Characterization of Pathogenesis of SCA17 Transgenic Mice
Ya-Chin Chang
1, Chen-Ming Hsu
1, Cheng-Yueh Lin
1, Chiung-Mei Chen
2, Hsiu-Mei Hsieh
1*
1 Department of Life Science, National Taiwan Normal University
Taipei, Taiwan
2 Department of Neurology, Chang Gung Memorial Hospital
Taipei, Taiwan
(Received: 3 Jun 2010, accepted: 5 July 2010) ABSTRACT
TATA binding protein (TBP) is a general transcription factor that plays an important role in initiation of transcription. TBP gene is located in chromosome 6q27 and contains a CAG/CAA trinucleotide repeat region in its 5’ end, which encodes a polyglutamine (polyQ) tract. Spinocerebellar ataxias type 17 (SCA17) is an autosomal dominant cerebellar ataxia caused by TBP gene with an expanded polyQ tract correlated to the disease onset and progression. To investigate the TBP trinucleotide expansion effect on neurodegeneration, we have generated transgenic mice expressing the human TBP gene with expanded CAA/CAG tracts under the control of Purkinje cell-specific promoter, Pcp2/L7 promoter. Our previous studies have shown that these hTBP109Q transgenic mice had ataxia and severe Purkinje cell loss in the cerebellum. In the present study, we found Bergmann glia surrounding the Purkinje cells were also increased as astrocytes that was identified in our previous study and suggesting a neurodegeneration occurred in the mouse cerebella. According to the result of cDNA microarray analysis, we found several calcium regulatory protein expression were differentiated expressed in the transgenic mouse cerebella. We further conducted western analysis and confirmed that the expressions of calbindin, inositol 1, 4, 5-triphosphate receptor 1 (IP3R1) and Cacnα1G were downregulated in the SCA17 mouse cerebellum. We suggest that hTBP109Q mutant protein in the mouse brain might result in the impairment of calcium homeostasis.
Key Words: Purkinje cell, SCA17, Bergmann glia
Introduction
Autosomal dominant spinocerebellar ataxias (SCAs) are a heterogeneous group of neurological disorders clinically characterized by progressive ataxia due to cerebellar dysfunction. Patients exhibit a slowly progressive cerebellar syndrome and usually combined with oculomotor disorders, dysarthria, kinetic tremor, gait ataxia, peripheral neuropathy, and even cognitive impairment. SCAs are named sequentially as currently SCA 1 through to SCA 31 (Sato et al., 2009). SCAs were initially classified according to clinical and neuropathological descriptions. Three patterns of atrophy could be identified through MRI images of patients’ brains: a pure cerebellar atrophy (in SCA4, 5, 6, 8, 9, 10, 11, 14, 15, 16, 18, 21, 22), a pattern of olivopontocerebellar atrophy (SCA1, 2, 3, 7, 13), and a pattern of global cerebral atrophy (SCA2, 17, 19, Dentatorubral-pallidoluysian atrophy (DRPLA))
(Manto, 2005). Among them nine SCAs, SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA17 and DRPLA, are caused by CAG expansion encoding a polyglutamine (polyQ) repeat.
SCA17 was identified as the form of SCAs in 4 Japanese pedigrees (Nakamura et al., 2001). The clinical and neuropathological spectrum associated with SCA17 mutation appears to be broad. The symptoms at onset, which occur at a mean age of 34.6±13.2 years (range: 3–75) (Stevanin and Brice, 2008), are predominantly gait instability (Silveira et al., 2002) or other movement disorders, such as focal dystonia (Bruni et al., 2004) or chorea (Bauer et al., 2004). Psychiatric disturbances such as behavioral changes, psychosis or depression as well as dementia can also represent the presenting symptoms (Bauer et al., 2004). Morphometric data showed that the maximum atrophy was found in the left cerebellar posterior lobe, and large regions with reduced grey matter volume were observed in the
occipito-parietal structures bilaterally (Lasek et al., 2006). SCA17 is a rare type of autosomal dominant SCA caused by a CAG/CAA expansion in the gene encoding the TATA-binding protein (TBP). Wild type TBP in humans contains a long polyQ stretch ranging in size from 29 to 42. Expansion composite repeat beyond 42 has been shown to cause SCA17 (van Roon-Mom et al., 2005). TBP is an important and general transcription factor ubiquitously expressed from a single gene on chromosome 6q27 and constitutes an integral component of the transcription initiation complexes of three RNA polymerases. TBP protein of the RNA polymerase II transcription factor D (TFIID) complex, is the DNA-binding subunit and anchors the complex to the TATA box upstream of the first codon. The N-terminus of the protein contains the long stretch of glutamines and modulates the DNA binding activity of the C-terminus (Lescure et al., 1994; Stevanin and Brice, 2008). However, the expanded polyQ stretch causes the transcriptional dysfunction and results in disease phenotypes (Friedman et al., 2007).
