Open Access
Methodology article
ProLoc-GO: Utilizing informative Gene Ontology terms for
sequence-based prediction of protein subcellular localization
Wen-Lin Huang
1, Chun-Wei Tung
2, Shih-Wen Ho
3, Shiow-Fen Hwang
1and
Shinn-Ying Ho*
2,4Address: 1Institute of Information Engineering and Computer Science, Feng Chia University, Taichung, Taiwan, 2Institute of Bioinformatics, National Chiao Tung University, Hsinchu, Taiwan, 3Department of Management Information System, Chin Min Institute of Technology, Miaoli, Taiwan and 4Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan
Email: Wen-Lin Huang - [email protected]; Chun-Wei Tung - [email protected];
Shih-Wen Ho - [email protected]; Shiow-Fen Hwang - [email protected]; Shinn-Ying Ho* - [email protected] * Corresponding author
Abstract
Background: Gene Ontology (GO) annotation, which describes the function of genes and gene products across
species, has recently been used to predict protein subcellular and subnuclear localization. Existing GO-based prediction methods for protein subcellular localization use the known accession numbers of query proteins to obtain their annotated GO terms. An accurate prediction method for predicting subcellular localization of novel proteins without known accession numbers, using only the input sequence, is worth developing.
Results: This study proposes an efficient sequence-based method (named ProLoc-GO) by mining informative
GO terms for predicting protein subcellular localization. For each protein, BLAST is used to obtain a homology with a known accession number to the protein for retrieving the GO annotation. A large number n of all annotated GO terms that have ever appeared are then obtained from a large set of training proteins. A novel genetic algorithm based method (named GOmining) combined with a classifier of support vector machine (SVM) is proposed to simultaneously identify a small number m out of the n GO terms as input features to SVM, where
m <<n. The m informative GO terms contain the essential GO terms annotating subcellular compartments such
as GO:0005634 (Nucleus), GO:0005737 (Cytoplasm) and GO:0005856 (Cytoskeleton). Two existing data sets SCL12 (human protein with 12 locations) and SCL16 (Eukaryotic proteins with 16 locations) with <25% sequence identity are used to evaluate ProLoc-GO which has been implemented by using a single SVM classifier with the m = 44 and m = 60 informative GO terms, respectively. ProLoc-GO using input sequences yields test accuracies of 88.1% and 83.3% for SCL12 and SCL16, respectively, which are significantly better than the SVM-based methods, which achieve < 35% test accuracies using amino acid composition (AAC) with acid pairs and AAC with dipedtide composition. For comparison, ProLoc-GO using known accession numbers of query proteins yields test accuracies of 90.6% and 85.7%, which is also better than Hum-PLoc (85.0%) and Euk-OET-PLoc (83.7%) using ensemble classifiers with hybridization of GO terms and amphiphilic pseudo amino acid composition for SCL12 and SCL16, respectively.
Conclusion: The growth of Gene Ontology in size and popularity has increased the effectiveness of GO-based
features. GOmining can serve as a tool for selecting informative GO terms in solving sequence-based prediction problems. The prediction system using ProLoc-GO with input sequences of query proteins for protein subcellular localization has been implemented (see Availability).
Published: 1 February 2008
BMC Bioinformatics 2008, 9:80 doi:10.1186/1471-2105-9-80
Received: 25 October 2007 Accepted: 1 February 2008 This article is available from: http://www.biomedcentral.com/1471-2105/9/80
© 2008 Huang et al; licensee BioMed Central Ltd.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Background
Gene Ontology (GO) [1] annotation, which describes the function of genes and gene products across species, has recently been utilized to predict protein subcellular and subnuclear localization. The prediction of protein locali-zation is important for elucidating protein functions involved in various cellular processes. Additionally, the accomplishment of the various genome sequencing projects causes the accumulation of massive amount of gene sequence information. For example, the percentage of large-scale eukaryotic proteins with subcellular loca-tions annotated in the Swiss-Prot database increased rap-idly from 52.4% (version 49.5, released on April 18, 2006) [2] to 69.4% (version 50.7, released Sep. 11, 2006) [3]. Meanwhile, the percentage of proteins with subcellu-lar locations annotated in the GO database increased from 44.9% [2] to 65.5% [3]. The growth of the GO data-base in size and popularity increases the effectiveness of GO-based features.
Some existing computation methods in literature for pre-dicting protein localization are described below according to the used classifiers and features.
a) Mining informative features. The prediction methods in this group focus on mining informative features con-sisting of GO terms [2-5], sorting signals [6,7], amino acid composition (AAC) [8-10], k-peptide encoding vector [7,11-14], physicochemical properties of amino acids [15-17], and fusing AAC and physicochemical properties [2,4,18,19].
b) Designing efficient classifiers. Most of the following prediction methods use effective classifiers based on sup-port vector machine (SVM) [5,10-12,14,16,17,20] or the k nearest neighbour (k-NN) classifiers [2,4,5,13,19,21]. c) Integrating informative features with efficient classifier. Methods in this group include pSLIP [17], ProLoc [18], Euk-OET-PLoc [2] and Hum-PLoc [4]. The pSLIP system utilizes five top-rank features of physicochemical proper-ties according to the prediction accuracy of SVM using a single feature [17]. The ProLoc system uses SVM with automatic selection from physicochemical properties to predict protein subnuclear localization [18]. The two ensemble classifiers Euk-OET-PLoc [2] and Hum-PLoc [4] fuse many basic individual classifiers operated by the engine of k-NN rules, where protein sequences are repre-sented by hybridizing the GO annotation and amphiphilic pseudo amino acid (Pse-AA) composition. Additionally, these two efficient GO-based systems Euk-OET-PLoc [2] and Hum-PLoc [4] predict subcellular local-ization of proteins using their known accession numbers. However, they cannot work for novel proteins without
known accession numbers. The GO-AA method [5], which uses GO terms of homologies retrieved by BLAST to assess protein similarity, can deal with novel proteins without known accession numbers for subnuclear locali-zation prediction. Besides, some SVM-based methods using only the features derived from input sequences, such as ProtLock with AAC [8], Ploc with AAC and acid pairs [10], and HSLPred with AAC and dipeptide compo-sition [11], predict subcellular localization inaccurately [4]. Therefore, this study would develop an accurate SVM-based method for predicting subcellular localization of novel proteins by using input sequences with BLAST. The Gene Ontology provided by the GO Consortium [1] has quickly grown in size and popularity. The newest ver-sion (UniProt 52.0 released in September 2007) of GO [22] contained 29,383 terms in the three branches, molec-ular function, biological process and cellmolec-ular component. The terms and relationships among them are represented by a directed acyclic graph in which vertices represent the GO terms, and edges represent the relationships among these terms. Genes can be annotated with GO terms creat-ing gene associations that can be used for whole genome analyses [23].
GO annotation has been successfully used in various sequence-based applications, which can be classified into two groups. 1) The first group uses the GO terms and their corresponding structure information of GO graph, such as grouping GO terms to improve the assessment of gene set enrichment [24]; using GO with probabilistic chain graphs for protein classification [25,26], and prediction of subnuclear localization [5]. 2) The second group uses GO terms only without structure information, such as predict-ing transcription factor DNA bindpredict-ing preference [27] and various predictions of subcellular and subnuclear locali-zation [2,4,5]. In the second group, protein sequences are often represented as high dimensional vectors of n binary features, where n is the total number of terms in the com-plete annotation set (a component of 1 if the annotation is hit, and 0 otherwise) [28]. This representation is valua-ble in well-known vector space clustering algorithms such as k-NN [2,4,13,19,21] and fuzzy k-NN [13,29,30]. How-ever, because n is often large, and each gene product is generally annotated by few GO terms, the vectors became long and sparse, making the clustering rather problematic [28].
