502 ORIGINAL RESEARCH
ORIGINAL RESEARCH
Y.-C. Chou et al.Biomed. Chromatogr. 22: 502–510 (2008)
Published online 18 January 2008 in Wiley InterScience (www.interscience.wiley.com) DOI: 10.1002/bmc.960
Determination of lipoprotein lipase activity in post
heparin plasma of streptozotocin-induced diabetic
rats by high-performance liquid chromatography
with fluorescence detection
Yu-Ching Chou,1 Yih-Chiao Tsai,1 Chien-Ming Chen,2 Shih-Ming Chen3 and Jen-Ai Lee1*
1Department of Pharmaceutical Analysis, School of Pharmacy, Taipei Medical University, No. 250, Wu-Hsing St, Taipei 110, Taiwan 2Department of Electro-Optical Engineering, National Taipei University of Technology, No. 1, Sec. 3, Chung-Hsiao E. Rd, Taipei 106, Taiwan 3Department of Clinical Pharmacy, School of Pharmacy, Taipei Medical University, 250 Wu-Hsing St., Taipei 110, Taiwan
Received 13 September 2007; accepted 29 September 2007
ABSTRACT: The activity of lipoprotein lipase (LPL), an enzyme responsible for lipoprotein metabolism, would vary in diseases and metabolic disorders. For determination of LPL activity, a highly sensitive high performance liquid chromatography (HPLC) method using a fluorescent reagent, 4-nitro-7-piperazino-2,1,3-benzoxadiazole (NBD-PZ) was applied to determinate the oleic acid (OA) generated from triolein by LPL activity without multiple solvents extraction step. We studied the optimal conditons of the reaction including the effect of emulsifiers, deproteinizing solvents, and the concentration of bovine serum albumin (BSA). Ten millimolar concentrations of triolein, 5% of BSA, 1% of Gum arabic (GA), and acetonitrile showed the optimum conditions for measuring the LPL activity. The accuracy values for the determination of LPL activity in 10μL of rat post heparin plasma were 108.73 ~ 114.36%, and the intra- and inter-day precision values were within 1.28% and 2.91%, respectively. The limit of detection was about 4.53 nM (signal-to-noise ratio 3).
The proposed method was applied to determination of LPL activity in post heparin plasma of normal and streptozotocin-induced diabetic rats associated with 52.3% reduction. The established assay system could be used for determining LPL activity in different physiological and pathological conditions to clarify the relationship between LPL activity and diabetes mellitus. Copyright © 2008 John Wiley & Sons, Ltd.
KEYWORDS: lipoprotein lipase; fluorescence; HPLC; NBD-PZ; emulsifiers; BSA; STZ-induced diabetic rats
*Correspondence to: Jen-Ai Lee, Department of Pharmaceutical Analysis, School of Pharmacy, Taipei Medical University, No. 250, Wu-Hsing St, Taipei 110, Taiwan.
E-mail: [email protected]
Abbreviations used: BSA, bovine serum albumin; BuOH, n-butanol; DPDS, 2,2′-dipyridyl disulfide; EtOH, ethanol; FFAs, free fatty acids; GA, gum arabic; HPLC, high performance liquid chromatography; LPL, lipoprotein lipase; MeCN, acetonitrile; MeOH, methanol; NBD-PZ, 4-nitro-7-piperazino-2,1,3-benzoxadiazole; OA, oleic acid; STZ, streptozotocin; TPP, triphenylphosphine; TFA, trifluoroacetic acid. Contract/grant sponsor: National Science Council of the Republic of China; Contract/grant number: NSC 94-2113-M-038-005.
INTRODUCTION
Lipoprotein lipase (LPL, EC 3.1.1.34), a 448-residue glycoprotein, is responsible for the metabolism of triglyceride-rich lipoproteins in plasma. It is synthesized in tissue parenchymal cells and is secreted and trans-ported to the capillary endothelium, where it binds to heparin sulfate (Goldberg, 1996; Santamarina-Fojo and Dugi, 1994). Abundant amounts of LPL are found mainly in organs with a high demand for free fatty
acids (FFAs) such as adipose tissues, heart and skeletal muscles (Semenkovich et al., 1989; Zechner, 1997). Studies have indicated that important roles of this rate-limiting enzyme exist in lipoprotein metabolism and clinical disorders, including diabetes mellitus, cardiovascular diseases, diet-related metabolic disorders and lipoprotein metabolism-related disorders such as hyperlipoproteinemia, and familial hypercholesterolemia (Eckel, 1989; Mahley et al., 1991; Scheen, 1999). These observations not only provide insights into the impor-tance of lipoprotein lipase, but also implicate the potential role of LPL activity in the pathogenesis of dyslipidemia in the condition associated with insulin resistance (Jeppesen et al., 1995; Nikkilä et al., 1977; Pulawa and Eckel, 2002; Pykalisto et al., 1975; Taskinen et al., 1982). A number of studies have shown various results that have been difficult to verify due to methodological differences such as the type of substrates measured, variable reaction medium and conditions, and different sample characteristics (Maheux et al., 1997; Sartippour and Renier, 2000; Yamazaki et al., 2002). Over the last
decade, the standard analytical system of LPL is still limited. In addition, recent studies have failed to examine the effect of the composition of reaction medium on the enzyme reaction, since changing the contents of the emulsion could theoretically produce results. However, this may cause methodological errors.