It has been proved that excitotoxin could cause nerve-cell death in a variety of neurodegenerative disorders including Alzheimer's disease, Parkinson's disease, Huntington's disease and amyotrophic lateral sclerosis (Shaw and Ince, 1997) Glutamate is the major excitatory neurotransmitter in the central nervous system (CNS), which was rapidly removed from the extracellular space by glutamate transporters localized in neuronal and glial membranes (Shigeri et al., 2004). Excitotoxicity could happen when excess glutamate accumulated outside cells after brain injury, which causes calcium ions entering cells via NMDA receptor channel, leading to neuronal damage and eventual cell death (Michaelis, 1998). Several studies have shown that deranged calcium signaling may play an important role in polyQ diseases including SCA3 (Chen et al., 2008) and Huntington’s disease (Tang et al., 2003; Bezprozvanny and Hayden, 2004). We therefore would like to study whether the calcium deregulation is involved in the pathogenesis of SCA17 transgenic mice.
Materials and methods
SCA17 transgenic mouse model
The construct of human TBP (hTBP) cDNA
with 109Q driven by Pcp2 promoter was used to generate the SCA17 transgenic mice. Characterization of these mice through observation and rotarod task indicated that SCA17 transgenic mice have ataxia and reduced motor coordination, Further pathological analyses showed that Purkinje cell loss and active gliosis occurred in the transgenic mouse cerebella.
All animal experimental procedures were approved by the Committee on Animal Research of National Taiwan Normal University and carried out in accordance with the guidelines of the Committee. Genotyping analysi
We genotyped the SCA17 transgenic mice by isolating genomic DNA from 0.5 cm length of mouse tails by lysis tissue in 10 mg/mL proteinase K (in 2% SDS, 5 M NaCl, 0.5 M EDTA (pH 8.0), 1 M Tris (pH 7.4)) at 65℃ overnight followed by salt extraction and ethanol precipitation. Transgenic mice were identified using polymerase chain reaction (PCR) with a forward primer from Pcp2/L7 promoter (forward: 5’-TAT GGT GAG AGC AGA GAT GG-3’), and a backward primer from hTBP cDNA (hTBP-3R: 5’-CTG CTG GGA CGT TGA CTG CTG-3’). A 769 bp fragment was amplified under 94 60 sec for denaturing, 54 60 sec for ℃ ℃ primer annealing, 72 60 sec for elongation, and ℃ repeated for 35 cycles.
Microarray analysis
RNA from mouse cerebella and brain stem was extracted by using the RNeasy total RNA kit (Qiagen), according to the manufacturer's protocol. All samples were treated with the RNase-free DNase set (Qiagen). The quality of total RNA was analyzed by using the RNA 6000 Nano LabChip kit on a 2100 Bioanalyzer (Agilent Technologies). Microarray analysis was performed by using U133A GeneChips (Affymetrix). Briefly, 4 μg of high-quality total RNA was reverse transcribed (Invitrogen), cleaned by using the QIAquick purification kit (Qiagen), and then used as a template for in vitro transcription by using T7 MEGA script reagents (Ambion) and biotin-11-UTP (PerkinElmer, NEN). Resulting biotin-labeled cRNA was recovered and purified with the RNeasy kit (Qiagen), hybridized to the chips, and fluorescently tagged and scanned according to the manufacturer’s protocol.