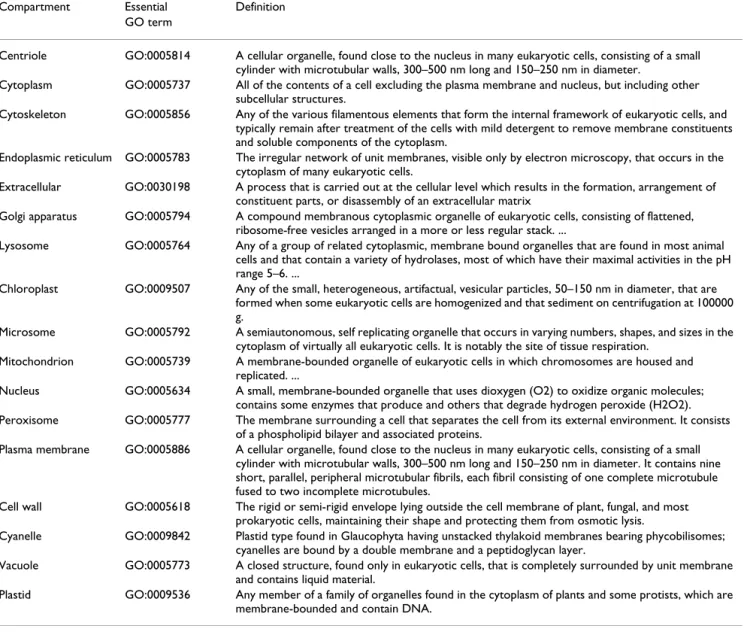
This study proposes an efficient method, named GOmin-ing, based on an intelligent genetic algorithm (IGA) [31,32] incorporating an SVM classifier to simultaneously identify a small number m out of a large number n of GO terms as input features, where m <<n. Some GO annota-tions corresponding to subcellular compartments are called essential GO terms for subcellular localization
pre-diction, such as GO:0005634 (Nucleus), GO:0005737 (Cytoplasm) and GO:0005856 (Cytoskeleton), shown in Table 1. These essential GO terms are regarded as domain knowledge to be included in the feature set of m informa-tive GO terms for subcellular localization prediction. A prediction method ProLoc-GO based on GOmining was implemented using the feature set of informative GO terms. This method performed well in predicting protein subcellular localization from input sequences only.
Results
Data sets
Two existing data sets SCL12 [4] and SCL16 [2] obtained from UniProtKB/Swiss-Prot database [33] were used to evaluate the proposed method ProLoc-GO. The SCL12 and SCL16 have 2041 human proteins localized in 12
human subcellular compartments and 4150 eukaryotic proteins in 16 subcellular compartments, respectively. The two data sets were operated by a culling program [34] so that those sequences had < 25% sequence identity. The proteins in SCL12 were screened strictly using the fol-lowing rules: 1) only those sequences annotated with "human" in the ID (identification) field were collected; 2) sequences annotated with ambiguous or uncertain terms, such as "potential", "probable", "probably", "maybe", or "by similarity", were excluded; 3) sequences annotated by two or more locations were excluded, and 4) sequences with less than 50 amino acid residues were removed [4]. The data set SCL12 was divided into two parts, SCL12L and SCL12T, with 919 and 1122 proteins, respectively.
Table 1: Essential GO terms and their definitions
Compartment Essential Definition GO term
Centriole GO:0005814 A cellular organelle, found close to the nucleus in many eukaryotic cells, consisting of a small cylinder with microtubular walls, 300–500 nm long and 150–250 nm in diameter.
Cytoplasm GO:0005737 All of the contents of a cell excluding the plasma membrane and nucleus, but including other subcellular structures.
Cytoskeleton GO:0005856 Any of the various filamentous elements that form the internal framework of eukaryotic cells, and typically remain after treatment of the cells with mild detergent to remove membrane constituents and soluble components of the cytoplasm.
Endoplasmic reticulum GO:0005783 The irregular network of unit membranes, visible only by electron microscopy, that occurs in the cytoplasm of many eukaryotic cells.
Extracellular GO:0030198 A process that is carried out at the cellular level which results in the formation, arrangement of constituent parts, or disassembly of an extracellular matrix
Golgi apparatus GO:0005794 A compound membranous cytoplasmic organelle of eukaryotic cells, consisting of flattened, ribosome-free vesicles arranged in a more or less regular stack. ...
Lysosome GO:0005764 Any of a group of related cytoplasmic, membrane bound organelles that are found in most animal cells and that contain a variety of hydrolases, most of which have their maximal activities in the pH range 5–6. ...
Chloroplast GO:0009507 Any of the small, heterogeneous, artifactual, vesicular particles, 50–150 nm in diameter, that are formed when some eukaryotic cells are homogenized and that sediment on centrifugation at 100000 g.
Microsome GO:0005792 A semiautonomous, self replicating organelle that occurs in varying numbers, shapes, and sizes in the cytoplasm of virtually all eukaryotic cells. It is notably the site of tissue respiration.
Mitochondrion GO:0005739 A membrane-bounded organelle of eukaryotic cells in which chromosomes are housed and replicated. ...
Nucleus GO:0005634 A small, membrane-bounded organelle that uses dioxygen (O2) to oxidize organic molecules; contains some enzymes that produce and others that degrade hydrogen peroxide (H2O2). Peroxisome GO:0005777 The membrane surrounding a cell that separates the cell from its external environment. It consists
of a phospholipid bilayer and associated proteins.
Plasma membrane GO:0005886 A cellular organelle, found close to the nucleus in many eukaryotic cells, consisting of a small cylinder with microtubular walls, 300–500 nm long and 150–250 nm in diameter. It contains nine short, parallel, peripheral microtubular fibrils, each fibril consisting of one complete microtubule fused to two incomplete microtubules.
Cell wall GO:0005618 The rigid or semi-rigid envelope lying outside the cell membrane of plant, fungal, and most prokaryotic cells, maintaining their shape and protecting them from osmotic lysis.
Cyanelle GO:0009842 Plastid type found in Glaucophyta having unstacked thylakoid membranes bearing phycobilisomes; cyanelles are bound by a double membrane and a peptidoglycan layer.
Vacuole GO:0005773 A closed structure, found only in eukaryotic cells, that is completely surrounded by unit membrane and contains liquid material.
Plastid GO:0009536 Any member of a family of organelles found in the cytoplasm of plants and some protists, which are membrane-bounded and contain DNA.
The SCL12L set was used for training and the SCL12T was used for independent testing, as shown in Table 2[4]. The proteins of SCL16 were screened according to four cri-teria. The first criterion is to exclude sequences annotated with "prokaryotic", because this study focused only on eukaryotic proteins. The other three criteria were the same as criteria 2–4 for SCL12 above. Table 3 shows the
num-bers of proteins within each compartment, where the SCL16 consists of two parts, SCL16L for training and SCL16T for independent testing. The sequences in the training and test data sets were obtained from the web servers of Euk-OET-PLoc [2] and Hum-PLoc [4].
GO annotation
This study applied the Gene Ontology Annotation (GOA) database [35], which includes GO annotations for non-redundant proteins from many species in the UniProtKB/ Swiss-Prot database [33]. The GOA database was down-loaded directly from [36] (UniProt 45.0 released in Jan. 2007). The accession numbers of proteins are required for querying the GOA database to obtain GO terms. BLAST [37,38] was used to obtain a homology with a known accession number to the protein for retrieving the GO terms. The corresponding accession numbers of all pro-tein sequences in SCL12 and SCL16 were obtained by using BLAST with h = 1 and e = 10-9.
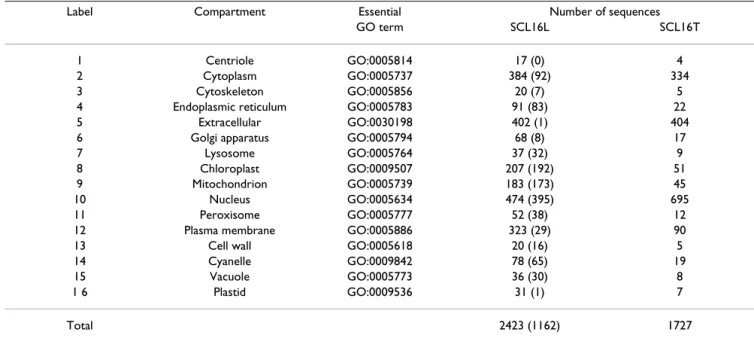
Table 4 shows the GO annotation results of all proteins in the training data sets SCL12L and SCL16L. For SCL12L, the size of the complete set of all GO terms that appeared was n = 1714 from the 919 human proteins. The smallest, largest and mean numbers of GO terms annotated for individual proteins were 0, 35 and 8.3, respectively. The percentage of training proteins whose homologies were not annotated by any GO term (that is, the number of GO terms annotated is zero) was 1.31%. For SCL16L, n = 2870 GO terms were obtained from 2423 eukaryotic proteins. The smallest, largest and mean numbers of GO terms Table 3: Data set SCL16. The data set SCL16 consists of SCL16L and SCL16T as the learning data set and testing data set,
respectively. There are 15 essential GO terms corresponding to eukaryotic subcellular compartments. Note that GO:0005814 is not appeared in the set of n = 2870 GO terms. The number t of (t) in SCL16L represents the number of sequences which are correctly annotated by only one essential GO term.