To accurately determine the LPL activity, several factors have been considered in developing appropriate analytical assays. These factors include choosing the substrate of LPL to be as physiologically specific as possible and the chemicals representing the end pro-ducts to have enough particle surfaces for interfacial acti-vation and co-enzymes, and the assay to be as sensitive as possible (Bauer, 1988; Gupta et al., 2003; Wang et al., 1982). To date, numerous methods, including titrimetric (Hoppe and Theimer, 1996), radiometric (Nilsson-Ehle and Schotz, 1976), fluorimetric (Duque et al., 1996) and HPLC assays (Eguchi, 2002), have been developed. Among these methods, the radioisotope-labeled sub-strate for LPL has been widely used to evaluate LPL activity in various physiological and pathological condi-tions. However, there are some drawbacks associated with this method, such as its inability to completely separate FFAs and radioactive substrates, which may affect the precision and accuracy of analysis (Belfrage and Vaughan, 1969). Other difficulties, including ex-posed to radiation hazards from radioactive materials, disposal of radioactive isotopes, restriction to licensed laboratories and the short half-life of the labeled reagents, have also been mentioned (Ball et al., 2002). Many commercial kits with fluorescent-labeled substrates may solve the hazard from radioisotopes but they exhibit time-limit reactivity and high background disturbances and are not suitable for long-term study (Beisson et al., 2000; Sartippour and Renier, 2000). A better analysis for LPL activity using HPLC has been developed by Eguchi (2002). However, adequate con-centrations of emulsifier or BSA, which factor is domi-nant in LPL reactions, have not been investigated.
Considering these problems, an assay system which is able to detect and quantify fatty acid generated by LPL using HPLC plus fluorimetric detection, without using the step of sample extraction and producing un-stable synthesized substrates, has been established. The present study was carried out to investigate the impor-tance of LPL reaction medium. More specifically, the goal of this study was to evaluate the influence of emul-sifiers, serum albumin, and solvent system on the pro-posed assay for plasma LPL activity. For this purpose, LPL activity was measured in reaction media and conditions. Subsequently, the proposed method was applied to determine the activity of LPL in normal and streptozotocin (STZ)-induced diabetic rats to establish an appropriate model for elucidating the character of LPL activity in plasma homeostasis of diabetes mellitus.
EXPERIMENTAL
Chemicals. LPL from bovine milk (0.94 mg protein/mL), GA,
sodium deoxycholate, Tween® 20, Tween® 40, Tween® 80,
BSA, heparin, streptozotocin (STZ), triolein, OA (C18:1), and
heptadecanoic acid (margaric acid, C17:0) were purchased
from Sigma Chemical (St Louis, MO, USA). Tris–HCl buffer was obtained from Calbiochem, which belongs to EMD Biosciences (Darmstadt, Germany). Sodium chloride, sodium hydroxide and citric acid monohydrate were purchased from Nacalai Tesque (Kyoto, Japan). 4-Nitro-7-piperazino-2,1,3-benzoxadiazole (NBD-PZ), triphenylphosphine (TPP) and 2,2′-dipyridyl disulfide (DPDS) were obtained from Tokyo Kasei Chemicals (Tokyo, Japan). Trifluoroacetic acid (TFA) was purchased from Riedel-de Haën (Seelze, Germany). HPLC-grade acetonitrile (MeCN), methanol (MeOH), ethanol (EtOH) and n-butanol (BuOH) were from Merck (Darmstadt, Germany).
Preparation of enzyme source from normal and STZ-induced diabetic rats. Animal experiments were approved
by the Laboratory Animal Research Committee of Taipei Medical University. Wistar male rats (Laboratory Animal Center of National Taiwan University, Taipei, Taiwan) weighing 200–250 g were used in the experiment and kept in an environmentally controlled room with food and tap water
ad libitum. To induce diabetic rats, streptozotocin dissolved in
50 mM citrate buffer (pH = 4.5) was administered
intraperi-toneally at a dose of 100 mg/kg after fasting for 12 h as de-scribed in previous study (Lee et al., 2005). Diabetes mellitus was confirmed by documenting persistent hyperglycemia that was measured using a commercial kit (Lifescan, CA, USA). Lipoprotein lipases that bound to heparin sulfate on the endothelial wall were released by injecting 100 IU/kg of heparin intravenously. After 10 min, the blood of experimental animals from normal and diabetic rats was drawn from the tail vein and centrifuged with Mikro 22R Centrifuge (Hettich Zentrifugen, Tuttlingen, Germany) at 10,000 rpm for 10 min at 4°C im-mediately. The supernatant was used as an enzyme source and stored at −20°C.