Brain fixation and histology
Mice were anesthetized with avertin (0.4 g/kg body weight) and then perfused with 100 ml of 0.9% sodium chloride followed by 150 ml of 4% paraformaldehyde (pH 7.4). The brain was removed, post-fixed for 4 h in 4% paraformaldehyde (pH 7.4), and then immersed in 10% sucrose for 1 h, 20% sucrose for 2 h, 30% sucrose overnight in the 4oC. Sagittal sections of brains were cut at 30 µm and stored in a bath of 0.2% sodium azide until use. Immunohistochemistry and immunofluorescence
Sections were incubated in 3% H2O2 for 30
min, rinsed, blocked with 5% normal goat serum (DAKO, X0907) for 1 h, incubated overnight at room temperature probed with anti-Cacnα1G (1:500; Santa Cruz Biotechnology), anti-MAP2 (1.5:1000; Chemicon), anti-S100 (1:400; Chemicon) or 1TBP18 (1:30000; Santa Cruz) in antibody diluent (1:100, DAKO S3022). After three times wash with 1X TBST for 10 min each, sections were then incubated with the secondary antibody (DAKO K0690) or anti-secondary antibody (green fluorescence, Alex Fluor® 488 Donkey anti-mouse IgG) for 1 hr at room temperature and light protection and counterstained with 4', 6-diamidino-2-phenylindole (DAPI, sigma) for 3 min. After three times wash with 1X TBST for 15 min each time, sections were reacted with DAB solution (DAKO K3468) for 2 min. The sections were then mounted on gelatin-coated slides and dried overnight. Light micrographs were taken using a Leica microscope (Leica DMIL,TYPE 090-135.002, Germany) equipped with a digital camera (SPOT, 15.2 64 Mp shifting Pixel, USA) and the image acquisition software (SPOT Advanced, Version 4.6). All data in this study are presented as means ± standard deviations. The SPSS–Independent T test or one way anova test was used for inter-group comparisons. A two-tailed probability value of < 0.05 was considered significant.
Western blotting analysis
Mice were anesthetized with anvertin (0.4 g/kg body weight) and then perfused with 100 ml of 0.9% sodium chloride followed by rapid removal of brains (n = 3 per group). Total protein extracts of the whole brains were collected by homogenization in RIPA buffer (10 mM Tris pH 7.5, 150 mM sodium chloride, 5 mM Ethylenedinitrilo-tetraacetic
acid (pH 8.0), 0.1% sodium dodecyl sulfate, 1% deoxycholic acid and 1% NP40) to which protease inhibitors (Pierce Biotechnology) had been freshly added. Homogenates of whole hemispheres were centrifuged for 30 min at 13400 × g, and 50 μg of the supernatant proteins was separated by electrophoresis on SDS-PAGE and transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membranes. Membranes were blocked in blocking buffer [5% skim milk in TBST (20 mM Tris (pH7.5), 140 mM NaCl, 0.05%Tween20)] for 2 h, and subsequently probed with anti-S100 (1:1000; Chemicon), anti-Cacnα1G (1:250; Santa Cruz Biotechnology), or anti-GFAP (1:2000; Chemicon) in TBST overnight at 4 °C. Anti-β-actin antibody (1:2000; Chemicon) was used as a control. Incubated membranes were then treated with secondary antibody conjugated with HRP in TBST for 1 h. Blots were developed by enhanced chemiluminescence (Millipore). The optical densities of each band were determined using an image analysis program (Multi Gauge®, FUJIFILM). Relative optical densities were calculated versus measured values of β-actin.
Statistical analysis
All data in this study are shown as means ± SE . The SPSS–Independent T test or one way Anova test was used for inter-group comparisons. A two-tailed probability value of p < 0.05 was considered significant.