Label Compartment Essential Number of sequences GO term SCL16L SCL16T 1 Centriole GO:0005814 17 (0) 4 2 Cytoplasm GO:0005737 384 (92) 334 3 Cytoskeleton GO:0005856 20 (7) 5 4 Endoplasmic reticulum GO:0005783 91 (83) 22 5 Extracellular GO:0030198 402 (1) 404 6 Golgi apparatus GO:0005794 68 (8) 17 7 Lysosome GO:0005764 37 (32) 9 8 Chloroplast GO:0009507 207 (192) 51 9 Mitochondrion GO:0005739 183 (173) 45 10 Nucleus GO:0005634 474 (395) 695 11 Peroxisome GO:0005777 52 (38) 12 12 Plasma membrane GO:0005886 323 (29) 90 13 Cell wall GO:0005618 20 (16) 5 14 Cyanelle GO:0009842 78 (65) 19 15 Vacuole GO:0005773 36 (30) 8 1 6 Plastid GO:0009536 31 (1) 7 Total 2423 (1162) 1727
Table 2: Data set SCL12. The data set SCL12 consists of SCL12L and SCL12T as the learning data set and testing data set, respectively. There are 12 essential GO terms corresponding to subcellular compartments. The number t of (t) in SCL12L represents the number of sequences which are correctly annotated by only one essential GO term.
Label Compartment Essential Number of sequences GO term SCL12L SCL12T 1 Centriole GO:0005814 20 (1) 25 2 Cytoplasm GO:0005737 155 (38) 377 3 Cytoskeleton GO:0005856 12 (6) 14 4 Endoplasmic reticulum GO:0005783 28 (19) 35 5 Extracellular GO:0030198 140 (0) 301 6 Golgi apparatus GO:0005794 33 (5) 42 7 Lysosome GO:0005764 32 (27) 40 8 Microsome GO:0005792 7 (0) 8 9 Mitochondrion GO:0005739 125 (111) 228 10 Nucleus GO:0005634 196 (179) 580 11 Peroxisome GO:0005777 18 (16) 23 12 Plasma membrane GO:0005886 153 (23) 368 Total 919 (425) 1122
annotated were 0, 50 and 7.7, respectively. The percentage of training proteins whose homologies were not anno-tated was 3.96%. The proteins annoanno-tated by GO are often represented as an n-dimensional binary feature vector, where the attribute value is 1 if the corresponding GO term is annotated, and 0 otherwise.
To know the prediction performance according to only the essential GO terms annotated, we calculated the num-bers of sequences annotated by g essential GO terms. Table 4 shows that 453 out of 919 (49.3%) sequences are annotated by only one essential GO term (g = 1) for SCL12L, where 425 sequences are correctly annotated and 28 sequences are incorrectly annotated. The other 466 sequences annotated by zero (g = 0) or more than one (g > 1) essential GO term can not be effectively predicted. Table 2 lists the numbers of sequences which are correctly annotated by only one essential GO term for every com-partment. The two GO terms, GO:0005634 (Nucleus) and GO:0005739 (Mitochondrion), made a great contribu-tion to the prediccontribu-tion accuracy of 46.2% (= 425/919),
which correctly annotate a large number of sequences, 179 and 111, respectively.
As for SCL16L, the number of sequences annotated by only one essential GO term is 1247 out of 2423 (51.5%). Table 3 lists the numbers of sequences which are correctly annotated by only one essential GO term for every com-partment. Only 48.0% (= 1162/2423) of the sequences with known accession numbers can be correctly predicted by using only the annotation of essential GO terms. According to Table 3, the three essential GO terms, GO:0005634 (Nucleus, 395 out of 474), GO:0009507 (Chloroplast, 192 out of 207) and GO:0005739 (Mito-chondrion, 173 out of 183), made a great contribution to prediction accuracy.
The analytic results reveal that it is not sufficient to use only essential GO terms for accurately predicting protein subcellular localization. However, the essential GO terms play an important role in designing GO-based prediction methods.
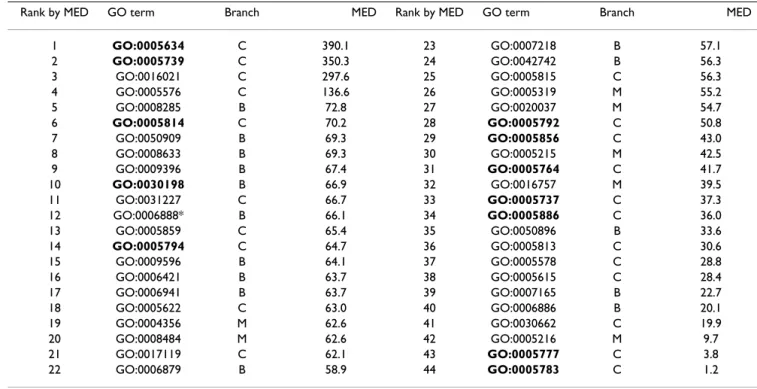
Table 5: The m = 44 informative GO terms by applying GOmining to SCL12L. The GO terms in bold style are essential GO terms.
Rank by MED GO term Branch MED Rank by MED GO term Branch MED
1 GO:0005634 C 390.1 23 GO:0007218 B 57.1 2 GO:0005739 C 350.3 24 GO:0042742 B 56.3 3 GO:0016021 C 297.6 25 GO:0005815 C 56.3 4 GO:0005576 C 136.6 26 GO:0005319 M 55.2 5 GO:0008285 B 72.8 27 GO:0020037 M 54.7 6 GO:0005814 C 70.2 28 GO:0005792 C 50.8 7 GO:0050909 B 69.3 29 GO:0005856 C 43.0 8 GO:0008633 B 69.3 30 GO:0005215 M 42.5 9 GO:0009396 B 67.4 31 GO:0005764 C 41.7 10 GO:0030198 B 66.9 32 GO:0016757 M 39.5 11 GO:0031227 C 66.7 33 GO:0005737 C 37.3 12 GO:0006888* B 66.1 34 GO:0005886 C 36.0 13 GO:0005859 C 65.4 35 GO:0050896 B 33.6 14 GO:0005794 C 64.7 36 GO:0005813 C 30.6 15 GO:0009596 B 64.1 37 GO:0005578 C 28.8 16 GO:0006421 B 63.7 38 GO:0005615 C 28.4 17 GO:0006941 B 63.7 39 GO:0007165 B 22.7 18 GO:0005622 C 63.0 40 GO:0006886 B 20.1 19 GO:0004356 M 62.6 41 GO:0030662 C 19.9 20 GO:0008484 M 62.6 42 GO:0005216 M 9.7 21 GO:0017119 C 62.1 43 GO:0005777 C 3.8 22 GO:0006879 B 58.9 44 GO:0005783 C 1.2
Table 4: Results of GO annotation for all sequences in SCL12L and SCL16
Data set Total GO terms n Number of GO terms Number of sequences annotated by g essential GO terms Smallest Largest Mean g = 0 g = 1 g > 1
SCL12L 1714 0 35 8.3 404 453 62 SCL16L 2870 0 50 7.7 1025 1247 151
Selected informative GO terms
Selecting a set of m informative GO terms out of n candi-date GO terms is a combinatorial optimization problem C(n, m), which can be solved by using the intelligent genetic algorithm with an inheritance mechanism (IGA) [31,32]. IGA can efficiently search for the solution Sr+1 to C(n, r+1) by inheriting a good solution Sr to C(n, r). This
study proposes an efficient algorithm based on IGA, called GOmining, to identify a small set of m informative GO terms including the essential GO terms as features to SVM. The GOmining algorithm incorporates LIBSVM [39] using series of binary classifiers. GOmining aims to maxi-mize the training accuracy of prediction using 10-fold cross-validation (10-CV) when identifying the m inform-ative GO terms.
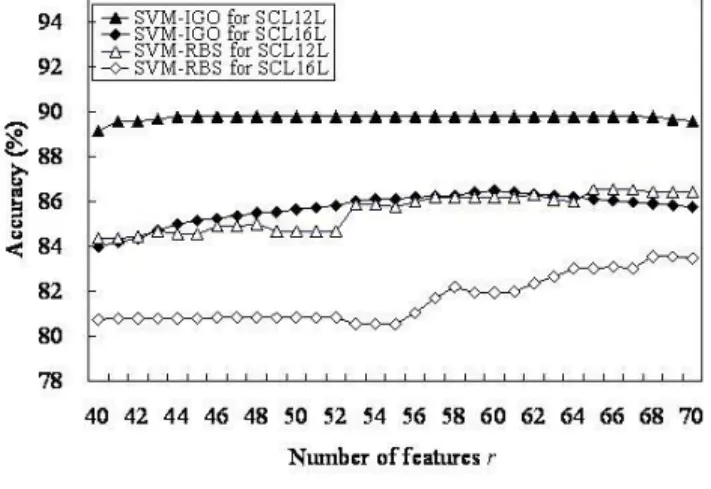
The SVM classifier based on the selected informative GO terms as features is called SVM-IGO. To evaluate a candi-date set of r informative GO terms accompanied with the SVM parameters, the prediction accuracy of 10-CV serves as a fitness function of IGA. Figure 1 shows the results of SVM-IGO from r = 40, 41,..., 70. Table 5 lists the m = 44 informative GO terms for SCL12L obtained from the
highest accuracy of 89.8% (r = 44), where the SVM param-eters (C, γ) = (23, 2-4). Table 6 lists the m = 60 informative
GO terms for SCL16L, where the highest accuracy was 86.5%, and (C, γ) = (25, 2-3).
