Title: Global Study of All-Oral Daclatasvir Plus Asunaprevir for HCV Genotype 1b
Authors: Michael Manns, MD1; Stanislas Pol, MD2; Ira M. Jacobson, MD3; Patrick Marcellin, MD4; Stuart C. Gordon, MD5; Cheng-Yuan Peng, MD6; Ting-Tsung Chang, MD7; Gregory T. Everson, MD8; Jeong Heo, MD9; Guido Gerken, MD10; Boris Yoffe, MD11; William J. Towner, MD12; Marc Bourliere, MD13; Sophie Metivier, MD14; Chi-Jen Chu, MD15; William Sievert, MD16; Jean-Pierre Bronowicki, MD17; Dominique Thabut, MD18; Youn-Jae Lee, MD19; Jia-Horng Kao, MD20; Fiona McPhee, PhD21; Justin Kopit, PhD21; Patricia Mendez, MD22; Misti Linaberry, MPH22; Eric Hughes, MD22; and Stephanie Noviello, MD22 on behalf of the HALLMARK DUAL Study Team
1Hannover Medical School, Hannover, Germany; 2Hôpital Cochin, Paris, France; 3Weill Cornell Medical College, New York, NY, USA; 4Hôpital Beaujon, Clichy, France; 5Henry Ford Health Systems, Detroit, MI, USA; 6School of Medicine,China Medical University, Taichung, Taiwan; 7National Cheng Kung
University Hospital, Tainan, Taiwan; 8University of Colorado Denver, Aurora, CO, USA; 9Department of Internal Medicine, College of Medicine,Pusan National University and Medical Research Institute, Pusan National University Hospital, Busan, Republic of Korea; 10University of Duisburg-Essen, Essen, Germany; 11VAMC,Baylor College of Medicine, Houston, TX, USA; 12Kaiser Permanente, Los Angeles, CA, USA; 13Hôpital Saint Joseph, Marseille, France; 14CHU Purpan, Toulouse, France; 15Taipei Veterans General Hospital and National Yang-Ming University, Taipei, Taiwan; 16Monash Health and Monash University, Melbourne, Australia; 17INSERM Unité 954, Centre Hospitalier Universitaire de Nancy and Université de Lorraine, Vandoeuvre-lès-Nancy, France; 18Hôpital Pitié-Salpêtrière, Paris, France; 19Inje University Busan Paik Hospital, Busan, Republic of Korea; 20National Taiwan University Hospital, Taipei, Taiwan; 21Bristol-Myers Squibb Research and Development, Wallingford, CT, USA; 22 Bristol-Myers Squibb Research and Development, Princeton, NJ, USA
Corresponding author: Michael P. Manns, MD
Department of Gastroenterology, Hepatology and Endocrinology Hannover Medical School, Carl-Neuberg Str. 1
Hannover 30625, Germany Telephone: +49 511 532 3305 Fax: +49 511 532 4896
e-mail: [email protected]
Target journal: The Lancet
Word count (Introduction through Discussion): 3000 (limit 3000) Figures and tables: 5 tables, 2 figures
References: 30 (limit 30)
Abstract (word count: 299, limit 300)
Background: An unmet need exists for interferon- and ribavirin-free treatments for chronic hepatitis C virus (HCV) infection. In this study, we evaluated all-oral therapy with daclatasvir (NS5A replication complex inhibitor) plus asunaprevir (NS3 protease inhibitor) in patients with genotype 1b infection, including those with high unmet need and/or cirrhosis.
Methods: This phase 3, multi-cohort study (HALLMARK-DUAL; ClinicalTrials.gov, NCT01581203) was conducted at 116 sites in 18 countries between May 11, 2012 and October 9, 2013. Patients were adults chronically infected with HCV genotype 1b who were treatment-naive, prior nonresponders to peginterferon/ribavirin, or medically ineligible for, or previously intolerant of, peginterferon/ribavirin. Treatment-naive patients were randomized (2:1; double-blinded) to receive daclatasvir 60 mg once daily plus asunaprevir 100 mg twice daily (n=203) or placebo (n=102) for 12 weeks. The
daclatasvir+asunaprevir group continued treatment through week 24; placebo recipients entered another daclatasvir+asunaprevir study. Nonresponders (n=205) and ineligible/intolerant patients (n=235) received open-label daclatasvir+asunaprevir for 24 weeks. The primary endpoint was sustained virologic response at posttreatment week 12.
Findings: Daclatasvir+asunaprevir provided sustained virologic response rates (95% confidence intervals) of 90% (85%–94%), 82% (77%–87%), and 82% (77%–87%) in treatment-naive, nonresponder, and ineligible/intolerant patients, respectively. Response rates were similar in noncirrhotic (85%; n=437) and cirrhotic (84%; n=206) patients, with no differences observed based on age, gender, race, or IL28B genotype. Serious adverse events occurred in 6% and adverse events leading to discontinuation (most commonly reversible alanine/aspartate aminotransferase
elevations) occurred in 2% of patients, with no deaths. Grade 3/4 laboratory abnormalities were uncommon, with low incidences of aminotransferase elevations during the first 12 weeks with daclatasvir+asunaprevir and placebo in treatment-naive patients (≤2% each).
Interpretation: Daclatasvir+asunaprevir provided high sustained virologic response rates in
treatment-naive, nonresponder, and ineligible/intolerant patients, including those with cirrhosis, and was well tolerated in patients with HCV genotype 1b infection.
Introduction
Chronic hepatitis C virus (HCV) infection affects approximately 170 million people worldwide, and remains a significant global health problem. HCV exists as seven genotypes; genotype 1
predominates in the United States (≈70% of infections), Europe, North Asia, Australia, and South America and was historically the most difficult to cure.1–4 Subtypes 1a and 1b represent
approximately 60% and 40%, respectively, of genotype 1 infections in the United States1; elsewhere (eg, Korea and Taiwan), subtype 1b generally predominates.4–6
Currently approved treatments for HCV genotype 1 infection are limited to combinations containing peginterferon alfa and ribavirin. The newest regimens, which include the direct-acting antivirals (DAAs) simeprevir or sofosbuvir, are substantially more effective than peginterferon/ribavirin alone, with response rates of ≥80% in treatment-naive patients7–9 and lower rates in peginterferon/ribavirin nonresponders.10 However, due to treatment-limiting adverse events (AEs), peginterferon/ribavirin-based regimens are perceived as poorly tolerated, leading many providers and patients to avoid initiating therapy. Moreover, many patients cannot tolerate, or are medically ineligible for, these therapies and those with genotype 1 have no viable options.11,12 Thus, therapeutic research has increasingly focused on developing effective and better-tolerated all-oral regimens, with several new ones recently reported.13–17
Daclatasvir is a potent pangenotypic NS5A inhibitor with antiviral activity across HCV genotypes 1 to 6 in vitro18; asunaprevir is a NS3 protease inhibitor active against genotypes 1, 4, 5, and 6 in vitro.19 In a phase 3 Japanese study in genotype 1b, all-oral dual therapy with daclatasvir+asunaprevir
demonstrated high sustained virologic response (SVR) rates and good tolerability in
peginterferon/ribavirin nonresponders (81%) and peginterferon/ribavirin ineligible/intolerant patients (87%).20 This global study evaluated the efficacy and safety of daclatasvir+asunaprevir in treatment-naive and treatment-experienced patients with HCV genotype 1b infection.