Results and Discussion
Cell degeneration in SCA17 transgenic mouse cerebellum
Our previous studies have shown that SCA17 mouse cerebellar weight is severely reduced. As the cerebellum is the most affected brain region in SCA17 patients, we used immunohistochemical analyses to examine the extent of neurodegeneration in our SCA17 mouse cerebellum. Our results showed that severe loss of Purkinje cells was identified by the calbindin staining (unpublished results). To further characterize whether TBP aggregation is a common feature in the SCA17 mice, we used 1TBP18 antibody (Friedman et al., 2008) for the immunofluorescence staining analysis. Compared to the wild-type littermate, we found that TBP-109Q was highly expressed and aggregated in the nuclei of Purkinje
cells (Fig. 1). These data suggest that the degeneration of Purkinje cells might be resulted from the toxicity of mutant TBP aggregation.
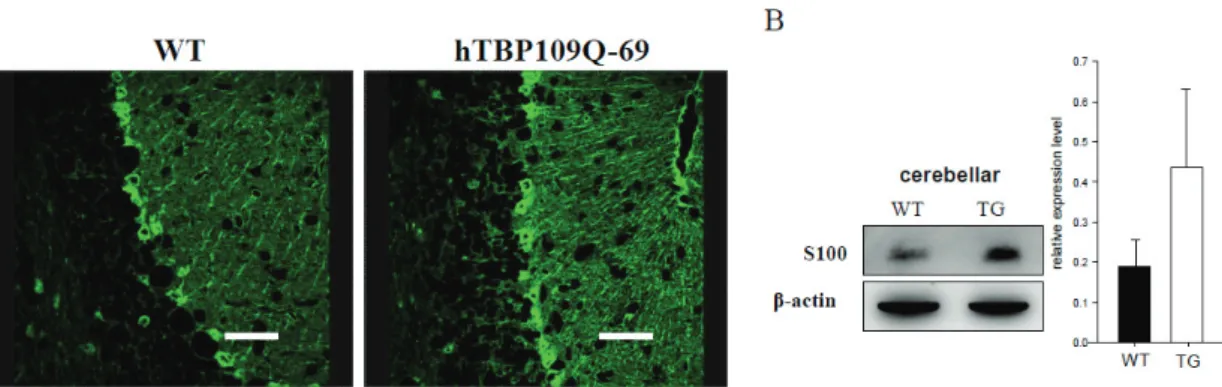
Reactive giosis, indicated by increased expression of the astrocytic marker GFAP, was identified in the SCA17 mouse cerebellum. We further used S100 antibody to examine Bergmann glia cells that ensheathe Purkinje cell dendrites, synapses and soma are involved in the protection of Purkinje cells (Rothermundt et al., 2003). We found that S100 was upregulated in the immunofluorescence staining (Fig. 2A). The western blot analysis using S100 antibody also confirmed this result (Fig. 2B). These data reveal that the Purkinje cell degeneration would result in the proliferation of surrounding astrocytes and Bergmann glia (Drake-Baumann and Seil, 1999; Fukaya et al., 1999; Custer et al., 2006). These results were consistent with the studies of other polyQ-mediated neurodenerative diseases, including SCA1, SCA3 and Huntington’s diseases (Kumada et al., 2000; Ishiguro et al., 2001; Vig et al., 2006).
Impairment of calcium homeostasis in SCA17 transgenic mice
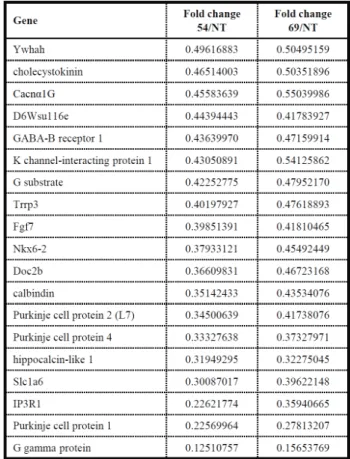
To study the underlying molecular pathogenesis of SCA17, we have conducted the cDNA microarray analysis on the SCA17 transgenic mice. The microarray array results with transcripts from mouse cerebella reveal the impairment of calcium homeostasis in the SCA17 animals. Compared to wild-type control animal, both lines 69 and 54 transgenic mice have several genes involved in calcium regulation were down-regulated in the cerebellar region. These transcripts include calbindin, hippocalcin-like 1, Cacnαlg, G substrate and IP3R1 (Table 1). We conducted western analysis and confirmed that calbindin, IP3R1 (unpublished data) and Cacnα1G were expressed in the Purkinje cells and downregulated in transgenic mice (Fig. 3).