Proceeding of the enzyme reaction. The assay of LPL
activity is based on fluorimetric measurement of derivatized fatty acids formed enzymatically from the external added substrate, triolein (Eguchi, 2002). Experiments for the selec-tion of substrate concentraselec-tions, emulsifiers, BSA concentra-tions and deproteinized solvents were conducted for optimizing the assay system. First of all, five types of emulsifiers were used for emulsifying the triolein substrate, including GA, sodium deoxycholate, Tween® 20, 40 and 80. Weighted
emul-sifiers were mixed and sonicated with the buffer containing 0.2M Tris–HCl, and 0.1M NaCl (pH adjusted to 8.2 with 1M
NaOH) until homogenization. The concentration of each emulsifier was set at 1 and 5% (w/v). Among the chosen emulsifiers, GA was commonly used for stabilized triolein emulsion. Consequently, GA concentrations of 0, 0.25, 0.5, 1, 2, 4, 6 and 8% were also evaluated. Pure triolein was then emulsified with the buffer made with different types and con-centrations of emulsifiers according to the descriptions above. Moreover, the proportion of BSA in the reaction buffer was investigated at six concentrations (0, 1.25, 2.5, 5, 7.5 and
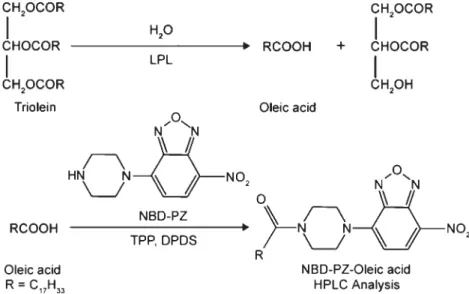
Figure 1. The principle for determination of LPL activity. OA, the product
generated after triolein hydrolyzed by LPL, was derivatized with NBD-PZ and quantified by a HPLC system.
10%). Ten micro-liters of enzyme source was combined with triolein emulsion to a total volume of 100μL and incubated for 30 min at 37°C. The enzyme reaction was terminated and deproteinized by addition of 900μL of acetonitrile and centri-fuged directly at 3300 rpm for 5 min at 25°C. Equal volumes of MeOH, EtOH, and BuOH were tested as well. The supernatant in each experiment was continually derivatized and analyzed as follows.
Derivatization of oleic acid with NBD-PZ. OA released
from triolein by LPL activity was derivatized with a fluo-rescence reagent, NBD-PZ, via the principle of the deriva-tive reaction, as shown in Fig. 1. The procedures were carried out according to our previous method with minor modifica-tions (Tsai et al., 2003). Briefly, 100μL of 2 mM NBD-PZ in
MeCN mixed with 10μL each of 280 mM TPP and DPDS in
MeCN was supplemented with the resulting reaction. Ten microliters of 0.5 mM margaric acid dissolved in MeCN was
added as an internal standard (IS) (Sparreboom et al., 1996). The derivatization reaction was incubated for 20 min at 40°C and then terminated by the addition of 450μL of 0.1% TFA in distilled deionized water. An Empore SDB-RPS cartridge (3M, St Paul, MN, USA) was used to remove the excess derivative reagents. The cartridge was preconditioned with 100μL of MeCN and loaded with 100 μL of the resulting solution. One-hundred microliters of MeCN were loaded subsequently for thorough elution. Two portions of the eluent were combined for HPLC analysis. All procedures were per-formed using disposable polypropylene brown Eppendorf tubes (1.5 mL) to speed the experiments.
HPLC conditions. The samples were subjected to a
reversed-phase HPLC system consisted of an intelligent auto-sampler (AS-950; Jasco Co., Tokyo, Japan) equipped with a 20μL sample loop and a pump (L-7100; Hitachi, Tokyo, Japan). The composition of the mobile phase was acetonitrile– methanol–water (50:35:15, v/v/v) performed for 30 min at a flow rate of 0.7 mL/min. NBD-PZ labeled fatty acids were
separated on an C8 column (YMC-Pack Pro C8, 150 × 4.6 mm
i.d., spherical 5μm, 120 Å, YMC Co., Kyoto, Japan) and detected with a fluorescence detector (F-1000; Hitachi, Tokyo, Japan) at 491 and 547 nm excitation and emission wavelengths, respectively. The results were analyzed using a D-2500 chromato-integrator (Hitachi, Tokyo, Japan).
Validation study. For quantitation of the OA formation by
the enzyme action, a calibration curve was constructed. One-hundred microliters of standard OA at concentrations of 0, 0.50, 1.00, 2.00, 4.00, 6.00 and 8.00 mM was brought up to
1000μL with MeCN and the same derivatization procedures (n = 5) were subsequently carried out. The calibration curve was plotted from the peak area ratio of derivative of OA to a derivative of margaric acid against the concentration of OA dissolved in MeCN. Spiked amounts of OA (0, 1, 2 and 3 mM) into the reaction mixture were performed to estimate
the precision and accuracy of the present analytical method. The intra- and inter-day precision expressed as the relative standard derivation (RSD) was assessed by analyzing six samples for one day and on six different days. The accuracy of this method was presented as the calculated recovery.