Methods
Trial Design and Patients
This multinational, phase 3, multi-cohort study of daclatasvir (60-mg tablet once daily) plus asunaprevir (100-mg softgel capsule twice daily) was conducted at 116 sites in 18 countries (including in North and South America, Europe, and Asia) between May 11, 2012 and October 9, 2013. Eligible patients were those aged ≥18 years with genotype 1b infection and HCV RNA ≥10,000 IU/mL who met inclusion criteria for one of three cohorts: treatment-naive, prior
peginterferon/ribavirin nonresponder (null or partial response), or peginterferon/ribavirin ineligible/intolerant (treatment-naive and treatment-experienced). Ineligible/intolerant patients included those with depression, anaemia/neutropenia, or compensated advanced fibrosis/cirrhosis (F3/F4) with thrombocytopenia. Anaemia, neutropenia, and thrombocytopenia were defined as haemoglobin 85 to <120 (female) or <130 (male) g/L, absolute neutrophils 0·5 to <1·5109 cells/L, and platelets 50 to <90109 cells/L, respectively, at screening and/or history of these conditions on peginterferon/ribavirin (see Table 1 and Supplementary Appendix). The protocol was approved by the institutional review board/independent ethics committee at each site and all patients provided written informed consent.
Randomization and Blinding
Treatment-naive patients were randomly assigned 2:1 (double-blinded; stratified by cirrhosis status) to receive daclatasvir+asunaprevir or matching placebo for 12 weeks (for comparison of safety and tolerability). Randomization was via an Interactive Voice Response System using a computer-generated random allocation sequence. Patients and investigator sites were blinded to treatment assignment and HCV RNA results through week 12; the sponsor was blinded to treatment
assignment through week 12. The daclatasvir+asunaprevir group continued open-label treatment through 24 weeks; placebo recipients entered another study and received daclatasvir+asunaprevir
for 24 weeks. Nonresponders and ineligible/intolerant patients received open-label
daclatasvir+asunaprevir for 24 weeks. Daclatasvir+asunaprevir-treated patients in all cohorts were followed for 24 weeks posttreatment.
Assessments and Endpoints
Serum HCV RNA levels were assayed using the COBAS TaqMan HCV test v2·0 (Roche Molecular Systems; lower limit of quantitation 25 IU/mL) at baseline; on-treatment weeks 1, 2, 4, 6, 8, 12, 16, 20, and 24 (or at early discontinuation); and posttreatment weeks 4, 12, and 24. HCV
genotype/subtype was assessed using the VERSANT HCV genotype 2·0 assay (Siemens). IL28B genotype (rs12979860 single-nucleotide polymorphism) was determined by PCR amplification and sequencing (Applied Biosystems TaqMan assay). Resistance testing used population sequencing of plasma samples from patients with virologic failure and HCV RNA ≥1000 IU/mL. Safety monitoring was via AE reports, clinical laboratory assessments, vital signs, and physical examinations.
Primary efficacy endpoints were the proportions of treatment-naive patients
(daclatasvir+asunaprevir-treated) and nonresponders with SVR (HCV RNA <25 IU/mL) at posttreatment week 12 (SVR12). Secondary efficacy endpoints included SVR12 rates among
ineligible/intolerant patients, SVR12 by IL28B genotype, and proportions with undetectable HCV RNA at weeks 4, 12, 4 and 12, and end of treatment. Virologic breakthrough was defined as confirmed >1-log10 increase from nadir in HCV RNA or confirmed HCV RNA ≥25 IU/mL after <25 IU/mL
measurement, futility as confirmed HCV RNA ≥25 IU/mL at week 8, and posttreatment relapse as confirmed HCV RNA 25 IU/mL following undetectable end-of-treatment measurement.
Safety endpoints included incidence of AEs, serious AEs, discontinuations due to AEs, anaemia (decline in haemoglobin to <100 g/L), rash (see Supplementary Appendix), and grade 3/4 laboratory
abnormalities. For the treatment-naive cohort, differences in rates of grade 3/4 laboratory
abnormalities between daclatasvir+asunaprevir and placebo during the first 12 weeks were assessed.
Statistical Analyses
Target sample sizes were 200, 200, and up to 225 patients in the treatment-naive
(daclatasvir+asunaprevir-treated), nonresponder, and ineligible/intolerant cohorts, respectively. These would ensure that safety events occurring at a rate ≥1·2% (≥1·1% for ineligible/intolerant cohort) would be detected with ≥90% probability; 100 treatment-naive placebo recipients would detect with ≥90% probability safety events occurring at a 2·3% rate. With these sample sizes, the width of the 95% CI for the SVR12 rate would be at most 14%.
Efficacy analyses were restricted to daclatasvir+asunaprevir-treated patients. In the primary analysis, missing SVR12 data were counted as treatment failures; analysis was also performed based on SVR12 documented on or after posttreatment week 12. Two-sided 95% CIs for response rates were computed using normal approximations to the binomial distribution. Primary objective for the treatment-naive cohort was to show that the lower bound of the 95% CI for daclatasvir+asunaprevir was >68% (based on historical results for telaprevir combined with peginterferon/ribavirin; see Supplementary Appendix); primary objective in other cohorts was to estimate SVR12 rates. Safety analyses were performed in daclatasvir+asunaprevir-treated patients, by cohort, and in the treatment-naive cohort by treatment (daclatasvir+asunaprevir vs placebo) during the 12-week blinded phase (see Supplementary Appendix).
Role of the Funding Source
The sponsor, in collaboration with the authors, participated in study design; data collection, analysis, and interpretation; and drafting of the manuscript. All authors had full access to the data and vouch
for the integrity and accuracy of the data reported. The corresponding author had final responsibility for the decision to submit for publication.
