem. 42, 731 共1991兲; H. S.
Johnston, ibid. 43, 1共1992兲.
3共a兲 M. A. A. Clyne, D. J. McKenney, and R. T. Watson, J. Chem. Soc.,
Faraday Trans. 1 71, 322共1975兲; 共b兲 M. A. A. Clyne, D. J. McKenney, and R. T. Watson, ibid. 73, 1169共1977兲.
4
N. Basco and J. Hunt, Int. J. Chem. Kinet. 11, 649共1979兲.
5R. A. Cox and R. G. Derwent, J. Chem. Soc., Faraday Trans. 1 75, 1635 共1979兲.
6J. P. Burrows and R. A. Cox, J. Chem. Soc., Faraday Trans. 1 77, 2465 共1981兲.
7
G. D. Hayman, J. M. Davies, and R. A. Cox, Geophys. Res. Lett. 13, 1347
共1986兲.
8L. T. Molina and L. J. Molina, J. Phys. Chem. 91, 43743共1987兲. 9R. A. Cox and G. D. HayMan, Nature共London兲 332, 796 共1988兲. 10
S. P. Sander, R. Friedl, and Y. L. Young, Science 245, 1095共1989兲.
11M. Trolier, R. L. Mauldin III, and A. R. Ravishankara, J. Phys. Chem. 94,
4896共1990兲.
12F. G. Simon, W. Schneider, G. K. Moortgat, and J. P. Burrows, J.
Photo-chem. Photobiol., A 94, 4896共1990兲.
13
S. L. Nickolaisen, R. R. Friedl, and S. P. Sander, J. Phys. Chem. 98, 155
共1994兲.
14A. Horowitz, J. N. Crowley, and G. K. Moortgat, J. Phys. Chem. 98,
11924共1994兲.
15
W. J. Bloss, S. L. Nickolaisen, R. J. Salawitch, R. R. Friedl, and S. P. Sander, J. Phys. Chem. A 105, 11226共2001兲.
16Z. F. Xu, R. S. Zhu, and M. C. Lin, J. Phys. Chem.共submitted兲.
FIG. 14. Comparison of the predicted k2⫹k4/2 with experimental values.
Solid line is the predicted result.䊉, Ref. 13. 䉭, Ref. 5.
17R. S. Zhu and M. C. Lin, PhysChemComm 25, 1共2001兲.
18R. S. Zhu, Z. F. Xu, and M. C. Lin, J. Chem. Phys. 116, 7452共2002兲. 19R. S. Zhu and M. C. Lin, J. Phys. Chem. A 106, 8386共2002兲. 20
A. D. Becke, J. Chem. Phys. 98, 5648共1993兲.
21A. D. Becke, J. Chem. Phys. 96, 2155共1992兲; 97, 9173 共1992兲. 22C. Lee, W. Yang, and R. G. Parr, Phys. Rev. B 37, 785共1988兲. 23C. Gonzalez and H. B. Schlegel, J. Phys. Chem. 90, 2154共1989兲. 24A. M. Mebel, K. Morokuma, and M. C. Lin, J. Chem. Phys. 103, 7414
共1995兲.
25M. J. Frisch, G. W. Trucks, H. B. Schlegel et al.,
GAUSSIAN 98, Revision A.1, Gaussian, Inc., Pittsburgh, PA, 1998.
26S. J. Klippenstein, A. F. Wagner, R. C. Dunbar, D. M. Wardlaw, and S. H.
Robertson,VARIFLEX: Version 1.00, 1999.
27
R. G. Gilbert and S. C. Smith, Theory of Unimolecular and
Recombina-tion ReacRecombina-tions共Blackwell Scientific, Carlton, Australia, 1990兲.
28K. A. Holbrook, M. J. Pilling, and S. H. Robertson, Unimolecular Reac-tions共Wiley, New York, 1996兲.
29
V. Mokrushin, V. Bedanov, W. Tsang, M. R. Zachariah, and V. D. Knyazev, CHEMRATE, Version 1.19, National Institute of Standards and Technology, Gaithersburg, MD 20899, 2002.