RESULTS AND DISCUSSION
Determination of LPL activity by the novel HPLC analysis
The existing assays for determination of lipoprotein lipase activity have been developed over three decades (Baginsky and Brown, 1977, 1979; Nilsson-Ehle and Schotz, 1976). Recently, some single nucleotide polymorphisms were discovered from human LPL gene (Hoh and Hodge, 2000; Nickerson et al., 1998; Templeton et al., 2000). Analysis for enzyme activity of mutated LPL gene needs more precise and accurate
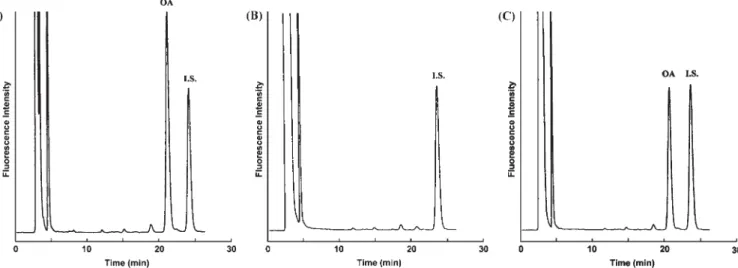
Figure 2. HPLC analysis of NBD-PZ labeled OA which was liberated by commercial LPL activity. (A) Chromatogram of
stand-ard OA. (B) Chromatogram of triolein only. (C) Chromatogram of triolein with the presence of commercial LPL.
Figure 3. HPLC analysis of NBD-PZ labeled OA which was liberated by LPL activity in rat post-heparin
plasma. (A) The peak representing the OA derivative on chromatogram could be found with the presence of LPL. (B) The peak disappeared with MeCN pretreatment since LPL was deproteinized by MeCN.
[Fig. 2(A)], whereas the blank with 10μL double-distilled deionized water instead of post heparin plasma exhib-ited no background interference [Fig. 2(B)]. As shown in Fig. 2(C), the derivatized OA by commercial bovine milk LPL was detected and completely separated from other impurities using the reversed-phase HPLC system described in the Experimental section. Figure 3(A) indicated that the successful detection of OA was produced by LPL activity in post-heparin plasma of normal rats. NBD–OA was not found when LPL was pre-denatured in post-heparin plasma of normal rats [Fig. 3(B)]. Enzyme activity was calculated according to the OA formation and expressed as milliunits in methods with good sensitivity. As pointed out in other
HPLC-based enzymatic assays, the direct analysis of enzyme action, once separated from substrate and other interfering substances, offers clear results (Eguchi, 2002). The NBD–oleic acid reaction was fast and started immediately on vortex mixing (data not shown). Figure 2 shows the chromatograms of the standard NBD–OA derivative, triolein only as a blank and NBD-PZ-labeled OA derived from sample. The NBD–OA and NBD–margaric acid derivatives were eluted at approximately 22 and 24 min, respectively. Under addition of 4 mM OA as standard control, it
observed in the study of Yadav, who demonstrated that anionic detergents that might repel the lipase from the interface or bind to the enzyme or even encapsulate the enzyme so that the substrate-binding site is blocked (Yadav et al., 1998). With 10 mM triolein as the
substrate, GA (1%) seemed to be the best emulsifier for determination of LPL activity.
1% Gum arabic provides the most efficient emulsion of the substrate
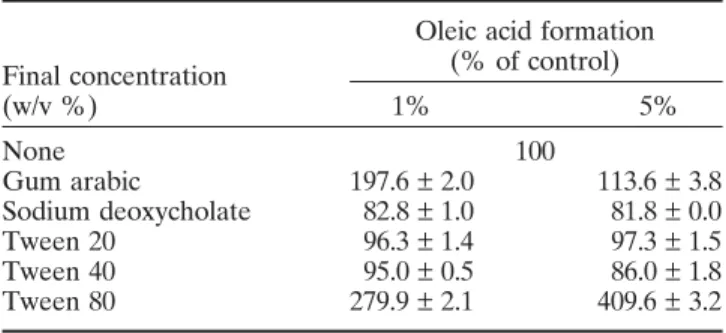
Since triolein dispersed in 1% GA instead of 5% results in the increase of enzyme activity, the optimization of GA concentrations for LPL activity in the assay emul-sion was further investigated. As shown in Fig. 4, LPL activity showed a rising tendency until GA concentra-tion reached 1%, then slowly decreased and remained stable from 2 to 8%. Using the same substrate concen-tration, identical conditions for emulsification of triolein and the same enzyme preparation, the LPL activity was clearly affected by concentration of emulsifier. Appar-ently, the enhanced emulsifying capacity of increased Table 1. The emulsification suitable for LPL by different
emulsifiers were evaluated by judging the formation of OA (n ===== 5)
Oleic acid formation
Final concentration (% of control)
(w/v %) 1% 5% None 100 Gum arabic 197.6 ± 2.0 113.6 ± 3.8 Sodium deoxycholate 82.8 ± 1.0 81.8 ± 0.0 Tween 20 96.3 ± 1.4 97.3 ± 1.5 Tween 40 95.0 ± 0.5 86.0 ± 1.8 Tween 80 279.9 ± 2.1 409.6 ± 3.2
Table 2. Precision and accuracy of oleic acid released by LPL activity (n ===== 6)
Oleic acid added (mM)
0.00 1.00 2.00 3.00 Intraday Measured (mM) 2.18 3.31 4.36 5.44 CV (%) 1.28% 1.20% 0.97% 0.90% Recovery (%) — 113.48% 108.75% 108.73% Interday Measured (mM) 2.19 3.33 4.36 5.54 CV (%) 2.91% 0.58% 0.50% 0.39% Recovery (%) — 114.36% 108.77% 111.61%
Figure 4. Influence of concentration of gum arabic. Maximum
activity of OA was observed in emulsification with 1% of gum arabic. Overloaded emulsifier caused a decrease in release of OA (n = 5).