Results
Patient Disposition and Characteristics
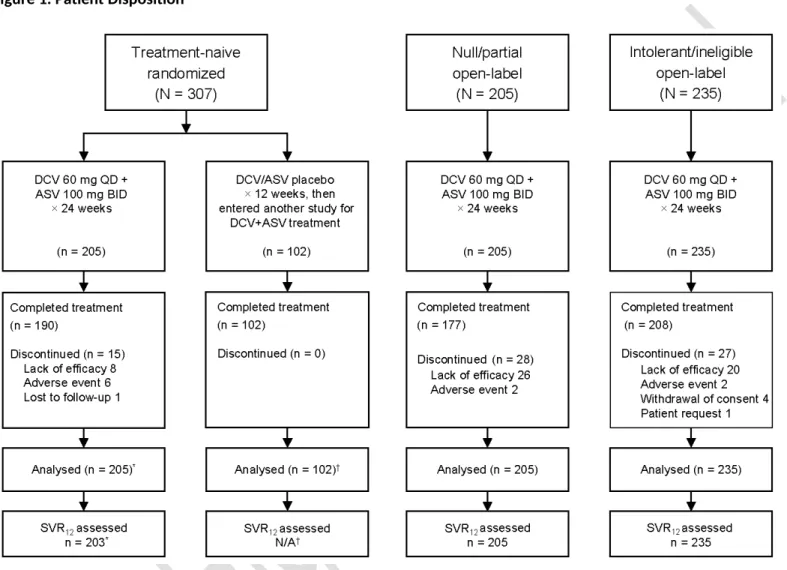
This study included 307 treatment-naive patients (205 daclatasvir+asunaprevir and 102 placebo; all randomized patients received intended treatment), 205 nonresponders, and 235 ineligible/intolerant patients (Figure 1); 93%, 86%, and 89%, respectively, completed daclatasvir+asunaprevir therapy. Overall, patients had a median age of 57 years, 51% were female, and 68% were white, 25% Asian, and 6% black/African American; 70% of daclatasvir+asunaprevir-treated patients with IL28B genotype assessments were non-CC (Table 1). Cirrhosis, assessed by liver biopsy or transient elastography (FibroScan; ≥14·6 kPa), was present in 16% of treatment-naive patients, 31% of
nonresponders, and 47% of ineligible/intolerant patients, which included a subcohort with advanced fibrosis/cirrhosis and thrombocytopenia. There were no notable differences in patient characteristics between treatment-naive patients randomized to daclatasvir+asunaprevir versus placebo.
Virologic response
Daclatasvir+asunaprevir achieved SVR12 rates of 90%, 82%, and 82% in treatment-naive,
nonresponder, and ineligible/intolerant patients, respectively (91%, 82%, and 83% documented on or after posttreatment week 12; Table 2). In treatment-naive patients, the lower bound of the 95% CI for the SVR12 rate (85%) exceeded the 68% specified in the primary objective. HCV RNA reductions from baseline were rapid and sustained; mean decreases at week 2 were 4·8 to 5·0 log10 IU/mL. In the treatment-naive, nonresponder, and ineligible/intolerant cohorts, respectively, HCV RNA was undetectable at week 4 in 83%, 73%, and 68% of patients and at the end of treatment in 93%, 85%, and 87%. SVR12 rates were higher among patients with undetectable (89% [425/477]) versus detectable (73% [117/161]) HCV RNA at week 4.
There were no apparent differences in SVR12 rates by gender, age, race, body mass index, or IL28B genotype (Table 3). Overall SVR12 rates were similar in cirrhotic (84%) and noncirrhotic (85%) patients. Among nonresponders, SVR12 rates were 82% and 81% in null and partial responders, respectively. Among ineligible/intolerant patients, SVR12 rates were 80%, 91%, and 73% in the depression, anaemia/neutropenia, and advanced fibrosis/cirrhosis with thrombocytopenia
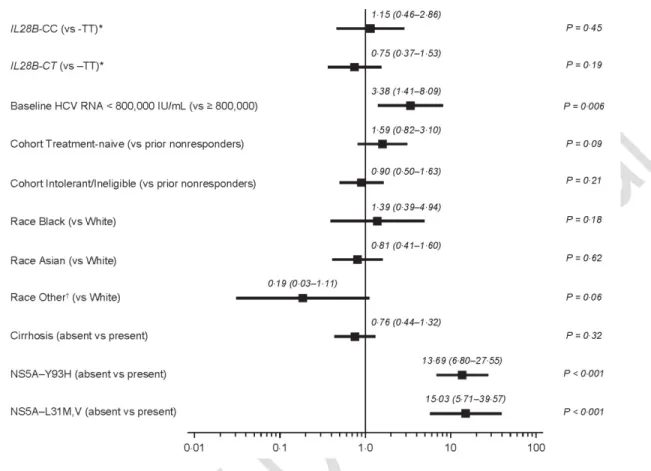
subcohorts, respectively. In analysis of SVR12 by baseline platelet count in the overall population, 71% of patients with <90109 cells/L achieved SVR12 (Supplementary Table 1). Multivariate regression analysis of baseline factors (Figure 2) identified only HCV RNA ≥800,000 IU/mL and presence of NS5A resistance-associated variants (RAVs; at positions L31 or Y93) as negative predictors of SVR12. Notably, SVR12 rates were high in patients with HCV RNA <800,000 and ≥800,000 IU/mL (92% and 82%, respectively). Baseline NS5A-L31 variants were present in 4·5% of patients, 41% of whom achieved SVR12; NS5A-Y93 variants were present in 8% of patients, 38% of whom achieved SVR12
(Supplementary Tables 2 and 3).
Virologic failure
Among daclatasvir+asunaprevir-treated patients, 16% failed to achieve SVR12; 6%, 14%, and 12% had on-treatment failures in the treatment-naive, nonresponder, and ineligible/intolerant cohorts, respectively (Table 2). NS5A and NS3 variants at amino acid positions associated with drug resistance were each observed at baseline in approximately 20% of genotypable isolates. Other than signature RAVs at NS5A positions L31 and Y93 (discussed earlier) and NS3 position D168, baseline variants in either protein did not appear related to virologic outcome (Supplementary Table 2). Signature RAVs at NS5A-L31, NS5A-Y93, and/or NS3-D168 were detected in 75 of 596 patients with both NS5A and NS3 baseline sequence. Among these 75 patients, 29 achieved SVR12. Among 521 patients who had both NS5A and NS3 baseline sequence and did not have NS5A-L31, NS5A-Y93, and/or NS3-D168E RAVs, 478 achieved SVR12. The most common treatment-emergent variants associated with virologic
failure were observed at NS5A-L31, NS5A-Y93, and NS3-D168 (Supplementary Table 4). These were observed together in the majority of patients with key NS5A and NS3 RAVs (61/79; 77%); the remaining patients had other combinations of NS5A and NS3 variants (Supplementary Table 5).