30M. P. McGrath, K. C. Clemitshaw, F. S. Rowland, and W. J. Hehre,
Geo-phys. Res. Lett. 15, 883共1988兲.
31
M. P. McGrath, K. C. Clemitshaw, F. S. Rowland, and W. J. Hehre, J. Phys. Chem. 94, 6126共1990兲.
32F. Jensen and J. Oddershede, J. Phys. Chem. 94, 2235共1990兲.
33J. F. Stanton, C. M. L. Rittby, R. J. Bartlett, and D. W. Toohey, J. Phys.
Chem. 95, 2107共1991兲.
34A. P. Rendell and T. J. Lee, J. Chem. Phys. 94, 6219共1991兲. 35Z. Slanina and F. Uhlik, J. Phys. Chem. 95, 5432共1991兲.
36Z. Slanina and F. Uhlik, Chem. Phys. Lett. 182, 51共1991兲; Geophys. Res.
Lett. 18, 1703共1991兲.
37
T. J. Lee, C. M. Rohlfing, and J. E. Rice, J. Chem. Phys. 97, 6593共1992兲.
38D. Christen, H. G. Mack, and H. S. P. Mu¨ller, J. Mol. Struct. 509, 137 共1999兲.
39M. Birk, R. R. Friedl, E. A. Cohen, H. M. Pickett, and S. P. Sander, J.
Chem. Phys. 91, 6588共1989兲.
40H. S. P. Mu¨ller and E. A. Cohen, J. Phys. Chem. A 101, 3049共1997兲. 41
H. S. P. Mu¨llerand H. Willner, Inorg. Chem. 31, 2527共1992兲.
42F. Jensen and J. Oddershede, J. Phys. Chem. 94, 2235共1990兲. 43B. M. Cheng and Y. P. Lee, J. Chem. Phys. 90, 5930共1989兲.
44J. Jacobs, M. Kronberg, H. S. P. Mu¨ller, and H. Willner, J. Am. Chem.
Soc. 116, 1106共1994兲.
45
W. K. Li and C. Y. Ng, J. Phys. Chem. A 101, 113共1997兲.
46
M. W. Chase, Jr., NIST-JANAF Thermochemical Tables, 4th ed. 共AIP, Woodbury, NY, 1998兲.
47J. O. Hirschfelder, C. F. Curtiss, and R. B. Bird, Molecular Theory of Gases and Liquids, 2nd ed.共Wiley, New York, 1964兲.
48H. Hippler, J. Troe, and H. J. Wendelken, J. Chem. Phys. 78, 6709共1983兲. 49C.-C. Hsu, A. M. Mebel, and M. C. Lin, J. Chem. Phys. 105, 2346共1996兲. 50D. Chakraborty, C.-C. Hsu, and M. C. Lin, J. Chem. Phys. 109, 8887
共1998兲. 51
R. S. Zhu, E. G. W. Diau, M. C. Lin, and A. M. Mebel, J. Phys. Chem. A 105, 11249共2001兲.
52
J. Troe, J. Phys. Chem. 83, 114共1979兲.
53J. Troe, Ber. Bunsenges. Phys. Chem. 87, 161共1983兲; R. G. Cilbert, K.
Luther, and J. Troe, ibid. 87, 169共1983兲.
54R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, Jr., J. A. Kerr, M.
J. Rossi, and J. Troe, J. Phys. Chem. Ref. Data 26, 521共1997兲.
55W. B. DeMore, S. P. Sander, D. M. Golden, R. F. Hampson, M. J. Kurylo,
C. J. Howard, A. R. Ravishankara, C. E. Kolb, and M. J. Molina, JPL Publication 97-4共1997兲.
56
C. Park, J. Phys. Chem. 80, 565共1976兲.
57
F. H. C. Edgecombe, R. G. W. Norrish, F. R. S. Thrush, and B. A. Thrush, Proc. R. Soc. London, Ser. A 243, 24共1957兲.
58F. J. Lipscomb, R. G. W. Norrish, and G. Porter, Nature共London兲 174,
785共1954兲.