the sample assayed. One milliunit of enzyme activity is defined as the release of 1 nmol of OA per microliter of plasma per minute at 37°C. The enzymatic reaction was linear for at least 2 h at 37°C (data not shown). Selection of emulsifiers
Numerous experimental and clinical studies have been conducted to evaluate the role of LPL activity in rela-tion to hyperlipoproteinemia, atherosclerosis and meta-bolic disorders such as obesity and diabetes. Since there are various assay systems and inconsistent components of substrate emulsion, it was difficult to evaluate and compare results for those researches (Maheux et al., 1997; Sartippour and Renier, 2000; Yamazaki et al., 2002). For improvement and optimization of the assay system, the effects of emulsifiers, BSA and depro-teinized solvents on sample were investigated. As shown in Table 1, using Tween® 80 as emulsifier
yielded the greatest enzyme activity at both concentra-tions (1 and 5%). Tween® 80 (polyoxyethylene sorbitan
monooleate) consists of monooleate and has been reported to be a substrate for lipase (Surinenaite et al., 2002); it may contribute to the OA determination but interferes with the major hydrolysis of triolein. Unlike Tween® 80, neither Tween® 20 nor 40 was suitable for
emulsification, although they did not cause interference (Archibald, 1946). LPL activity was not detected when emulsifying with the anionic detergent, sodium deoxycholate (Table 2). This phenomenon was also
such as apolipoprotein CII was necessary for LPL reac-tion (LaRosa et al., 1970). To provide the co-factor, apolipoprotein CII, serum obtained from animal or human was heated to eliminate endogenous lipolytic activity (Nilsson-Ehle and Schotz, 1976) or directly purified apolipoprotein CII from serum by complex pro-cedures (Lobo and Wilton, 1997). This heat-inactivated serum was added to the reaction emulsion in our experiment; however, the effect of apolipoprotein CII on LPL activity was limited (data not shown). Lobo and Wilton (1997) indicated that LPL activity in rat serum was still detected without heat-inactivated serum in
vitro. To simplify the assay system, heat-inactivated
serum was not used in this study. In addition, we pro-posed that the BSA might provide the synergic effect which the apolipoprotein CII had on enzyme action. Therefore, the presence of BSA was a key factor for determination of LPL activity.
Solvent effects
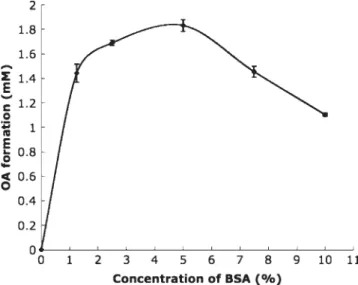
Deproteinized solvents were tested as another impor-tant parameter in establishing the assay system of LPL activity. Figure 6 showed that the value of OA liber-ated from the enzyme reaction was lower in alcoholic solvents than in MeCN (p < 0.001). It may result from the esterification of OA and alcoholic solvent mole-cules activated by the plasma lipases (Sharma et al., 2001). The usage of NBD-PZ dissolved in MeCN as the fluorescent agent also gave a better reactivity Figure 5. Influence of concentration of BSA. Maximum
activ-ity of OA was observed in emulsification with the presence of 5% of BSA. BSA is a cofactor in LPL reaction but over-loaded BSA might interfere adsorption of enzyme (n = 5).
Figure 6. Optimization of the deproteinized agents. Four
types of solvents including MeOH, EtOH, BuOH and MeCN were used. MeCN was found to dissolve the most amount of OA released by LPL compared with other three solvents. Analysis was performed by a two-tailed independent stu-dent’s t-test: * p < 0.001 compared with other types (n = 6).
GA concentrations increased the surface area of the oil–water interface, and, therefore, LPL activity is at optimum. It was surprising that continuously increased GA concentrations did not increase the enzyme activity as expected. In contrast, overloaded GA caused a decrease in activity of LPL. Possibly, OA produced by LPL when GA was overloaded in high concentrations may retain in emulsified triolein particles. After adding deproteinized solvent to terminate the LPL reaction, some OA was also removed with the emulsified triolein particle by centrifugation (Hoppe and Theimer, 1996). 5% BSA is critical for effective determination of LPL activity
The dependence of enzyme activity for BSA was also studied. LPL activity was undetectable with triolein emulsions stabilized only by the emulsifying agent, GA, in the absence of BSA in the test solution (Fig. 5). The greatest catalytic activity was observed while 5% of BSA existed in the reaction buffer. However, excessive addition of BSA (up to 10%) caused a decrease in LPL activity. Traditionally, serum albumin has been shown to interact with the lipase-emulsified substrate reaction in two different ways. It protects the lipase from irreversible inactivation at low concentration. On the other hand, it inhibits enzyme activity by blocking the substrate surface (Tsujita et al., 1996). Hence, BSA prevents the accumulation of the released OA at the water–oil interface of the emulsion which has been reported to inhibit enzyme action (Blanchette-Mackie and Scow, 1976; Karpe et al., 1992). Thus, the concen-tration of serum albumin was important for determina-tion of LPL activity. It was reported that activators
(Schneider et al., 1991) and stability compared with other derivative agents or solvents (Eguchi, 2002). The fatty acid produced during the incubations was conventionally isolated using a modification of liquid– liquid partition system described by Belfrage and Vaughan (1969). The reactions were stopped by addi-tion of methanol-chloroform-heptane 1.41/1.25/1 (v/v/v) followed by adding potassium carbonate-borate buffer (pH = 10.5) (Belfrage and Vaughan, 1969). The extrac-tion and all subsequent procedures were complicated, whereas the deproteinized solvent used in our assay was not only employed easily and eliminated contami-nation but also gave good recovery results. After opti-mization of the assay system, three components of the assays should be considered: the selection for type and concentration of emulsifiers, the proportion of BSA and the deproteinization of extraction solvents. Three of them cannot be left out of discussion because the complex mechanism of lipolysis at the interface is between the emulsified substrate and the water-soluble enzyme.