Safety
The most common AEs (≥10% in any cohort) were headache, fatigue, diarrhoea, nausea, and asthenia (Table 4). Among daclatasvir+asunaprevir-treated patients, 2% (12/642) had anaemia (treatment-naive, 1% [2/203]; nonresponder, 1% [3/205]; ineligible/intolerant, 3% [7/234]) and 7% (46/645) had rash (treatment-naive, 8% [16/205]; nonresponder, 5% [11/205]; ineligible/intolerant, 8% [19/235]). All rash-related events were of grade 1/2 intensity, with no treatment
discontinuations. AE-related discontinuations were uncommon (10 [2%]) with
daclatasvir+asunaprevir (treatment-naive, 6 [3%]; nonresponder and ineligible/intolerant, 2 [≈1%] each). The few discontinuations were most commonly related to alanine/aspartate aminotransferase (ALT/AST) elevations (seven patients, five with grade 4 events; six of seven achieved SVR12), which resolved off-treatment. All AEs occurring in ≥5% in any cohort and events causing discontinuation are summarized in Supplementary Tables 6 and 7, respectively.
Serious AEs on-treatment were reported in 39 patients (6%), with similar incidences (5%–7%) across cohorts (Table 4; Supplementary Table 8). Four events were considered related to daclatasvir and/or asunaprevir by investigators: two atrial fibrillation events in the ineligible/intolerant cohort (one each at week 16 and follow-up week 4), and one ALT increased and one event reported as “hepatic enzyme increased” in the treatment-naive cohort. The “hepatic enzyme increased” event occurred at week 24 (after last dose of daclatasvir+asunaprevir) in a 26-year-old male with confirmed Gilbert’s syndrome (UGT1A1*28 homozygous). Although the patient met prespecified laboratory criteria for potential drug-induced liver injury (pDILI; ALT 5 baseline or nadir and 10 the upper limit of normal [ULN], and total bilirubin 2 the ULN), due to the impact of the history of Gilbert’s
syndrome on the total bilirubin laboratory criterion, he did not meet the clinical criteria for pDILI that there be no alternative explanation for either the transaminase or bilirubin elevations. In this patient, ALT was 690 U/L and total bilirubin was 64 µmol/L at week 24 (46 U/L and 26 µmol/L, respectively, at day 1); direct bilirubin did not increase (7 µmol/L at both times), consistent with Gilbert’s syndrome rather than pDILI as the cause of hyperbilirubinaemia. These elevations were asymptomatic and declined 9 days after treatment completion and returned to baseline levels within 4 weeks posttreatment. The patient achieved SVR12.
Grade 3/4 aminotransferase elevations were uncommon, with transient elevations in ALT observed in 15 (2%) patients (including the 2 patients with serious AEs of “ALT increased” and “hepatic enzyme increased” described above) and those in AST observed in 12 (2%) patients (Table 4). Patients with and without cirrhosis had similar incidences of grade 3/4 ALT (1% vs 3%) and AST (1% vs 2%) elevations. There was one grade 3/4 haemoglobin reduction; incidences of other grade 3/4
hematologic abnormalities were ≤2% in each cohort, except for a 4% incidence of platelet reductions in ineligible/intolerant patients, which included a subcohort with low platelets at screening (50 to <90109 cells/L).
Daclatasvir+asunaprevir was well tolerated among the subcohorts of ineligible/intolerant patients (Supplementary Table 9), with no cohort-level increase in neuropsychiatric events in the depression subcohort, no increase in measures of bone marrow suppression in the anaemia/neutropenia subcohort, and no worsening of liver synthetic parameters in the advanced fibrosis/cirrhosis subcohort.
During the 12-week blinded phase, rates of overall AEs and grade 3/4 laboratory abnormalities were similar in treatment-naive patients receiving daclatasvir+asunaprevir or placebo; grade 3/4
aminotransferase and total bilirubin elevations were observed in 2% of patients in each arm (Table 5).
Discussion
Daclatasvir+asunaprevir was the first regimen to demonstrate proof-of-concept for the effectiveness of an all-oral, interferon-free, DAA therapy in curing chronic HCV infection.21 In this global phase 3 study in patients with genotype 1b infection, including a high proportion with cirrhosis, all-oral, interferon- and ribavirin-free dual therapy with daclatasvir+asunaprevir provided SVR rates of up to 91% at posttreatment week 12 or later. High SVR12 rates were achieved in treatment-naive patients (91%) and those with high unmet need, such as peginterferon/ribavirin nonresponders (82%) or peginterferon/ribavirin ineligible/intolerant patients (83%). These findings are consistent with those from a phase 3 study in Japanese nonresponder or ineligible/intolerant patients with genotype 1b infection (SVR12 rates of 81% and 87%, respectively).20 Of note, ineligible patients in the Japanese study were naive, whereas those in this study were naive or treatment-experienced. Additionally, the Japanese study included fewer cirrhotic patients (10% versus 32% [daclatasvir+asunaprevir-treated]) and more patients aged 65 years (40% vs 21%).
Cirrhosis is associated with decreased SVR rates with peginterferon/ribavirin alone or in combination with telaprevir, boceprevir, simeprevir, or sofosbuvir.7,10,22 Notably, in this study, treatment response was similar in patients with or without cirrhosis. The SVR12 rate among patients with baseline platelet counts of 50 to <90109 cells/L was high (71%), but slightly lower than that in patients without thrombocytopenia, although the sample size was small. This difference may be related to cirrhosis with portal hypertension, which may result in lower hepatic drug exposure.
Treatment response was generally similar between patients with CC or non-CC IL28B genotypes, consistent with previous observations with interferon-free regimens.20,23–26 Gender, age, and race also
had no notable effects on treatment outcome. Most virologic failures on daclatasvir+asunaprevir were on-treatment breakthroughs, contrasting with cross-study data for sofosbuvir+ribavirin in similar patient groups with HCV genotypes 1 to 3, wherein the majority of failures were
posttreatment relapses.27 Patients who did not achieve SVR12 with daclatasvir+asunaprevir (16%) had a higher frequency of baseline NS5A variants at positions L31 and Y93; however, some patients who had these baseline variants still achieved SVR12.