Training accuracies of SVM-IGO and SVM-RBS performed by using SVM with a number r of selected informative GO terms
Figure 1
Training accuracies of SVM-IGO and SVM-RBS per-formed by using SVM with a number r of selected informative GO terms.
Table 6: The m = 60 informative GO terms by applying GOmining to SCL16L. The GO terms in bold style are essential GO terms.
Rank by MED GO term Branch MED Rank by MED GO term Branch MED
1 GO:0005634 C 331.7 31 GO:0005525 M 56.5 2 GO:0009507 C 244.2 32 GO:0005789 C 55.6 3 GO:0016020 C 148.1 33 GO:0004725 M 55.6 4 GO:0005739 C 147.8 34 GO:0008270 M 54.6 5 GO:0001844 B 70.7 35 GO:0015031 B 54.1 6 GO:0005212 M 70.5 36 GO:0005813 C 52.2 7 GO:0005886 C 68.5 37 GO:0005524 M 51.8 8 GO:0006094 B 67.9 38 GO:0051536 M 51.8 9 GO:0045261 C 67.5 39 GO:0016702 M 51.1 10 GO:0009536 C 66.6 40 GO:0005887 C 48.4 11 GO:0007010 B 65.8 41 GO:0005905 C 46.1 12 GO:0030198 B 65.4 42 GO:0005794 C 43.6 13 GO:0009626 B 65.4 43 GO:0005622 C 43.4 14 GO:0005047 M 64.6 44 GO:0005759 C 43.1 15 GO:0017134 M 64.1 45 GO:0005856 C 42.1 16 GO:0000287 M 63.3 46 GO:0016757 M 41.7 17 GO:0006888 B 61.7 47 GO:0005618 C 41.3 18 GO:0030234 M 61.7 48 GO:0005783 C 35.7 19 GO:0007323 B 61.0 49 GO:0005773 C 31.2 20 GO:0008083 M 60.8 50 GO:0009842 C 27.2 21 GO:0004521 M 60.3 51 GO:0006811 B 25.3 22 GO:0003723 M 60.2 52 GO:0006350 B 24.3 23 GO:0009514 C 59.9 53 GO:0016740 M 23.1 24 GO:0020015 C 59.4 54 GO:0005737 C 20.2 25 GO:0000922 C 59.0 55 GO:0005829 C 19.6 26 GO:0005681 C 58.9 56 GO:0016798 M 15.5 27 GO:0030149 B 57.9 57 GO:0007186 B 14.8 28 GO:0000917 B 56.9 58 GO:0019843 M 13.0 29 GO:0009405 B 56.8 59 GO:0005764 C 11.8 30 GO:0005615 C 56.5 60 GO:0005777 C 10.2
The orthogonal experimental design with orthogonal array and factor analysis used in IGA is an efficient method for simultaneously examining the individual effect of several factors on the evaluative function [40,41]. The factors are the parameters (GO terms) that manipu-late the evaluation function, and a setting of a parameter is regarded as a level of the factor. In this study, the two levels of one factor are the inclusion and exclusion of the ith GO term in the feature selection using IGA. The factor analysis can quantify the effects of individual factors on the evaluation function, rank the most effective factors and determine the best level for each factor to optimize the evaluation function. The most effective factor has the largest main effect difference (MED). Tables 5 and 6 show that the essential GO term GO:0005634 (Nucleus) having the largest values of MED is the most effective feature of discrimination. The only essential GO term GO:0030198 (Extracellular matrix organization and biogenesis) belongs to biologic process branch and the other essential GO terms belong to cellular component branch. The abbreviations M, B and C represent the three branches molecular function, biological process, and cellular com-ponent, respectively.
Evaluation of feature selection
SVM-IGO was implemented by using the m informative GO terms and the SVM classifier using (C, γ) = (23, 2-4)
and (C, γ) = (25, 2-3) for SCL12L and SCL16L, respectively.
To evaluate the effectiveness of SVM-IGO, four additional classifiers were implemented for comparison. Three clas-sifiers SVM-GO, k-NN-GO and fuzzy k-NN-GO in order based on SVM, k-NN and fuzzy k-NN were implemented by using all the n GO terms as features without GO term selection. The classifier SVM-RBS used SVM with a subset of n GO terms selected by the rank-based selection (RBS) method [17,42]. The best values of parameters C and γ determined using a step-wise approach were employed to the SVM-based methods SVM-GO and SVM-RBS, where γ ∈ {2-7, 2-6,..., 28} and C ∈ {2-7, 2-6,..., 28}. The best values
(C, γ) = (23, 2-4) and (24, 2-6) were applied to SVM-GO for
SCL12L and SCL16L, respectively. As for SVM-RBS, (C, γ) = (21, 2-3) and (22, 2-2) were used for SCL12L and SCL16L,
respectively. The Methods section describes SVM-RBS, k-NN-GO and fuzzy k-k-NN-GO in detail. Table 7 lists all pre-diction accuracies using 10-CV for both data sets SCL12L and SCL16L.
The highest accuracies of SVM-RBS are 86.5% and 83.5% using 65 and 68 selected GO terms for SCL12L and SCL16L, respectively, shown in Fig. 1. Table 7 shows that the three SVM-based classifiers (SVM-GO, SVM-RBS and SVM-IGO), with accuracies >80%, were better than the two k-NN based classifiers (GO and fuzzy k-NN-GO), with accuracies <75%, for both data sets. SVM-IGO had the highest accuracies 89.8% and 86.5% for SCL12L
and SCL16L, respectively. The GO term selection method based on GOmining was more effective than RBS and the method without selection of GO terms. Furthermore, SVM uses the selected GO terms as features, making it bet-ter than the k-NN classifier.
Performance comparison
The proposed ProLoc-GO method predicts the subcellular localization of an input sequence using either SVM-IGO or SVM-GO, depending on its annotation on the inform-ative GO terms (see Methods for detail). Tables 8, 9, 10, 11 list the results of ProLoc-GO using SCL12 and SCL16. Some existing AAC-based prediction methods, such as ProtLock [8], Least Euclidean distance [9], Ploc [10] and HSLPred [11], use only the query sequence as input data for their classifiers. Hum-PLoc [4] and Euk-OET-PLoc [2] use both the sequence and its accession number as input data. For comparison with these predictors, the method ProLoc-GO was performed using the two kinds of input data separately. The first test used only the sequence and used BLAST to obtain annotated GO terms. The second test used the known accession number of proteins directly. For the accuracy on both SCL12L and SCL16L, ProLoc-GO used leave-one-out cross-validation (LOOCV) for comparison with the other methods (see Methods sec-tion).
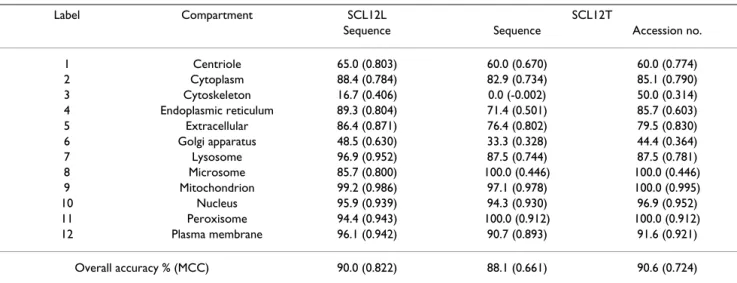
The test accuracies for ProLoc-GO performed on the human protein data set SCL12L and SCL12T were 90.0% and 88.1%, respectively, where m = 44, and (C, γ) = (23, 2-4) for SVM-IGO and SVM-GO. These results were much better than those (<35%) of the four sequence-based pre-diction methods [8-11] using only input sequences, shown in Table 8. Ploc [10] had the highest test accuracy of 34.3% among the four AAC-based methods.