Calbindin, a calcium-binding protein, has been suggested to controlcalcium buffering in Purkinje cells and may control neuronal behavior signal integration (Schwaller et al., 2002). A selective genetic deletion of calbindin from Purkinje cells results in mutant mice displaymarked deficits of motor coordination and sensory processing (Schwaller et al., 2002). Hippocalcin-like-1 (also named visinin-like protein 3) belongs to a family of
Figure 1. Mutant TBP aggregation in transgenic SCA17 mice. Immunofluorescence analysis of 1TBP18 (red) expression in Purkinge cells of 3-month-old wild-type littermate (A) and transgenic mice (B). IP3R1 (green) staining is used to identify the Purkinje cells (white arrowhead). The highly expressed mutant TBP is aggregated in the Purkinje cell nuclei of transgenic mice. Scale bar = 8 μm.
Table 1. Genes identified from microarray analysis with transcripts down-regulated more than 0.5-fold in both SCA 17 transgenic mouse lines 69 and 54 compared to the wild-type (NT) animal.
neuron-specific Ca2+-binding proteins found in
brain (Spilker et al., 2002). It has been reported to involve in a Ca2+ -dependent manner to regulate the
neuronal signaling in the brain (Spilker et al., 2002). Cacnαlg is one of the low voltage-activated, T-type, calcium channels and involve in several neuronal
Figure 2. Upregulation of S100 in SCA17 transgenic mice. (A) Immunofluorescence analysis of S100 expression in wild type (5 months old) and line 69 (5 months old) transgenic mouse cerebella. Scale bar = 40 μm. (B) Western blot result of the upregulation of S100 staining in the transgenic mice (n = 3).
Figure 3. Deregulation of calcium channel protein Cacnα1G in SCA17 transgenic mice. (A) Western blot analysis of Cacnα1G expression in different brain regions of transgenic mice and control littermates. (B) Quantification of western blot analysis shows significant decreasing of Cacnα1G staining in transgenic cerebella (n =3). *, p < 0.05. (C) IHC analyses show the Cacnα1G signals in the Purkinje cells that is down-regulated in the Purkinje layer of both transgenic lines 54 and 69 compared to wild-type mice. Scale bar = 50 μm. Arrows indicate the Purkinje cells.
activities, including the low threshold Ca2+ spikes,
neuronal oscillations and resonance, and rebound burst firing (Perez-Reyes, 2004). It was also reported that Ca2+ influx through T-channels could
promote axonal and dendritic outgrowth (McCobb and Kater, 1988). G-substrate is expressed exclusively in the Purkinje cells and will be activated through phosphorylation by cGMP- dependent protein kinase (Detre et al., 1984). The phosphorylated G-substrate has been reported as an inhibitor of protein phosphatase 2A, PP2A, a phosphotase involved in several neuronal signaling regulations (Khodakhah and Ogden, 1993; Ogden et al., 1993) (Endo et al., 1999; Endo et al., 2003). IP3R1 is abundantly expressed in Purkinje cells, and is an effector of type I mGluRs signaling pathway (Furuichi et al., 1989). IP3R1 is a Ca2+-release channel that elevates the cytoplasmic
Ca2+ level upon the binding of IP3, generated
through a G protein-coupled mGluR receptors (Berridge, 1993). The functional importance of IP3R1 is demonstrated that it is required for long-term depression (LTD) in Purkinje cells (Inoue et al., 1998), and reduction of IP3R1 results in the impairment of motor coordination of animals (Ogura et al., 2001)). Although S100 transcript alteration was not identified from the microarray analysis, the elevated S100 protein level was observed in SCA17 mouse cerebellum from both the western and immunofluorescence staining. S100 is also involved in regulation of Ca2+ homeostasis
(Zimmer et al., 1995; Donato, 2001; Zimmer et al., 2005).