Validation study
The established fluorescent HPLC assay system for the measurement of OA was sensitive and highly reproduc-ible, associated with an efficient chromatographic sepa-ration of OA from triolein and other similar fatty acids. The calibration curve for OA showed a good linearity within 8 mM. Slope and intercept, the standard error
mean of the slope and intercept, and the coefficients of determination were as follows: y = (0.2909 ± 0.0046)x − (0.0085 ± 0.0069), r2 = 0.9997. The slope and the
y-intercept values introduced in the equation represent the mean of five different curves on the five different days. The limit of detection was found to be 4.53 nM for
OA in our analytical system. The precision and accuracy of data for OA spiked into the reaction mixture are shown in Table 2. The recoveries (mean ± SEM, n = 6) at three different concentrations were 108.73–114.36%. The intra- and inter-day precision values expressed as RSD were smaller than 3%. The statistical validation indicates that the assay is satisfactory for the practical use.
Determination of LPL activity in post-heparin plasma of diabetic rats
Eguchi (2002) reported an HPLC method, in which the LPL activity was detected by reacting 9-anthryldiazomethane (ADAM) with enzyme-generated OA, successfully deter-mining the LPL activity in human plasma. However, the half-life of ADAM solution is only one week at −20°C, and an extraction step is necessary for the sample preparation (Eguchi, 2002). The short half-life may have some influences on accuracy and precision. In
our proposed method, NBD-PZ is used as a derivative reagent, which is more stable than ADAM. Further-more, the NBD-PZ derivative was monitored at excita-tion 491 nm and emission 547 nm. Both of them are long wavelengths which could avoid the interferences caused by autofluorescence in biological samples. The conditions of enzyme reaction were also optimized in this study. Eguchi’s method indicated that 4.5% of GA inhibited the enzyme activity, whereas 1% of GA emul-sifier expressed the greatest efficacy.
Finally, the proposed HPLC method was applied to determine LPL activity in the post-heparin plasma of normal and diabetic rats. Because much lower amounts or activity of total LPL were found in the diabetic stage than in the normal situation (Kovar et al., 2004), even in the transgenic mice with mutant LPL (Hoh and Hodge, 2000; Nickerson et al., 1998; Templeton et al., 2000). Thus, a highly sensitive method for the determi-nation of circulation LPL activity should be useful. OA formation by LPL activity in normal and diabetic rat plasma was derivatized with NBD-PZ, separated on the C8 column, and determined fluorimetrically. Using our
assay system, 10μL of plasma volume was sufficient for LPL activity determination. After receiving STZ for 1 week, significant hyperglycemia determined by the meter and strips was observed in the diabetic rats (494.7 ± 5.3 vs 115.0 ± 6.2 mg/dL in the normal group,
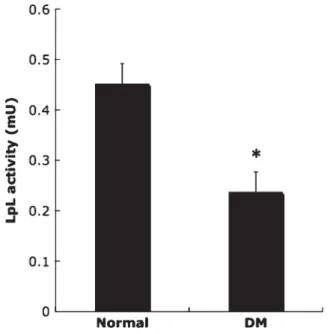
p < 0.001). Figure 7 showed that the LPL activity of
STZ-induced diabetic rats was markedly decreased compared with the normal rats (0.236 ± 0.041 vs 0.451 ± 0.041 mU in the normal group, p < 0.01). These
Figure 7. The LPL activity of normal and STZ-induced
dia-betic rats. LPL activity of STZ-induced diadia-betic rats was markedly decreased compared with normal rats. Analysis was performed by two-tailed independent Student’s t-test: * p< 0.01 compared with the normal group (n = 5).
phenomena observed in diabetic rats confirmed the previous report by Rodrigues and co-workers, who de-termined LPL activity using their radioisotope method. Their results show that LPL activity in the post-heparin plasma is significantly decreased due to hyperglycemia with insufficient secretion of insulin (Rodrigues et al., 1997). These data suggest that hyperglycemia or insulin resistance causes down-regulation activity of circulation LPL. This may account for hypertriglyceridemia, a complication of diabetes. Considering these results together, determination of LPL activity may help us understand the pathological conditions of complications of patients suffering from diabetes mellitus-associated diseases.