The Matthews correlation coefficient (MCC) [5,12,18] values are usually employed while evaluating the per-formance on unbalanced datasets. In addition to the over-all accuracy, the MCC values were also recorded due to the unbalance of numbers of proteins localized in the com-partments, such as 196 of Nucleus vs. 7 of Microsome (Table 2). The MCC is defined as follows [5]:
where pc is the number of correctly predicted proteins of the location c, sc is the number of correctly predicted pro-teins not in the location c, uc is the number of under-pre-dicted proteins, oc is the number of over-predicted proteins, and Nc is the number of locations. The test MCC performances of ProLoc-GO were 0.822 and 0.661 for SCL12L and SCL12T, respectively. Table 10 presents the
MCC pcsc ucoc pc uc pc oc sc uc sc oc c N c= − c +
(
)
(
+)
(
+)
(
+)
, =1 2 …, , , , (1)detailed results for individual compartments. The results of the five sequence-based methods reveal that the set of informative GO terms is more useful for protein subcellu-lar localization than the AAC-based features.
Performance of using known accession numbers
The accession number of each protein sequence in SCL12 and SCL16 was available in querying the GOA database. For comparison with the methods [2,4] based on the pro-teins with known accession numbers, ProLoc-GO using the known accession numbers of proteins as input data obtained test accuracies of 91.1% and 90.6% (MCC = 0.724) performed on SCL12L and SCL12T, respectively, where m = 56, (C, γ) = (22, 2-1) for SVM-IGO and (C, γ) =
(22, 2-4) for SVM-GO. Hum-PLoc [4] using hybridization
of GO terms and Pse-AA composition obtained training and test accuracies of 81.1% and 85.0% for SCL12L and SCL12T, respectively. The performance of ProLoc-GO using sequences or accession numbers as the input data was better than that of Hum-PLoc [4] using the ensemble classifiers with features of both sequence and accession number.
Tables 9 and 11 show the performance results of ProLoc-GO and Euk-OET-PLoc [2] using SCL16. ProLoc-ProLoc-GO using input sequences yielded test accuracies 86.6% (MCC = 0.799) and 83.3% (MCC = 0.706) for SCL16L and SCL16T, respectively, where m = 60, (C, γ) = (25, 2-3) for
SVM-IGO, and (C, γ) = (24, 2-6) for SVM-GO. ProLoc-GO
is significantly better than all the AAC-based methods with test accuracies smaller than 35%. ProLoc-GO yields the test accuracies 89.0% and 85.7% (MCC = 0.710) for SCL16L and SCL16T, respectively, using the known acces-sion numbers of proteins, where m = 60, (C, γ) = (22, 2-3)
for SVM-IGO and (C, γ) = (23, 2-5) for SVM-GO.
Euk-OET-PLoc [2] using the ensemble classifiers with features of both sequence and accession number obtains training and test accuracies of 81.6% and 83.7%, respectively. Pro-Loc-GO performed better than Euk-OET-PLoc on SCL16 using either sequences or accession numbers as the input data [2].
Analysis of informative GO terms
The GOmining method identifies a feature set of m effec-tive GO terms, called informaeffec-tive GO terms, to design an accurate SVM-based prediction method. Table 12 shows the distribution of the m informative GO terms in the GO graph. For SCL12L with m = 44, GOmining selected 12 essential GO terms and 32 instructive GO terms. The 32 instructive GO terms consist of 7 GO terms from the molecular function branch, 14 terms from the biological process branch, and 11 terms from the cellular compo-nent branch, denoted as 7(M), 14(B) and 11(C), respec-tively. Analytical results reveal that all the three branches contain instructive GO terms.
Due to the high correlation among GO terms in the GO graph, the feature selection of SVM should consider simultaneously a set of informative GO terms, rather than individual GO terms. Since the essential GO terms are always included, GOmining benefits from a confined search space of candidate instructive GO terms. Consider-ing the position relationships between instructive and essential GO terms in the GO graph, instructive GO terms belonged to one of the three classes: (a) offspring but not ancestor of some essential GO term; (b) between two essential GO terms, and (c) not offspring of any essential GO term. Of the 32 instructive GO terms, 4, 2 and 26 GO Table 7: Comparison of prediction accuracy (%) using 10-CV. Performance comparison uses prediction accuracy (%) of 10-CV.
Data set k-NN-GO Fuzzy k-NN-GO SVM-GO SVM-RBS SVM-IGO SCL12L 74.3 71.4 88.5 86.5 89.8 SCL16L 66.0 59.3 84.8 83.5 86.5
Table 8: Comparison of prediction accuracy (%) for SCL12. Prediction accuracies (%) for using leave-one-out cross-validation (LOOCV) on SCL12L and independent test on SCL12T are obtained from the paper [4]. The input data is sequence only (S) or sequence with accession number (AN).
Method Input data Features LOOCV SCL12L Independent test SCL12T ProtLock [8] S AAC 29.7 26.4
Least Euclidean distance [9] S AAC 30.1 29.5 Ploc [10] S AAC and amino acid pairs 30.5 34.3 HSLPred [11] S AAC and dipeptide composition 30.7 33.3 ProLoc-GO S GO terms using BLAST 90.0 88.1 Hum-PLoc [4] S with AN Hybridization of GO terms and Pse-AA 81.1 85.0 ProLoc-GO S with AN GO terms (No BLAST) 91.1 90.6
terms belonged to the classes (a), (b) and (c), respectively. The 26 GO terms consist of 7(M), 14(B) and 5(C). The GO terms near the root of the GO graphs are considered to be more generic while terms near the leaves are more specific [23]. Of the instructive GO terms, 81.2% (26/32) were not offspring of any essential GO term. These analyt-ical results reveal that the essential GO terms are informa-tive enough in predicting subcellular localization, and are effective in confining the space of searching instructive GO terms. The other six instructive GO terms from the cel-lular component branch have more specific functions than the essential GO terms in discrimination of the sub-cellular localization.
Figures 2, 3, 4 illustrate some of the instructive GO terms belonging to the three classes. Three instructive GO terms were found to belong to class (a), namely SCL12L: GO:0031227 (Intrinsic to endoplasmic reticulum mem-brane, rank 11), GO:30662 (Coated vesicle memmem-brane, rank 41) and GO:0017119 (Golgi transport complex, rank 21), according to Fig. 2. The two terms belonging to class (b), namely GO:0005815 (Microtubule organizing center, rank 25) and GO:0005813 (Centrosome, rank 36), were found between the essential GO terms GO:0005856 (Cytoskeleton) and GO:0005814 (Centriole), as shown
in Fig. 3. According to Fig. 4, five instructive GO terms belonging to the class (c) were not offspring of essential GO terms, GO:0016021 (Integral to membrane, rank 3), GO:0005576 (Extracellular region, rank 4), GO:0005622 (intracellular, rank 18), GO:0005578 (Proteinaceous extracellular matrix, rank 37) and GO:0005615 (Extracel-lular space, rank 38).
The m = 60 informative GO terms for SCL16L comprises 15 essential GO terms and 45 instructive GO terms. The 45 instructive GO terms consisted of 18(M), 13(B) and 14(C). The numbers of instructive GO terms coming from each branch were not significantly different. However, the numbers of instructive GO terms belonging to the three classes (a), (b) and (c) are 9, 0 and 36, respectively, which are very different. 80% (36/45) of the instructive GO terms were not offspring of any essential GO term. The 9 instructive GO terms belonging to the class (a) had 5, 2 and 2 terms, respectively, as shown in Figs. 2, 3 and 4. Class (c) has five GO terms with a dot-pattern box: GO:0005622 (intracellular), GO:0005615 (Extracellular space), GO:0020015 (Glycosome), GO:0016020 (Mem-brane) and GO:0045261 (Proton-transporting ATP syn-thase complex, catalytic core F(1)), as revealed by Fig. 4.
Some of the selected GO terms which are offspring of essential GO terms
Figure 2
Some of the selected GO terms which are offspring of essential GO terms. For SCL12L, there are three terms
shown: GO:0031227, GO:0030662 and GO:0017119. For SCL16L, five GO terms are shown: GO:0009514, GO:0005681, GO:0005789, GO:0005759 and GO:0005829.
The statistical results of instructive GO terms distributed in the three classes for both SCL12L and SCL16L reveal that the inclusion of essential GO terms can be regarded as using domain knowledge for GOmining to mine a fea-ture set of informative GO terms. The heuristic approach (using domain knowledge) of GOmining is efficient when the GO database grows fast. Therefore, GOmining can be easily applied to other applications of
sequence-based predictions using SVM with the features of inform-ative GO terms.
Discussion
The GO database has grown in size recently, increasing the effectiveness of GO-based features. Meanwhile, the per-centage of proteins with subcellular locations annotated in the GO database increased from 44.9% [2] to 65.5% [3]
Some of the selected GO terms which are between two essential GO terms
Figure 3
Some of the selected GO terms which are between two essential GO terms. For SCL12L, the two instructive GO
terms GO:0005815 and GO:0005813 are between the essential GO terms GO:0005856 and GO:0005814. For SCL16L, GO:0005813 and GO:0000922 are offspring of the essential GO term GO:0005856, belonging to the class (a). GO:0005814 is not an essential GO term for SCL16L.