It was reported that genes involved in Ca2+
homeostasis and signaling, is downregulated in SCA1 transgenic mouse at early postnatal ages, including calbindin (Vig et al., 1998) and parvalbumin (Vig et al., 1996). Some of the other downregulated genes identified in the SCA1 animals are IP3R1 and sarcoplasmic endoplasmic reticulum calcium ATPase type 2 (SERCA2) (a Ca2+-pump) (Lin et al., 2000). The decreased
expression of Homer-3, G-substrate, the glutamate transporter EAAT4, IP3R1, and CARP was proposed to cause alteration in cerebellar LTD of SCA1 mice (Serra et al., 2004). Several of these genes can be positioned as regulators of the glutamate signaling pathway in Purkinje cells (Serra et al., 2004). Intracellular Ca2+ regulates
virtually all cellular processes, including
proliferation, differentiation, growth and cell death (Berridge, 1998). From these results, we proposed that hTBP-109Q expression in the mouse cerebellum might affect calcium regulatory proteins and maybe cause impaired Ca2+ homeostasis.
Whether the deregulated calcium involved in the pathomechanism of SCA17 transgenic mice needs further study.
Acknowledgement
We thank Wei-Yi Chen and Fong-So Jang for their technical assistance. Our gratitude is also extended to Molecular Imaging Core Facility of National Taiwan Normal University under the auspices of the National Science Council. This work was supported by research grants from the National Taiwan Normal University (96TOP001) and National Science Council (NSC96-2311-B- 003-002 and NSC-97-2311-B-003-007-MY3).
References
Bauer P, Laccone F, Rolfs A, Wullner U, Bosch S, Peters H, Liebscher S, Scheible M, Epplen JT, Weber BH, Holinski-Feder E, Weirich- Schwaiger H, Morris-Rosendahl DJ, Andrich J, Riess O (2004) Trinucleotide repeat expansion in SCA17/TBP in white patients with Huntington's disease-like phenotype. J Med Genet 41:230-232.
Berridge MJ (1993) Inositol trisphosphate and calcium signalling. Nature 361: 315-325. Berridge MJ (1998) Neuronal calcium signaling.
Neuron 21: 13-26.
Bezprozvanny I, Hayden MR (2004) Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun 322: 1310-1317.
Bruni AC, Takahashi-Fujigasaki J, Maltecca F, Foncin JF, Servadio A, Casari G, D'Adamo P, Maletta R, Curcio SA, De Michele G, Filla A, El Hachimi KH, Duyckaerts C (2004) Behavioral disorder, dementia, ataxia, and rigidity in a large family with TATA box-binding protein mutation. Arch Neurol 61: 1314-1320.
Chen X, Tang TS, Tu H, Nelson O, Pook M, Hammer R, Nukina N, Bezprozvanny I (2008) Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia
type 3. J Neurosci 28: 12713-12724.
Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR (2006) Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci 9: 1302-1311.
Detre JA, Nairn AC, Aswad DW, Greengard P (1984) Localization in mammalian brain of G-substrate, a specific substrate for guanosine 3',5'-cyclic monophosphate-dependent protein kinase. J Neurosci 4: 2843-2849.
Donato R (2001) S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol 33: 637-668.
Drake-Baumann R, Seil FJ (1999) Influence of functional glia on the electrophysiology of Purkinje cells in organotypic cerebellar cultures. Neuroscience 88: 507-519.
Endo S, Nairn AC, Greengard P, Ito M (2003) Thr123 of rat G-substrate contributes to its action as a protein phosphatase inhibitor. Neurosci Res 45: 79-89.
Endo S, Suzuki M, Sumi M, Nairn AC, Morita R, Yamakawa K, Greengard P, Ito M (1999) Molecular identification of human G-substrate, a possible downstream component of the cGMP-dependent protein kinase cascade in cerebellar Purkinje cells. Proc Natl Acad Sci U S A 96: 2467-2472.