In conclusion, we have developed a HPLC method for the highly sensitive determination of circulation LPL activity in 10μL of rat post-heparin plasma. In addition, we also found that LPL activity in the circula-tion was drastically decreased in the diabetic stage induced by streptozotocin. The proposed assay offers some advantages as compared with previous reports. First, the most important advantage of this method is that it uses a non-radioactive substrate. This makes the substrate and product separate cleanly with HPLC. Second, our method provides a simple and fast measure-ment for LPL activity determination. Third, more accurate quantities of products and better qualities of reproduction are guaranteed in our method using margaric acid as an internal standard.
Acknowledgments
This work was financially supported by the National Science Council of the Republic of China (NSC 94-2113-M-038-005). The authors express their sincere appreciation to Dr T. Fukushima (University of Shizuoka, Tokyo, Japan) and Dr Lu-Tai Tien (Fu-Jen Catholic University, Taipei, Taiwan) for their helpful suggestions.
REFERENCES
Archibald RM. Determination of lipase activity. Journal of Biological
Chemistry 1946; 165: 443–448.
Baginsky ML and Brown WV. Differential characteristics of purified hepatic triglyceride lipase and lipoprotein lipase from human postheparin plasma. Journal of Lipid Research 1977; 18: 423–437. Baginsky ML and Brown WV. A new method for the measurement
of lipoprotein lipase in postheparin plasma using sodium dodecyl sulfate for the inactivation of hepatic triglyceride lipase. Journal of
Lipid Research 1979; 20: 548–556.
Ball SW, Bailey JR, Stewart JM, Vogels CM and Westcott SA. A fluorescent compound for glucose uptake measurements in isolated rat cardiomyocytes. Canadian Journal of Physiology &
Pharmaco-logy 2002; 80: 205–209.
Bauer JE. Substrate specificity studies of partially purified rabbit heart lipoprotein lipase. Artery 1988; 15: 272–291.
Beisson F, Tiss A, Rivière C and Verger R. Methods for lipase detec-tion and assay: a critical review. European Journal of Lipid Science
and Technology 2000; 102: 133–153.
Belfrage P and Vaughan M. Simple liquid–liquid partition system for isolation of labeled oleic acid from mixtures with glycerides.
Journal of Lipid Research 1969; 10: 341–344.
Blanchette-Mackie EJ and Scow RO. Retention of lipolytic products in chylomicrons incubated with lipoprotein lipase: electron micro-scope study. Journal of Lipid Research 1976; 17: 57–67.
Duque M, Graupner M, Stutz H, Wicher I, Zechner R, Paltauf F and Hermetter A. New fluorogenic triacylglycerol analogs as substrates for the determination and chiral discrimination of lipase activities.
Journal of Lipid Research 1996; 37: 868–876.
Eckel RH. Lipoprotein lipase. A multifunctional enzyme relevant to common metabolic diseases. New England Journal of Medicine 1989; 320: 1060–1068.
Eguchi Y. Analysis of lipoprotein lipase activity using high-performance liquid chromatography. Biomedical Chromatography 2002; 16: 500–503.
Goldberg IJ. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. Journal of Lipid
Research 1996; 37: 693–707.
Gupta R, Rathi P, Gupta N and Bradoo S. Lipase assays for conventional and molecular screening: an overview. Biotechnology
& Applied Biochemistry 2003; 37: 63–71.
Hoh J and Hodge SE. A measure of phase ambiguity in pairs of SNPs in the presence of linkage disequilibrium. Human Heredity 2000; 50: 359–364.
Hoppe A and Theimer RR. Titrimetric test for lipase activity using stabilized triolein emulsions. Phytochemistry 1996; 42: 973–978. Jeppesen J, Hollenbeck CB, Zhou MY, Coulston AM, Jones C, Chen
YD and Reaven GM. Relation between insulin resistance, hyperinsulinemia, postheparin plasma lipoprotein lipase activity, and postprandial lipemia. Arteriosclerosis, Thrombosis & Vascular
Biology 1995; 15: 320–324.
Karpe F, Olivecrona T, Walldius G and Hamsten A. Lipoprotein lipase in plasma after an oral fat load: relation to free fatty acids.
Journal of Lipid Research 1992; 33: 975–984.
Kovar J, Fejfarova V, Pelikanova T and Poledne R. Hyperglycemia downregulates total lipoprotein lipase activity in humans.
Physi-ological Research 2004; 53: 61–68.
LaRosa JC, Levy RI, Herbert P, Lux SE and Fredrickson DS. A specific apoprotein activator for lipoprotein lipase. Biochemical &
Biophysical Research Communications 1970; 41: 57–62.
Lee JA, Tsai YC, Chen HY, Wang CC, Chen SM, Fukushima T and Imai K. Fluorimetric determination of D-lactate in urine of normal and diabetic rats by column-switching high-performance liquid chromatography. Analytica Chimica Acta 2005; 534: 185–191. Lobo LI and Wilton DC. Effect of lipid composition on lipoprotein
lipase activity measured by a continuous fluorescence assay: effect of cholesterol supports an interfacial surface penetration model.
Biochemical Journal 1997; 321: 829–835.
Maheux P, Azhar S, Kern PA, Chen YD and Reuven GM. Relation-ship between insulin-mediated glucose disposal and regulation of plasma and adipose tissue lipoprotein lipase. Diabetologia 1997; 40: 850–858.