Table 9: Comparison of prediction accuracy (%) for SCL16. Prediction accuracies (%) of using LOOCV on SCL16L and independent test on SCL16T are obtained from the paper [2]. The input data is sequence only (S) or sequence with accession number (AN).
Method Input data Features LOOCV SCL16L Independent test SCL16T ProtLock [8] S AAC 28.7 25.3
Least Euclidean distance [9] S AAC 25.8 20.4 Ploc [10] S AAC and amino acid pairs 35.1 32.8 HSLPred [11] S AAC and dipeptide composition 33.1 34.5 ProLoc-GO S GO terms using BLAST 86.6 83.3 Euk-OET-PLoc [2] S with AN Hybridization of GO terms and Pse-AA 81.6 83.7 ProLoc-GO S with AN GO terms (No BLAST) 89.0 85.7
fast. It is indicated that there is a linkage in the GO anno-tation process between molecular function annoanno-tation and subcellular localization annotation [43]. Therefore, the GO-based prediction method for protein subcellular localization is increasingly efficient. Because the accession number of proteins is necessary for retrieving GO terms from GO databases, existing efficient GO-based systems Euk-OET-PLoc [2] and Hum-PLoc [4] directly utilize the accession numbers of proteins and a large number n of GO terms annotated in a complete set where n = 9918 for SCL12L [4] and n = 9567 for SCL16L [2].
To predict subcellular localizations for novel proteins, ProLoc-GO uses a good homology, rather than the query protein itself, to retrieve annotated GO terms using BLAST. To use GO term features effectively, ProLoc-GO uses only a homology with annotated GO terms to reduce
n. Thus, n = 1714 for SCL12L and n = 2870 for SCL16L.
Furthermore, a small set of m informative GO terms is selected simultaneously by GOmining. GOmining can consider internal relevant-feature correlation, instead of individual features by using an efficient global optimiza-tion method. The distribuoptimiza-tion analysis of informative GO terms in the GO graph is consistent with the properties of Table 10: Accuracies and MCC preformed on SCL12
Label Compartment SCL12L SCL12T
Sequence Sequence Accession no. 1 Centriole 65.0 (0.803) 60.0 (0.670) 60.0 (0.774) 2 Cytoplasm 88.4 (0.784) 82.9 (0.734) 85.1 (0.790) 3 Cytoskeleton 16.7 (0.406) 0.0 (-0.002) 50.0 (0.314) 4 Endoplasmic reticulum 89.3 (0.804) 71.4 (0.501) 85.7 (0.603) 5 Extracellular 86.4 (0.871) 76.4 (0.802) 79.5 (0.830) 6 Golgi apparatus 48.5 (0.630) 33.3 (0.328) 44.4 (0.364) 7 Lysosome 96.9 (0.952) 87.5 (0.744) 87.5 (0.781) 8 Microsome 85.7 (0.800) 100.0 (0.446) 100.0 (0.446) 9 Mitochondrion 99.2 (0.986) 97.1 (0.978) 100.0 (0.995) 10 Nucleus 95.9 (0.939) 94.3 (0.930) 96.9 (0.952) 11 Peroxisome 94.4 (0.943) 100.0 (0.912) 100.0 (0.912) 12 Plasma membrane 96.1 (0.942) 90.7 (0.893) 91.6 (0.921) Overall accuracy % (MCC) 90.0 (0.822) 88.1 (0.661) 90.6 (0.724)
Table 11: Accuracies and MCC preformed on SCL16
Label Compartment SCL16L SCL16T
Sequence Sequence Accession no. 1 Centriole 61.1 (0.747) 66.7 (0.729) 50.0 (0.577) 2 Cytoplasm 72.9 (0.706) 74.6 (0.676) 72.8 (0.659) 3 Cytoskeleton 20.0 (0.363) 0.0 (-0.002) 0.0 (-0.003) 4 Endoplasmic reticulum 90.1 (0.841) 77.3 (0.707) 72.7 (0.678) 5 Extracellular 89.6 (0.778) 84.4 (0.738) 84.9 (0.796) 6 Golgi apparatus 63.2 (0.653) 41.2 (0.508) 41.2 (0.486) 7 Lysosome 97.3 (0.973) 100.0 (0.865) 100.0 (0.865) 8 Chloroplast 98.6 (0.989) 100.0 (0.990) 100.0 (0.990) 9 Mitochondrion 99.5 (0.963) 91.1 (0.879) 95.6 (0.877) 10 Nucleus 89.0 (0.913) 86.3 (0.865) 93.5 (0.924) 11 Peroxisome 98.1 (0.971) 100.0 (0.960) 100.0 (0.925) 12 Plasma membrane 86.4 (0.851) 88.9 (0.773) 82.2 (0.798) 13 Cell wall 75.0 (0.655) 60.0 (0.422) 80.0 (0.631) 14 Cyanelle 88.5 (0.939) 94.7 (0.973) 100.0 (1.000) 15 Vacuole 86.1 (0.847) 62.5 (0.790) 50.0 (0.706) 16 Plastid 51.6 (0.595) 42.9 (0.426) 42.9 (0.311) Overall accuracy % (MCC) 86.6 (0.799) 83.3 (0.706) 85.7 (0.710)
GO annotation. Additionally, ProLoc-GO using input sequences is slightly worse than using the accession num-bers of proteins, with accuracies of 88.1% vs. 90.6% for SCL12T, and 83.3% vs. 85.7% for SCL16T, as shown in Tables 10 and 11.
Conclusion
Computational prediction methods from primary protein sequences are fairly economical in terms of identifying large-scale eukaryotic proteins with unknown functions. The GO annotation, which describes the function of genes and gene products across species, has been used to improve the prediction of protein subcellular localiza-tion. The accession numbers of proteins are necessary to query the GOA database to obtain GO terms. Since novel proteins have no known accession numbers, BLAST was used to obtain homologies with known accession num-bers to the proteins for the retrieval of GO terms.
GO annotation has grown in size and popularity. How-ever, few studies have explored informative GO terms from the over 20,000 annotations available at present for sequence-based prediction problems. This study proposes a genetic algorithm based method, GOmining, which combines SVM to simultaneously identify a small number
m out of the n GO terms as features to SVM, where m <<n.
The m GO terms include the essential GO terms annotat-ing subcellular compartments such as GO:0005634 (Nucleus), GO:0005737 (Cytoplasm) and GO:0005856 (Cytoskeleton). ProLoc-GO was evaluated using SVM with the GO-based features from two kinds of input data, sequence and known accession numbers of proteins. ProLoc-GO yields test accuracies of 88.1% and 83.3% from SCL12 and SCL16, respectively, when using only input sequences. These results are significantly superior to those of the other SVM-based methods, which have accu-racies <35% using AAC with acid pairs, and using AAC with dipedtide composition. ProLoc-GO using known accession numbers of proteins has accuracies 90.6% and 85.7% for SCL12 and SCL16, which is also slightly better than Hum-PLoc and Euk-OET-PLoc, which have 85.0% and 83.7%, respectively.
Analysis of m informative GO terms in the GO graph reveals that GOmining can consider internal relevant-fea-ture correlation, rather than individual fearelevant-fea-tures, by using an efficient global optimization method. GOmining can serve as an efficient tool for mining informative GO terms for various sequence-based predictions of proteins, espe-cially when the GO database grows fast. The prediction system using ProLoc-GO with protein sequence as input data for protein subcellular localization has been imple-mented (see Availability).
Methods
Proposed GOmining algorithm
An efficient genetic-algorithm-based method, called GOmining, is proposed for selecting informative GO terms. GOmining uses an intelligent genetic algorithm with an inheritable mechanism (IGA) [31,32], combined with an SVM classifier, to simultaneously identify a small number m out of a large number n of GO terms as input features, where m <<n. The exploration of the m informa-tive GO terms from n candidate GO terms is a combinato-rial optimization problem C(n, m) with a huge search space of size C(n, m) = n!/(m!(n-m)!)). An IGA based on orthogonal experimental design using a divide-and-con-quer strategy and systematic reasoning method can effi-ciently solve this large combinatorial optimization problem.
The leave-one-out cross-validation (LOOCV) is consid-ered to be the most rigorous and objective test. Although bias-free, this test is very computationally demanding and is often impractical for large data sets. The N-fold cross-validation not only provides a bias-free estimation of the accuracy at a much reduced computational cost, but is also considered as an acceptable test for evaluating predic-tion performance of an algorithm [44]. Therefore, GOmining uses the prediction accuracy of 10-CV as the fitness function to perform IGA on the entire training sets of proteins under considering the computation cost. The input of the algorithm GOmining is composed of 1) a training set of protein sequences categorized into a number of compartments (classes), and 2) the essential GO terms corresponding to the compartments. The out-put comprises a set of m informative GO terms and the associated parameter settings of an SVM classifier. Since the novel sequences without known accession numbers use BLAST to obtain annotated GO terms, all training sequences use the same BLAST to obtain GO terms for consistence.