Friedman MJ, Wang CE, Li XJ, Li S (2008) Polyglutamine expansion reduces the association of TATA-binding protein with DNA and induces DNA binding-independent neurotoxicity. J Biol Chem 283: 8283-8290. Friedman MJ, Shah AG, Fang ZH, Ward EG,
Warren ST, Li S, Li XJ (2007) Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci 10: 1519-1528.
Fukaya M, Yamada K, Nagashima M, Tanaka K, Watanabe M (1999) Down-regulated expression of glutamate transporter GLAST in Purkinje cell-associated astrocytes of reeler and weaver mutant cerebella. Neurosci Res 34: 165-175.
Furuichi T, Yoshikawa S, Miyawaki A, Wada K,
Maeda N, Mikoshiba K (1989) Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature 342: 32-38.
Ishiguro H, Yamada K, Sawada H, Nishii K, Ichino N, Sawada M, Kurosawa Y, Matsushita N, Kobayashi K, Goto J, Hashida H, Masuda N, Kanazawa I, Nagatsu T (2001) Age-dependent and tissue-specific CAG repeat instability occurs in mouse knock-in for a mutant Huntington's disease gene. J Neurosci Res 65: 289-297.
Khodakhah K, Ogden D (1993) Functional heterogeneity of calcium release by inositol trisphosphate in single Purkinje neurones, cultured cerebellar astrocytes, and peripheral tissues. Proc Natl Acad Sci U S A 90: 4976-4980.
Kumada S, Hayashi M, Mizuguchi M, Nakano I, Morimatsu Y, Oda M (2000) Cerebellar degeneration in hereditary dentatorubral- pallidoluysian atrophy and Machado-Joseph disease. Acta Neuropathol 99: 48-54.
Lasek K, Lencer R, Gaser C, Hagenah J, Walter U, Wolters A, Kock N, Steinlechner S, Nagel M, Zuhlke C, Nitschke MF, Brockmann K, Klein C, Rolfs A, Binkofski F (2006) Morphological basis for the spectrum of clinical deficits in spinocerebellar ataxia 17 (SCA17). Brain 129: 2341-2352.
Lescure A, Lutz Y, Eberhard D, Jacq X, Krol A, Grummt I, Davidson I, Chambon P, Tora L (1994) The N-terminal domain of the human TATA-binding protein plays a role in transcription from TATA-containing RNA polymerase II and III promoters. EMBO J 13: 1166-1175.
Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY (2000) Polyglutamine expansion down- regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci 3: 157-163.
Manto MU (2005) The wide spectrum of spinocerebellar ataxias (SCAs). Cerebellum 4:2-6.
McCobb DP, Kater SB (1988) Membrane voltage and neurotransmitter regulation of neuronal growth cone motility. Dev Biol 130: 599-609. Michaelis EK (1998) Molecular biology of
glutamate receptors in the central nervous system and their role in excitotoxicity,
oxidative stress and aging. Prog Neurobiol 54: 369-415.
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10: 1441-1448.
Ogden DC, Khodakhah K, Carter TD, Gray PT, Capiod T (1993) Mechanisms of intracellular calcium release during hormone and neurotransmitter action investigated with flash photolysis. J Exp Biol 184: 105-127.
Ogura H, Matsumoto M, Mikoshiba K (2001) Motor discoordination in mutant mice heterozygous for the type 1 inositol 1,4,5-trisphosphate receptor. Behav Brain Res 122: 215-219.
Rothermundt M, Peters M, Prehn JH, Arolt V (2003) S100B in brain damage and neurodegeneration. Microsc Res Tech 60: 614-632.
Sato N et al. (2009) Spinocerebellar ataxia type 31 is associated with "inserted" penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet 85: 544-557.
Schwaller B, Meyer M, Schiffmann S (2002) 'New' functions for 'old' proteins: the role of the calcium-binding proteins calbindin D-28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum 1: 241-258.
Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT (2004) Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet 13: 2535-2543.
Shaw PJ, Ince PG (1997) Glutamate, excitotoxicity and amyotrophic lateral sclerosis. J Neurol 244 Suppl 2: S3-14.
Shigeri Y, Seal RP, Shimamoto K (2004) Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Brain Res Rev 45: 250-265.