Daclatasvir+asunaprevir was well tolerated, with low incidences of serious AEs, AEs leading to discontinuation, and grade 3/4 laboratory abnormalities. Although the most common AEs leading to discontinuation were related to aminotransferase elevations, these were reversible and easily managed, and six of seven patients who discontinued for this reason achieved SVR12. Among ineligible/intolerant patients, no exacerbation of subcohort-specific conditions was seen. In treatment-naive patients, incidences of grade 3/4 laboratory abnormalities, including
aminotransferase and total bilirubin elevations, were similar with daclatasvir+asunaprevir and placebo over the first 12 weeks of treatment. AE rates also appeared similar with
daclatasvir+asunaprevir and placebo.
This was a global study enrolling a wide range of patients, including those with cirrhosis; the
similarity of results reported here to those in a Japanese study20 is consistent with generalizability of these findings. Limitations of this study include the absence of a placebo arm for safety/tolerability comparisons in the nonresponder and ineligible/intolerant cohorts, and the absence of patients who relapsed on prior peginterferon/ribavirin therapy. This study also did not evaluate
daclatasvir+asunaprevir in combination with ribavirin, although a study assessing the combination of daclatasvir with another protease inhibitor, simeprevir, showed no substantial differences with or without addition of ribavirin in genotype 1b infection.28
In the context of approved therapies, the interferon- and ribavirin-free regimen of
daclatasvir+asunaprevir provided SVR rates in genotype 1b infection that were similar to, or higher than, those reported for combinations of sofosbuvir or simeprevir with peginterferon/ribavirin in treatment-naive and treatment-experienced patients.7–10 Daclatasvir+asunaprevir showed an improved safety/tolerability profile compared with peginterferon/ribavirin-based therapies, particularly with respect to hematologic toxicities and systemic AEs7,29,30; the safety of
daclatasvir+asunaprevir was further demonstrated by comparison to placebo in treatment-naive patients. Recent publications on other interferon-free regimens have reported SVR rates of 94% to 99% in treatment-naive and treatment-experienced patients with genotype 1 infection.13–17 Based on its favourable safety and drug interaction profile, and high response rates with good tolerability in ineligible/intolerant patients with more comorbidities and/or concomitant medications,
daclatasvir+asunaprevir may be a treatment option for this patient population, particularly where genotype 1b is highly prevalent. Studies are underway to evaluate addition of a non-nucleoside polymerase inhibitor to daclatasvir+asunaprevir following promising early results in genotypes 1a and 1b,25 and to assess daclatasvir-containing all-oral combinations in multiple patient populations with high unmet need.
In conclusion, 24 weeks of interferon- and ribavirin-free oral therapy with daclatasvir+asunaprevir demonstrated high and sustained antiviral activity and favourable safety and tolerability in treatment-naive, interferon-based treatment-experienced, and interferon-ineligible/intolerant patients (including those with cirrhosis) with chronic HCV genotype 1b, the most common HCV genotype in many parts of the world.
Research in Context
We consulted a recent systematic review of HCV therapies13 and conducted a PubMed search (through April 17, 2014) for reports of clinical trials evaluating interferon-free treatments for
genotype 1 infection (search terms of “HCV” or “hepatitis C,” disregarding reports in other genotypes or of interferon-based regimens). We identified several relevant studies cited herein.13–17
Interpretation
Treatment for HCV genotype 1 infection is evolving rapidly, with a focus on developing interferon- and ribavirin-free regimens that provide higher response rates with improved safety and tolerability. In this study, high response rates were achieved in treatment-naive patients and
peginterferon/ribavirin nonresponders and ineligible/intolerant patients, who have a high unmet medical need. Response rates were similar in patients with and without cirrhosis, and
daclatasvir+asunaprevir was well tolerated. Although interferon-free regimens with higher SVR rates have recently been reported,13–17 the favourable safety and drug interaction profile of
daclatasvir+asunaprevir supports its potential as a treatment option for patients with genotype 1b infection, particularly those with comorbidities and/or concomitant medications and in regions where genotype 1b is prevalent.
Contributors
J. Kopit, P. Mendez, and E. Hughes designed the study. M. Manns, S. Pol, I. M. Jacobson, P. Marcellin, S. C. Gordon, C.-Y. Peng, T.-T. Chang, G. T. Everson, J. Heo, G. Gerken, B. Yoffe, W. J. Towner, M. Bourliere, S. Metivier, C.-J. Chu, W. Sievert, J.-P. Bronowicki, D. Thabut, Y.-J. Lee, and J.-H. Kao recruited patients and obtained data. F. McPhee, J. Kopit, M. Linaberry, E. Hughes, and S. Noviello analysed the data. All authors interpreted the data, participated in writing the manuscript, and approved the final version of the manuscript.
Acknowledgments
This study was supported by Bristol-Myers Squibb. The authors thank Meghan Lovegren, Gail Denisky, and Mahnaz Mohebbian for their contributions to study execution. Editorial support was provided by Joy Loh, PhD, of Articulate Science and was funded by Bristol-Myers Squibb.
Results from this study were previously presented, in part, in oral and poster presentations at The International Liver Congress™ 2014: The 49th Annual Meeting of the European Association for the Study of the Liver, April 9–13, London, United Kingdom.
Declaration of interests
M. Manns has received research funding from Bristol-Myers Squibb, Roche, Gilead, Boehringer Ingelheim, Novartis, Merck, Janssen, Idenix, and GlaxoSmithKline. S. Pol has received research funding from Bristol-Myers Squibb, Gilead, Roche, and Merck Sharp & Dohme; and served as a speaker and board member for Bristol-Myers Squibb, GlaxoSmithKline, Boehringer Ingelheim, Janssen, Gilead, Roche, Merck Sharp & Dohme, Sanofi, Novartis, Vertex Pharmaceuticals, and AbbVie. I. M. Jacobson has received research funding from Bristol-Myers Squibb, AbbVie, Achillion, Boehringer Ingelheim, Gilead, Novartis, Genentech, Merck, Janssen, and Vertex Pharmaceuticals; served as a consultant/advisor for Bristol-Myers Squibb, AbbVie, Achillion, Boehringer Ingelheim, Gilead, Genentech, Merck, Janssen, Vertex Pharmaceuticals, and Idenix; and served on a Speaker’s Bureau for Bristol-Myers Squibb, Gilead, and Vertex Pharmaceuticals. S. C. Gordon has received research funding from Bristol-Myers Squibb, AbbVie, Gilead, GlaxoSmithKline, Intercept
Pharmaceuticals, Kadmon, Merck, and Vertex Pharmaceuticals; served as a consultant/advisor for Bristol-Myers Squibb, AbbVie, Amgen, CVS Caremark, Gilead, Merck, Novartis, and Vertex
Pharmaceuticals; and served on a data monitoring board for Tibotec/Janssen. G. T. Everson has received research funding from Bristol-Myers Squibb, Gilead, AbbVie, Janssen, Roche/Genentech, and Vertex Pharmaceuticals; and has an issued patent (United States) and a pending patent (United
States, European Union, Canada, and Australia) for liver function testing. B. Yoffe has received research funding from Bristol-Myers Squibb, Gilead, and Vertex Pharmaceuticals. W. J. Towner has received research funding from Bristol-Myers Squibb, Gilead, Pfizer, ViiV, Merck, and Vertex Pharmaceuticals. M. Bourliere has received research funding from Bristol-Myers Squibb and Bayer, and personal fees from Bristol-Myers Squibb, Merck Sharp & Dohme, Janssen, Boehringer Ingelheim, Gilead, AbbVie, GlaxoSmithKline, Roche, and Vertex Pharmaceuticals. W. Sievert has served on advisory boards for Bristol-Myers Squibb, Merck, AbbVie, Gilead, and Janssen. J.-P. Bronowicki has received research funding from Bristol-Myers Squibb and lecture and/or consultancy fees from Bristol-Myers Squibb, Janssen, Merck Sharp & Dohme, Gilead, AbbVie, Boehringer Ingelheim, and Roche. F. McPhee, J. Kopit, P. Mendez, M. Linaberry, and S. Noviello are employees of Bristol-Myers Squibb; E. Hughes is an employee and stockholder of Bristol-Myers Squibb. P. Marcellin, C.-Y. Peng, T.-T. Chang, J. Heo, G. Gerken, S. Metivier, C.-J. Chu, D. Thabut, Y.-J. Lee, and J.-H. Kao have no conflicts of interest to disclose.