Step 1: (preparation of SVM) The multi-classification problem is solved by using a series of binary classifiers of LIBSVM [39]. In this study, the kernel parameter γ and
cost parameter C are tuned where γ ∈ {2-7, 2-6,..., 28} and
C ∈ {2-7, 2-6,..., 28}.
Step 2: (sequence representation) Obtain annotated GO terms from the GOA database for all training proteins using BLAST with h = 1 and e = 10-9. Let n be the total
number of GO terms that appear among all proteins in the training data set. For example, n = 1714 and n = 2870 were derived for SCL12L and SCL16L, respectively. The protein is represented as an n-dimensional binary feature vector.
Step 3: (inclusion of essential GO terms) Identify d essen-tial GO terms out of n GO terms and number them from 1 to d. For example, d = 12 and d = 15 were found from SCL12L and SCL16L, respectively.
Step 4: (chromosome encoding) The IGA-chromosome comprises n binary IGA-genes fi for selecting informative GO terms and two 4-bit IGA-genes for encoding γ and C,
where fi = 1, i = 1,..., d. The ith GO term is included in the feature set of the SVM classifier if fi = 1; otherwise, the ith GO term is excluded (fi = 0). Figure 5 shows the sequence representation and IGA-chromosome encoding method. Step 5: (initial solution) Perform IGA to select rstart out of
n GO terms, i.e., the solution to C(n, rstart), where the d GO terms are always selected. Table 13 shows the param-eter settings of IGA, such as crossover probability pc = 0.8. The procedure of IGA is described in detail in the work [18].
Step 6: (inheritance mechanism) The inheritance mecha-nism of IGA can efficiently search for the solution to C(n,
r+1) by inheriting a good solution Sr to C(n, r). Obtain all solutions Sr from r = rstart+1,..., rend one by one using IGA [31,32]. For example, rstart = 40 and rend = 70 according to former experience.
Step 7: (decoding chromosome) Let Sm be the most accu-rate solution with m selected GO terms among all solu-tions Sr. Obtain the m informative GO terms and parameter values of γ and C.
Step 8: (robust performance) Perform Steps 5–7 for N independent runs to obtain the best one of N solutions Sm and the associated parameter settings of the SVM parame-ters. The best solution considers both high prediction accuracy and high mean frequency of the m selected GO terms appeared in the N runs. In this study, N = 30.
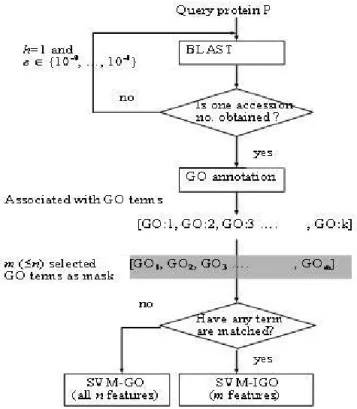
ProLoc-GO
As shown in Fig. 6, each query protein is first BLASTed with h = 1 and e = 10-9 against the Swiss-Prot database to
obtain a homology with a known accession number. If no such homology exists, then adjust the threshold value e of BLAST until the desired homology is obtained, where h =
1 and e ∈ {10-9, 10-8,..., 10-1}. The accession number of
the homology of each protein sequence in SCL12 and SCL16 was obtained by using BLAST with h = 1 and e = 10 -9. This accession number is used as input to the GOA
data-base for retrieving the corresponding k (>1) GO terms: GO:1, GO:2,... GO:k. If none of the k GO terms belongs to the set of m informative GO terms, then the sequence is represented using an n-dimensional binary vector and is predicted by the SVM-GO classifier. Otherwise, the sequence is represented as an m-dimensional binary vec-tor and is predicted by the SVM-IGO classifier. Notably, the SVM-GO classifier predicts only a very small percent-age of input sequences. ProLoc-GO is derived from the two major classifiers SVM-GO and SVM-IGO for subcellu-lar localization prediction.
Fuzzy k-NN
The protein is represented as an n-dimensional binary vec-tor and the generalized distance between two proteins P and Pi [2] is denoted as :
D(P, Pi) = 1 - P·Pi/||P|| ||Pi||, (2)
where P·Pi is the dot product of vectors P and Pi, and ||P|| and ||Pi|| are their moduli.
This study determined the best value of k by using a step-wise approach where k ∈ {1, 2,..., 10}.
The fuzzy k-NN classifier [13,29,30] is a variation of k-NN, which assign fuzzy membership values rc(P) of a query sequence P to each class c as follows:
where the distance is calculated by according to (1). In this study, the best values of parameters (k, w) are tuned iteratively from k ∈ {1, 2,..., 10} and w ∈ {1.05, 1.10,..., 1.95} for the fuzzy k-NN classifier.
r rc j j w j k j w j k c N c( ) C / , , ,..., P P P P P P =
( )
− −(
−)
= ∑ − −(
−)
= ∑ = 2 1 1 2 1 1 1 2 ,, (3) Table 12: Distribution of the m informative GO terms. Most instructive GO terms (80%) are not offspring of the essential GO terms that the ratios are 26/32 and 36/45 for SCL12L and SCL16L, respectively.SCL12L (m = 44) SCL16L (m = 60) Essential GO terms 12: 1 (B), 11 (C) 15: 1(B), 14(C) Instructive GO terms: 32: 45: (a) offspring of some essential GO term 4 (C) 9 (C)
(b) between two essential GO terms 2 (C) 0 (c) not offspring of any essential GO term 7(M), 14(B), 5 (C) 18(M), 13(B), 5(C)
SVM-RBS
To evaluate the proposed IGA-based feature selection method GOmining, this study implements a classifier SVM-RBS by using SVM with a subset of the n GO terms by the rank-based selection (RBS) method [17,42]. One previous work on ProLoc [18] showed that this univariate method RBS is inferior to the multivariate feature selec-tion by IGA for selecting physicochemical properties. First, each of all n GO terms (for example, n = 1714 for SCL12L) is ranked according to the accuracy of SVM with the evaluated single feature, where the best values of parameters (C, γ) were determined using a step-wise approach where γ ∈ {2-7, 2-6,..., 28} and C ∈ {2-7, 2-6,...,
28}. The top-ranking 70 features a
i, i = 1,..., 70 are then
picked, and the top-ranking 40 features with r = 40 are used as an initial feature set {b1,..., b40}. Consequently, the feature set with size r+1 is incrementally established by adding the best feature br+1 (having the highest
accu-Some of the selected GO terms are NOT offspring of any essential GO terms
Figure 4
Some of the selected GO terms are NOT offspring of any essential GO terms. For SCL12L, five instructive GO
terms are shown belonging to cellular component branch: GO:0016021, GO:0005576, GO:0005622, GO:0005578 and GO:0005615, which are not offspring of essential GO terms. For SCL16L, five GO terms belonging to the class (c) are shown: GO:0005622, GO:0005615, GO:0020015, GO:0016020 and GO:0045261. GO:0005905 and GO:0005887 belong to the class (a).
Sequence representation and IGA-chromosome encoding method
Figure 5
Sequence representation and IGA-chromosome encoding method.
racy of SVM using 10-CV) from the remaining 70-r fea-tures into the current feature set.
Authors' contributions
WLH designed the system, implemented programs, partic-ipated in manuscript preparation and carried out the detail study. CWT designed the system and implemented programs. SWH, SFH and SYH conceived the idea of this work. Additionally, SYH supervised the whole project and participated in manuscript preparation. All authors have read and approved the final manuscript.
Availability
The prediction system using ProLoc-GO with input sequences of query proteins for protein subcellular
locali-zation has been implemented at http:// iclab.life.nctu.edu.tw/prolocgo.
Acknowledgements
The authors would like to thank the National Science Council of Taiwan for financially supporting this research under the contract numbers NSC 96-2628-E-009-141-MY3 and NSC 96-2627-B-009-002.
References
1. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification
of biology. The Gene Ontology Consortium. Nat Genet
2000:25-29.
2. Chou KC, Shen HB: Predicting Eukaryotic Protein Subcellular
Location by Fusing Optimized Evidence-Theoretic K-Near-est Neighbor Classifiers. J Proteome Res 2006, 5(8):1888 -11897.
3. Chou KC, Shen HB: Euk-mPLoc: A Fusion Classifier for
Large-Scale Eukaryotic Protein Subcellular Location Prediction by Incorporating Multiple Sites. J Proteome Res 2007, 6(5):1728-34.