Mahley RW, Weisgraber KH, Innerarity TL and Rall SC, Jr. Genetic defects in lipoprotein metabolism. Elevation of atherogenic lipoproteins caused by impaired catabolism. JAMA 1991; 265: 78– 83.
Nickerson DA, Taylor SL, Weiss KM, Clark AG, Hutchinson RG, Stengard J, Salomaa V, Vartiainen E, Boerwinkle E and Sing CF. DNA sequence diversity in a 9.7-kb region of the human lipo-protein lipase gene. Nature Genetics 1998; 19: 233–240.
Nikkilä EA, Huttunen JK and Ehnholm C. Postheparin plasma lipoprotein lipase and hepatic lipase in diabetes mellitus. Relation-ship to plasma triglyceride metabolism. Diabetes 1977; 26: 11–21. Nilsson-Ehle P and Schotz MC. A stable, radioactive substrate
emul-sion for assay of lipoprotein lipase. Journal of Lipid Research 1976; 17: 536–541.
Pulawa LK and Eckel RH. Overexpression of muscle lipoprotein lipase and insulin sensitivity. Current Opinion in Clinical Nutrition
& Metabolic Care 2002; 5: 569–574.
Pykalisto OJ, Smith PH and Brunzell JD. Determinants of human adipose tissue lipoprotein lipase. Effect of diabetes and obesity on basal- and diet-induced activity. Journal of Clinical Investigation 1975; 56: 1108–1117.
Rodrigues B, Cam MC, Kong J, Goyal RK and McNeill JH. Strain differences in susceptibility to streptozotocin-induced diabetes: effects on hypertriglyceridemia and cardiomyopathy. Cardiovascular
Research 1997; 34: 199–205.
Santamarina-Fojo S and Dugi KA. Structure, function and role of lipoprotein lipase in lipoprotein metabolism. Current Opinion in
Lipidology 1994; 5: 117–125.
Sartippour MR and Renier G. Upregulation of macrophage lipoprotein lipase in patients with type 2 diabetes: role of peripheral factors.
Diabetes 2000; 49: 597–602.
Scheen AJ. The epidemic of metabolic diseases, a major problem of public health. Revue Medicale de Liege 1999; 54: 87–94.
Schneider S, Schramm U, Schreyer A, Buscher HP, Gerok W and Kurz G. Fluorescent derivatives of bile salts. I. Synthesis and pro-perties of NBD-amino derivatives of bile salts. Journal of Lipid
Research 1991; 32: 1755–1767.
Semenkovich CF, Chen SH, Wims M, Luo CC, Li WH and Chan L. Lipoprotein lipase and hepatic lipase mRNA tissue specific expres-sion, developmental regulation, and evolution. Journal of Lipid
Research 1989; 30: 423–431.
Sharma R, Chisti Y and Banerjee UC. Production, purification, characterization, and applications of lipases. Biotechnology
Advances 2001; 19: 627–662.
Sparreboom A, van Tellingen O, Huizing MT, Nooijen WJ and Beijnen JH. Determination of polyoxyethyleneglycerol triricinoleate 35 (Cremophor EL) in plasma by pre-column derivatization and reversed-phase high-performance liquid chromatography. Journal
of Chromatography B: Biomedical Applications 1996; 681: 355–362.
Surinenaite B, Bendikiene V, Juodka B, Bachmatova I and Marcinkevichiene L. Characterization and physicochemical
pro-perties of a lipase from Pseudomonas mendocina 3121-1.
Biotechnology & Applied Biochemistry 2002; 36: 47–55.
Taskinen MR, Nikkilä EA, Kuusi T and Harmo K. Lipoprotein lipase activity and serum lipoproteins in untreated type 2 (insulin-independent) diabetes associated with obesity. Diabetologia 1982; 22: 46–50.
Templeton AR, Weiss KM, Nickerson DA, Boerwinkle E and Sing CF. Cladistic structure within the human Lipoprotein lipase gene and its implications for phenotypic association studies. Genetics 2000; 156: 1259–1275.
Tsai YC, Liao TH and Lee JA. Identification of L-3-hydroxybutyrate as an original ketone body in rat serum by column-switching high-performance liquid chromatography and fluorescence derivatiza-tion. Analytical Biochemistry 2003; 319: 34–41.
Tsujita T, Matsuura Y and Okuda H. Studies on the inhibition of pancreatic and carboxylester lipases by protamine. Journal of Lipid
Research 1996; 37: 1481–1487.
Wang CS, Kuksis A and Manganaro F. Studies on the substrate specificity of purified human milk lipoprotein lipase. Lipids 1982; 17: 278–284.
Yadav RP, Saxena RK, Gupta R and Davidson WS. Purification and characterization of a regiospecific lipase from Aspergillus terreus.
Biotechnology & Applied Biochemistry 1998; 28: 243–249.
Yamazaki H, Arai M, Matsumura S, Inoue K and Fushiki T. Intracranial administration of transforming growth factor-beta3 increases fat oxidation in rats. American Journal of Physiology—
Endocrinology & Metabolism 2002; 283: E536–544.
Zechner R. The tissue-specific expression of lipoprotein lipase: im-plications for energy and lipoprotein metabolism. Current Opinion