References (reference count: 30, limit 30)X
1. Manos MM, Shvachko VA, Murphy RC, Arduino JM, Shire NJ. Distribution of hepatitis C virus genotypes in a diverse US integrated health care population. J Med Virol 2012; 84: 1744–50.2. Ghany MG, Strader DB, Thomas DL, Seeff LB, American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 2009; 49: 1335–74.
3. European Association for the Study of the Liver. Clinical practice guidelines: management of hepatitis C virus infection. J Hepatol 2014; 60: 392–420.
4. Negro F, Alberti A. The global health burden of hepatitis C virus infection. Liver Int 2011; 31 (suppl 2): 1–3.
5. Cornberg M, Razavi HA, Alberti A, et al. A systematic review of hepatitis C virus epidemiology in Europe, Canada and Israel. Liver Int 2011; 31 (suppl 2): 30–60.
6. Sievert W, Altraif I, Razavi HA, et al. A systematic review of hepatitis C virus epidemiology in Asia, Australia and Egypt. Liver Int 2011; 31 (suppl 2): 61–80.
7. Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368: 1878–87.
8. Jacobson I, Dore GJ, Foster GR, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from QUEST-1, a phase III trial [abstract]. J Hepatol 2013; 58 (suppl 1): S574.
9. Manns M, Marcellin P, Poordad FP, et al. Simeprevir with pegylated interferon alfa 2a or 2b plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-2): a randomised, double-blind, placebo controlled phase 3 trial. The Lancet 2014. In press.
10. Olysio (simeprevir) [package insert]. Titusville, NJ: Janssen Therapeutics; 2013.
11. Manns MP, von Hahn T. Novel therapies for hepatitis C—one pill fits all? Nat Rev Drug Discov 2013; 12: 595–610.
12. Melia MT, Muir AJ, McCone J, et al. Racial differences in hepatitis C treatment eligibility.
Hepatology 2011; 54: 70–8.
13. Pawlotsky JM. New hepatitis C therapies: the toolbox, strategies, and challenges.
Gastroenterology 2014; 146: 1176–92.
14. Feld JJ, Kowdley KV, Coakley E, et al. Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med 2014; 370: 1594–603.
15. Zeuzem S, Jacobson IM, Baykal T, et al. Retreatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med 2014; 370: 1604–14.
16. Afdhal N, Reddy KR, Nelson DR, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med 2014; 370: 1483–93.
17. Afdhal N, Zeuzem S, Kwo P, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370: 1889–98.
18. Gao M. Antiviral activity and resistance of HCV NS5A replication complex inhibitors. Curr Opin
19. McPhee F, Sheaffer AK, Friborg J, et al. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob Agents Chemother 2012; 56: 5387–96.
20. Kumada H, Suzuki Y, Ikeda K, et al. Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology 2014. doi: 10.1002/hep.27113.
21. Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366: 216–24.
22. Bourliere M, Khaloun A, Wartelle-Bladou C, et al. Future treatment of patients with HCV cirrhosis. Liver Int 2012; 32 (suppl 1): 113–9.
23. Suzuki Y, Ikeda K, Suzuki F, et al. Dual oral therapy with daclatasvir and asunaprevir for patients with HCV genotype 1b infection and limited treatment options. J Hepatol 2013; 58: 655–62.
24. Chayama K, Takahashi S, Toyota J, et al. Dual therapy with the nonstructural protein 5A inhibitor, daclatasvir, and the nonstructural protein 3 protease inhibitor, asunaprevir, in hepatitis C virus genotype 1b-infected null responders. Hepatology 2012; 55: 742–8.
25. Everson GT, Sims KD, Rodriguez-Torres M, et al. Efficacy of an interferon- and ribavirin-free regimen of daclatasvir, asunaprevir, and BMS-791325 in treatment-naive patients with HCV genotype 1 infection. Gastroenterology 2014; 146: 420–9.
26. Sulkowski MS, Gardiner DF, Rodriguez-Torres M, et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med 2014; 370: 211–21.
28. Zeuzem S, Hezode C, Bronowicki J, et al. Daclatasvir in combination with simeprevir ± ribavirin for hepatitis C virus genotype 1 infection [abstract]. Top Antivir Med 2014; 22(e-1): 34–5.
29. Zeuzem S, Andreone P, Pol S, et al. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364: 2417–28.
30. Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364: 1207–17.
Figure Legends
Figure 1. Patient Disposition
DCV, daclatasvir; ASV, asunaprevir; SVR12, sustained virologic response at posttreatment week 12; N/A, not assessed.
*Two patients were inadvertently assigned, rather than randomized, to daclatasvir+asunaprevir therapy and achieved SVR12. These patients were excluded from efficacy analyses but were included in safety analyses.
†Treatment-naive patients who received placebo were not included in efficacy analyses.
Figure 2. Odds Ratios and 95% CIs from Multivariate Logistic Regression of SVR12 on Baseline Covariates
HCV, hepatitis C virus; SVR12, sustained virologic response at posttreatment week 12. *rs12979860 single-nucleotide polymorphism.