Epub 2007 Mar 31. 4. Chou KC, Shen HB: Hum-PLoc: A novel ensemble classifier for
predicting human protein subcellular localization. Biochem Biophys Res Commun 2006, 347:150-157.
5. Lei Z, Dai Y: Assessing protein similarity with Gene Ontology
and its use in subnuclear localization prediction. BMC Bioinfor-matics 2006:491-590.
6. Emanuelsson O, Nielsen H, Brunak S, von Heijne G: Predicting
Subcellular Localization of Proteins Based on their N-termi-nal Amino Acid Sequence. J Mol Biol 2000, 300(4):1005-1016.
7. Nakai K, Horton P: PSORT: a program for detecting sorting
signals in proteins and predicting their subcellular localiza-tion. Trends Biochem Sci 1999, 24:34-35.
8. Cedano J, Aloy P, P’erez-Pons JA, Querol E: Relation between
amino acid composition and cellular location of proteins. J Mol Biol 1997, 266:594-600.
9. Nakashima H, Nishikawa K: Discrimination of intracellular and
extracellular proteins using amino acid composition and res-idue-pair frequencies. J Mol Biol 1994, 238:54-61.
10. Park KJ, Kanehisa M: Prediction of protein subcellular locations
by support vector machines using compositions of amino acid and amino acid pairs. Bioinformatics 2003, 19:1656-1663.
11. Garg A, Bhasin M, Raghava GP: Support vector machine-based
method for subcellular localization of human proteins using amino acid compositions, their order, and similarity search. J Biol Chem 2005, 280:14427-14432.
12. Hua S, Sun Z: Support vector machine approach for protein
subcellular localization prediction. Bioinformatics 2001, 17:721-728.
13. Huang Y, Li Y: Prediction of protein subcellular locations using
fuzzy k-NN method. Bioinformatics 2004, 20:21-28.
14. Yu CS, Lin CJ, Hwang JK: Predicting subcellular localization of
proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci 2004, 13:1402-1406.
15. Bhasin M, Garg A, Raghava GPS: PSLpred: prediction of
subcellu-lar localization of bacterial proteins. Bioinformatics 2005, 21:2522-2524.
16. Bhasin M, Raghava GPS: ESLpred: SVM-based method for
sub-cellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucleic Acids Res 2004, 32:W414-419.
17. Sarda D, Chua G, Li KB, Krishnan A: pSLIP: SVM based protein
subcellular localization prediction using multiple physico-chemical properties. BMC Bioinformatics 2005, 6:152-163.
18. Huang WL, Tung CW, Huang HL, Hwang SF, Ho SY: ProLoc:
Pre-diction of protein subnuclear localization using SVM with automatic selection from physicochemical composition fea-tures. BioSystems 2007, 90(2):573-581.
19. Nanni L, Lumini A: An ensemble of K-local hyperplanes for
pre-dicting protein-protein interactions. Bioinformatics 2006, 22:1207-1210.
Prediction flowchart of ProLoc-GO using both classifiers SVM-IGO and SVM-GO
Figure 6
Prediction flowchart of ProLoc-GO using both classi-fiers SVM-IGO and SVM-GO.
Table 13: The used control parameters of IGA
Parameter Value Population size Npop 50
Selection probability ps 0.2 Crossover probability pc 0.8 Mutation probability pm 0.05 Factor number of orthogonal arrays 7 Maximum generations Gmax 60
Publish with BioMed Central and every scientist can read your work free of charge "BioMed Central will be the most significant development for disseminating the results of biomedical researc h in our lifetime."
Sir Paul Nurse, Cancer Research UK
Your research papers will be:
available free of charge to the entire biomedical community peer reviewed and published immediately upon acceptance cited in PubMed and archived on PubMed Central yours — you keep the copyright
Submit your manuscript here:
http://www.biomedcentral.com/info/publishing_adv.asp
BioMedcentral 20. Cai YD, Liu XJ, Xu XB, Chou KC: Support vector machines for
prediction of protein subcellular location by incorporating quasi-sequence-order effect. J Cell Biochem 2002, 84:343-348.
21. Chou KC, Shen HB: Predicting protein subnuclear location
with optimized evidence-theoretic K-nearest classifier and pseudo amino acid composition. Biochem Biophys Res Commun
2005, 337:752-756.
22. http://www.geneontology.org/. .
23. Yi G, Sze SH, Thon MR: Identifying clusters of functionally
related genes in genomes. Bioinformatics, 2007, 23(9):1053-60.
Epub 2007 Jan 19.
24. Lewin A, Grieve I: Grouping Gene Ontology terms to improve
the assessment of gene set enrichment in microarray data. BMC Bioinformatics 2006, 7(1):426.
25. Carroll S, Pavlovic V: Protein classification using probabilistic
chain graphs and the Gene Ontology structure. Bioinformatics
2006, 22(15):1871-1878.
26. Wolstencroft K, Lord P, Tabernero L, Brass A, Stevens R: Protein
classification using ontology classification. Bioinformatics 2006, 22(14):e530-538.
27. Qian Z, Cai YD, Li Y: A novel computational method to predict
transcription factor DNA binding preference. Biochem Biophys Res Commun 2006, 348(3):1034-1037.
28. Popescu M, Keller JM, Mitchell JA: Fuzzy Measures on the Gene
Ontology for Gene Product Similarity. IEEE/ACM Trans Comput Biol Bioinformatics 2006, 3(3):263-74.
29. Huang WL, Chen HM, Hwang SF, Ho SY: Accurate prediction of
enzyme subfamily class using an adaptive fuzzy k-nearest neighbor method. BioSystems, 2007, 90(2):405-418. Epub 2006
Oct 26.
30. Keller JM, Gray MR, Givens JA: A fuzzy k-nearest neighbours
algorithm. IEEE Trans Syst Man Cybern 1985, 15:580-585.
31. Ho SY, Chen JH, Huang MH: Inheritable genetic algorithm for
biobjective 0/1 combinatorial optimization problems and its applications. Systems, Man and Cybernetics, Part B, IEEE Transactions on 2004, 34(1):609-620.
32. Ho SY, Shu LS, Chen JH: Intelligent evolutionary algorithms for
large parameter optimization problems. Evolutionary Computa-tion, IEEE Transactions on 2004, 8:522-541.
33. Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS: UniProt: the Universal
Pro-tein knowledgebase. Nucleic Acids Res 2004, 32:D115-D119.
34. Wang GL, Dunbrack Jr. RL: PISCES: a protein sequence culling
server. Bioinformatics 2003, 19:1589-1591.
35. Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R: The Gene Ontology Annotation
(GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res 2004, 32(Database issue):D262-266.
36. GOA. [ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/UNIPROT/ ]. .
37. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local
alignment search tool. J Mol Biol 1990, 215(3):403-410.
38. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lip-man DJ: Gapped BLAST and PSIBLAST:a new generation of
protein database search programs. Nucleic Acids Res 1997, 25:3389-3402.
39. Chang CC, Lin CJ: LIBSVM : a library for support vector
machines, Software available at http://www.csie.ntu.edu.tw/ ~cjlin/libsvm. 2001.
40. Ho SY, Hsieh CH, Chen HM, Huang HL: Interpretable gene
expression classifier with an accurate and compact fuzzy rule base for microarray data analysis. BioSystems 2006, 85:165-176.
41. Tung CW, Ho SY: POPI: Predicting immunogenicity of MHC
class I binding peptides by mining informative physicochem-ical properties. Bioinformatics 2007, 23(8):942-949.
42. Li T, Zhang C, Ogihara M: A comparative study of feature
selec-tion and multiclass classificaselec-tion methods for tissue classifi-cation based on gene expression. Bioinformatics 2004, 20:2429-2437.
43. Lu Z, Hunter L: GO Molecular Function Terms Are Predictive
of Subcellular Localization. Pac Symp Biocomput 2005:151-161.
44. Stone M: Cross-validatory choice and assessment of statistical
![Table 8: Comparison of prediction accuracy (%) for SCL12. Prediction accuracies (%) for using leave-one-out cross-validation (LOOCV) on SCL12L and independent test on SCL12T are obtained from the paper [4]](https://thumb-ap.123doks.com/thumbv2/9libinfo/7779038.150751/8.918.74.833.151.226/comparison-prediction-accuracy-prediction-accuracies-validation-independent-obtained.webp)
![Table 9: Comparison of prediction accuracy (%) for SCL16. Prediction accuracies (%) of using LOOCV on SCL16L and independent test on SCL16T are obtained from the paper [2]](https://thumb-ap.123doks.com/thumbv2/9libinfo/7779038.150751/10.918.82.835.924.1098/table-comparison-prediction-accuracy-prediction-accuracies-independent-obtained.webp)