Silveira I et al. (2002) Trinucleotide repeats in 202 families with ataxia: a small expanded (CAG)n allele at the SCA17 locus. Arch Neurol 59: 623-629.
Spilker C, Gundelfinger ED, Braunewell KH (2002) Evidence for different functional properties of the neuronal calcium sensor proteins VILIP-1 and VILIP-3: from subcellular localization to cellular function. Biochim Biophys Acta 1600: 118-127.
Stevanin G, Brice A (2008) Spinocerebellar ataxia 17 (SCA17) and Huntington's disease-like 4 (HDL4). Cerebellum 7: 170-178.
Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I (2003) Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39: 227-239.
van Roon-Mom WM, Reid SJ, Faull RL, Snell RG (2005) TATA-binding protein in neurodegenerative disease. Neuroscience 133: 863-872.
Vig PJ, Fratkin JD, Desaiah D, Currier RD, Subramony SH (1996) Decreased parvalbumin immunoreactivity in surviving Purkinje cells of patients with spinocerebellar ataxia-1. Neurology 47: 249-253.
Vig PJ, Subramony SH, Burright EN, Fratkin JD, McDaniel DO, Desaiah D, Qin Z (1998) Reduced immunoreactivity to calcium-binding proteins in Purkinje cells precedes onset of ataxia in spinocerebellar ataxia-1 transgenic mice. Neurology 50: 106-113.
Vig PJ, Lopez ME, Wei J, D'Souza DR, Subramony S, Henegar J, Fratkin JD (2006) Glial S100B Positive Vacuoles In Purkinje Cells: Earliest Morphological Abnormality In SCA1 Transgenic Mice. J Neurol Sci Turk 23: 166-174.
Zimmer DB, Cornwall EH, Landar A, Song W (1995) The S100 protein family: history, function, and expression. Brain Res Bull 37: 417-429.
Zimmer DB, Chaplin J, Baldwin A, Rast M (2005) S100-mediated signal transduction in the nervous system and neurological diseases. Cell Mol Biol (Noisy-le-grand) 51: 201-214.
脊髓小腦萎縮症第十七型基因轉殖小鼠之分子特性探討
張雅津
1許振銘
1林承岳
1陳瓊美
2謝秀梅
1*
1 國立臺灣師範大學生命科學系 2長庚紀念醫院林口總院神經內科 (收稿日期:2010.6.3,接受日期:2010.7.5) 摘 要TATA binding protein (TBP)為細胞中一種主要的轉錄因子,其在主導基因轉錄的起始過程中扮
演著重要的角色。人類TBP基因位於染色體6q27,其5’端包含一段CAG三核苷酸重複序列,轉譯出 的蛋白質N端上會形成一段多麩醯胺(polyglutamine, polyQ)的片段。SCA17為一種體染色體顯性遺 傳之神經退化性疾病,目前已知SCA17致病原因與TBP基因之CAG重複序列擴增有關,為了探討 TBP基因N端CAG三核苷重複擴增與神經退化的關係,我們之前已經利用小腦Purkinje細胞專一性 表現之Pcp2/L7啟動子,建立了帶有109個CAG重複之TBP基因之轉殖小鼠,作為研究SCA17之疾病 動物模式。由外觀及分子生物之分析中,我們已確認此基因轉殖小鼠有步態不穩、小腦中Purkinje cells明顯退化及缺失之病徵。在目前的研究中,我們進一步發現此基因轉殖小鼠小腦中與Purkinje cells相鄰的Bergmann glia有增加之現象,此結果與之前研究看到的小腦中astrocyte之增加有類似之 意義,代表神經退化之結果。此外,進行微陣列實驗並分析資料後,我們發現有許多與鈣離子調控 相關之基因表現量改變,經由西方墨點法進一步確認,發現基因轉殖小鼠小腦中calbindin、Inositol 1,4,5-Triphosphate Receptor 1 (IP3R1)及Cacnα1G表現量明顯較正常小鼠為低。我們認為此結果顯示 hTBP 109Q突變蛋白於基因轉殖小鼠腦細胞中可能造成鈣離子恆定失調的情形。