Table 1. Baseline Characteristics. Characteristic Treatment-naive (DCV+ASV) N = 205 Treatment-naive (Placebo) N = 102 Prior nonresponder N = 205 Ineligible/intolerant N = 235 Median age, yr 55 54 58 60 Male sex, n (%) 101 (49·3) 54 (52·9) 111 (54·1) 98 (41·7) Race, n (%) White 135 (65·9) 59 (57·8) 148 (72·2) 169 (71·9) Black 14 (6·8) 8 (7·8) 10 (4·9) 10 (4·3) Asian 52 (25·4) 33 (32·4) 45 (22·0) 56 (23·8) Other 4 (2·0) 2 (2·0) 2 (1·0) 0 HCV RNA level, n (%) <800,000 IU/mL 53 (25·9) 26 (25·5) 27 (13·2) 48 (20·4) ≥800,000 IU/mL 152 (74·1) 76 (74·5) 178 (86·8) 187 (79·6) Cirrhosis, n (%) 33 (16·1) 16 (15·7) 63 (30·7) 111 (47·2) IL28B genotype, n (%) CC 76 (37·1) N/A 29 (14·1) 82 (34·9) CT 101 (49·3) N/A 123 (60·0) 102 (43·4) TT 28 (13·7) N/A 50 (24·4) 41 (17·4)
Not reported 0 N/A 3 (1·5) 10 (4·3)
Prior response to P/R, n (%)
Null N/A N/A 119 (58·0) N/A
Partial N/A N/A 84 (41·0) N/A
Relapse* N/A N/A 2 (1·0) N/A
Ineligible/intolerant reason, n (%)
Depression N/A N/A N/A 71 (30·2)
Anaemia†/neutropenia‡ N/A N/A N/A 87 (37·0)
Compensated advanced fibrosis/cirrhosis with
thrombocytopenia§,|,¶
ASV, asunaprevir; DCV, daclatasvir; HCV, hepatitis C virus; N/A, not applicable; P/R, peginterferon/ribavirin. *Protocol violations.
†Haemoglobin 85 to <120 (female) or <130 (male) g/L at screening and/or decline to <100 g/L during prior peginterferon/ribavirin therapy. ‡Absolute neutrophils 0·5 to <1·5109 cells/L at screening and/or decline to <0·75109 cells/L during prior peginterferon/ribavirin therapy. §Platelets 50 to <90109 cells/L at screening and/or decline to <50109 cells/L during prior peginterferon/ribavirin therapy.
|Includes 6 (7·8%) with stage F3, 70 (90·9%) with stage F4, and 1 (1·3%) not reported (percentages based on number of patients in the subcohort with compensated advanced fibrosis/cirrhosis with thrombocytopenia).
Table 2. Virologic Responses. Patients, n (%) [95% CI]* Treatment-naive (DCV+ASV) N = 203† Prior nonresponder N = 205 Ineligible/intolerant N =235 SVR12‡ 182 (89·7) [85·5–93·8] 168 (82·0) [76·7–87·2] 192 (81·7) [76·8–86·6] SVR12 (documented on or after posttreatment week 12)‡,§ 184 (90·6) [86·6–94·6] 169 (82·4) [77·2–87·6] 194 (82·6) [77·7–87·4] HCV RNA undetectable during treatment
Week 4 168 (82·8) [77·6–88·0] 150 (73·2) [67·1–79·2] 159 (67·7) [61·7–73·6] Weeks 4 and 12 163 (80·3) [74·8–85·8] 140 (68·3) [61·9–74·7] 149 (63·4) [57·2–69·6] Week 12 191 (94·1) [90·8–97·3] 182 (88·8) [84·5–93·1] 205 (87·2) [83·0–91·5] End of treatment 189 (93·1) [89·6–96·6] 174 (84·9) [80·0–89·8] 204 (86·8) [82·5–91·1] Non-SVR12, n (%) All 21 (10·3) 37 (18·0) 43 (18·3) On-treatment failures Virologic breakthrough¶ 9 (4·4) 26 (12·7) 20 (8·5) Futilityǁ 0 0 1 (0·4)
Detectable or missing RNA at end of treatment 4 (2·0) 3 (1·5) 8 (3·4)
Relapse** 5/189 (2·6) 7/174 (4·0) 12/204 (5·9) Missing RNA at posttreatment week 12†† 3/189 (1·6) 1/174 (0·6) 2/204 (1·0) ASV, asunaprevir; DCV, daclatasvir; HCV, hepatitis C virus; SVR12, sustained virologic response at posttreatment week 12. *Response rates and two-sided 95% CIs by normal approximation are presented.
†Excludes two patients who were inadvertently assigned, rather than randomized, to daclatasvir+asunaprevir therapy; these patients achieved SVR12.
‡HCV RNA <25 IU/mL at posttreatment week 12.
§SVR12 status of patients with a missing HCV RNA measurement at posttreatment week 12 was determined using the next available measurement.
¶Confirmed >1-log10 increase from nadir in HCV RNA or confirmed HCV RNA ≥25 IU/mL after <25 IU/mL measurement. ǁConfirmed HCV RNA 25 IU/mL at week 8.
**Confirmed HCV RNA 25 IU/mL following end-of-treatment undetectable measurement; percentages based on the number of patients with undetectable HCV RNA at end of treatment.
††Includes those lost to follow-up, missing a critical visit, or with consent withdrawn; percentages based on the number of patients with undetectable HCV RNA at end of treatment.
Table 3. SVR12 by Subgroup. Patients, n/N (%) Treatment-naive (DCV+ASV) N = 203* Prior nonresponder N = 205 Ineligible/intolerant N = 235 Gender Male 89/99 (89·9) 92/111 (82·9) 81/98 (82·7) Female 93/104 (89·4) 76/94 (80·9) 111/137 (81·0) Age, yr <65 153/174 (87·9) 134/161 (83·2) 138/175 (78·9) ≥65 29/29 (100·0) 34/44 (77·3) 54/60 (90·0) Race White 118/133 (88·7) 121/148 (81·8) 140/169 (82·8) Black 13/14 (92·9) 10/10 (100·0) 8/10 (80·0) Asian 48/52 (92·3) 36/45 (80·0) 44/56 (78·6)
Body mass index, kg/m2
<25 96/105 (91·4) 73/88 (83·0) 80/98 (81·6)
25 to <30 62/69 (89·9) 67/85 (78·8) 76/94 (80·9)
≥30 24/29 (82·8) 28/32 (87·5) 36/43 (83·7)
HCV RNA level, IU/mL
<800,000 51/53 (96·2) 25/27 (92·6) 42/48 (87·5) ≥800,000 131/150 (87·3) 143/178 (80·3) 150/187 (80·2) Cirrhosis status Absent 153/171 (89·5) 113/142 (79·6) 104/124 (83·9) Present 29/32 (90·6) 55/63 (87·3) 88/111 (79·3) IL28B genotype CC 68/76 (89·5) 22/29 (75·9) 66/82 (80·5) CT 87/99 (87·9) 100/123 (81·3) 83/102 (81·4) TT 27/28 (96·4) 43/50 (86·0) 36/41 (87·8) Prior response to P/R
Null N/A 98/119 (82·4) N/A
Ineligible/intolerant reason
Depression N/A N/A 57/71 (80·3)
Anaemia/neutropenia N/A N/A 79/87 (90·8)
Compensated advanced fibrosis/cirrhosis with thrombocytopenia†
N/A N/A 56/77 (72·7)
ASV, asunaprevir; DCV, daclatasvir; HCV, hepatitis C virus; N/A, not applicable; P/R, peginterferon/ribavirin; SVR12, sustained virologic response at posttreatment week 12.
*Excludes two patients who were inadvertently assigned, rather than randomized, to daclatasvir+asunaprevir therapy. †SVR12 rates were 53/70 (75·7%) among those with cirrhosis (F4) and 2/6 (33·3%) among those with advanced fibrosis (F3).
Table 4. Summary of Daclatasvir+Asunaprevir On-Treatment Safety.* Patients, n (%) Treatment-naive (DCV+ASV) N = 205 Prior nonresponder N = 205 Ineligible/intolerant N = 235 Any AE 176 (85·9) 167 (81·5) 204 (86·8) Serious AEs† 12 (5·9) 11 (5·4) 16 (6·8)
AEs leading to discontinuation‡ 6 (2·9) 2 (1·0) 2 (0·9)
AEs in ≥10% of patients in any cohort§
Headache 50 (24·4) 50 (24·4) 59 (25·1)
Fatigue 43 (21·0) 45 (22·0) 52 (22·1)
Diarrhoea 24 (11·7) 28 (13·7) 51 (21·7)
Nausea 25 (12·2) 22 (10·7) 28 (11·9)
Asthenia 4 (2·0) 12 (5·9) 25 (10·6)
Grade 3/4 laboratory abnormalities|
Haemoglobin 0 1 (0·5) 0
Absolute neutrophil count 2 (1·0) 2 (1·0) 5 (2·1)
Lymphocytes 1 (0·5) 2 (1·0) 5 (2·1)
Platelet count 0 1 (0·5) 10 (4·3)
ALT 7 (3·4) 4 (2·0) 4 (1·7)
AST 7 (3·4) 2 (1·0) 3 (1·3)
Total bilirubin 1 (0·5) 0 2 (0·9)
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ULN, upper limit of normal. *Includes the time period from the first dose of study therapy through 7 days after the end of treatment. †A summary of serious AEs is provided in Supplementary Table 8.
‡A summary of AEs leading to discontinuation is provided in Supplementary Table 7. §A summary of all-grade, all-cause AEs is provided in Supplementary Table 6.
|Criteria were: haemoglobin, <90 g/L; absolute neutrophils, <0·75109 cells/L; lymphocytes, <0·5109 cells/L; platelets, <50109 cells/L; ALT, >5 the ULN; AST, >5 the ULN; total bilirubin, >2·5 the ULN.
Table 5. Summary of Safety in Treatment-Naive Patients Receiving Daclatasvir+Asunaprevir Versus Placebo (12-Week Blinded Phase). Patients, n (%) DCV+ASV N = 205* Placebo N = 102 Difference vs placebo (95% CI) Any AE 164 (80.0) 74 (72.5) N/A
Serious AEs 7 (3.4) 1 (1.0) N/A
AEs leading to discontinuation 3 (1.0) 0 N/A
AEs in ≥10% of patients in either group
Headache 42 (20.5) 17 (16.7) N/A
Fatigue 35 (17.1) 18 (17.6) N/A
Nausea 23 (11.2) 12 (11.8) N/A
Diarrhoea 22 (10.7) 10 (9.8) N/A
Grade 3/4 laboratory abnormalities†
Haemoglobin 0 1 (1.0) –1.0 (–2.9, 0.9)
Absolute neutrophil count 2 (1.0) 1 (1.0) 0 (–2.3, 2.4)
Lymphocytes 0 0 0
Platelet count 0 0 0
ALT 4 (2.0) 2 (2.0) 0 (–3.3, 3.3)
AST 3 (1.5) 1 (1.0) 0.5 (–2.0, 3.0)
Total bilirubin 0 1 (1.0) –1.0 (–2.9, 0.9)
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ASV, asunaprevir; DCV, daclatasvir; N/A, not applicable; ULN, upper limit of normal.
*N = 203 for grade 3/4 laboratory abnormalities,due to exclusion of two patients who discontinued from the study prior to the first laboratory test.
†Criteria were: haemoglobin, <90 g/L; absolute neutrophils, <0·75109 cells/L; lymphocytes, <0·5109 cells/L; platelets, <50109 cells/L; ALT, >5 ULN; AST, >5 ULN; total bilirubin, >2·5 ULN.
Figure 1. Patient Disposition
ASV, asunaprevir; DCV, daclatasvir; N/A, not assessed; SVR12, sustained virologic response at posttreatment week 12.
*Two patients were inadvertently assigned, rather than randomized, to daclatasvir+asunaprevir therapy and achieved SVR12. These patients were excluded from efficacy analyses but were included in safety analyses.
Figure 2. Odds Ratios and 95% CIs from Multivariate Logistic Regression of SVR12 on Baseline Covariates
HCV, hepatitis C virus; SVR12, sustained virologic response at posttreatment week 12. *rs12979860 single-nucleotide polymorphism.


![Table 2. Virologic Responses. Patients, n (%) [95% CI] * Treatment-naive(DCV+ASV)N = 203† Prior nonresponderN = 205 Ineligible/intolerantN =235 SVR 12 ‡ 182 (89·7) [85·5–93·8] 168 (82·0) [76·7–87·2] 192 (81·7) [76·8–86·6] SVR 12 (documented on or after](https://thumb-ap.123doks.com/thumbv2/9libinfo/8933575.268408/26.1188.89.995.127.809/virologic-responses-patients-treatment-nonrespondern-ineligible-intolerantn-documented.webp)



![TraditionalMLCalgorithmsmainlytacklethebatchMLCproblem,wheretheinputdataarepresentedinabatch[24,28].Nevertheless,inmanyMLCapplicationssuchase-mailcategorization[22],multi-labelexamplesarriveasastream.Onlineanalysisistherefore dimensionreducermotivatedbyma](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)