Influences of substitution on electronic structures of oligofluorenes
Chih-I Wu,a兲Guan-Ru Lee, Chan-Tin Lin, and Yu-Hung Chen
Department of Electrical Engineering and Graduate Institute of Electro-optical Engineering, National Taiwan University, Taipei, Taiwan 106, Republic of China
Yun-Hua Hong, Wei-Guan Liu, and Chung-Chih Wu
Department of Electrical Engineering, Graduate Institute of Electro-optical Engineering,
and Graduate Institute of Electronics Engineering, National Taiwan University, Taipei, Taiwan 106, Republic of China
Ken-Tsung Wong and Teng-Chih Chao
Department of Chemistry, National Taiwan University, Taipei, Taiwan 106, Republic of China
共Received 20 September 2005; accepted 18 October 2005; published online 8 December 2005兲 The influences of substitutions on the electronic structures of oligofluorenes were systematically investigated. Electronic states of oligofluorenes with various types of C9 substitutions were studied by both ultraviolet photoemission spectroscopy and theoretical calculation. It is found that the substituents could have significant interaction with the backbone and substantially modify the electronic structures of oligofluorenes, depending on the types of substitutions. Consequently, the electronic properties that would affect the operations of electronic devices also considerably depend on the substitutions of the oligofluorenes. © 2005 American Institute of Physics.
关DOI:10.1063/1.2140587兴
Organic semiconducting materials have attracted a lot of attention in the past two decades due to their applications in various optoelectronic devices, such as light-emitting diodes, solar cells, transistors, etc.1–4Among various classes of ma-terials, fluorene-based polymers and oligomers are one of the most widely studied classes, due to their many distinguished characteristics, such as highly efficient blue fluorescence,5–7 intriguing and efficient bipolar charge-transport properties,8 excellent electrochemical stability,9 and richness in confor-mational and morphological characteristics.10,11 In spite of their technical importance, experimental studies of electronic structures of fluorene-based materials, such as molecular or-bitals, corresponding energy levels, and densities of states 共DOS兲, etc., through photoemission spectroscopy are surpris-ingly sparse.12–14 In particular, one distinguished feature of fluorene-based materials is their structural variety provided by convenient substitution on the C9 atom, which renders fluorene-based materials superb tunability in physical char-acteristics. Yet, there is general lack of systematic investiga-tion of the dependence of electronic structures on such sub-stitutions, although such an understanding would be invaluable and indispensable in designing functional mol-ecules for the organic devices. In this letter, we present a systematic study of electronic structures of oligofluorenes with three types of substitutions most widely implemented onto fluorenes, i.e., the alkyl, aryl, and spiro-linked conju-gated substitutions.
Figure 1 shows three terfluorenes with dialkyl substitu-ents 共E3兲, diaryl substituents 共T3兲, and spiro-linked conju-gated substituents共B3兲 studied in this work. All three mate-rials were purified by gradient sublimation before analysis. The electronic structures, including the DOS and the energy levels of highest occupied molecular orbitals 共HOMOs兲, were studied both by ultraviolet photoemission spectroscopy
共UPS兲 of thin-film samples of terfluorenes and by quantum chemical calculation of molecules. The deposition and the UPS measurements of thin-film samples were performed in two interconnected ultrahigh-vacuum chambers. Organic thin films were deposited on gold-coated silicon substrates by thermal evaporation in the deposition chamber, and then transferred in situ to the analysis chamber. In the analysis chamber with a base pressure less than 1⫻10−10Torr, UPS was carried out using the He I 共21.22 eV兲 and He II 共40.8 eV兲 photon lines of the gas discharge lamp as the excitation sources and using the double-pass cylindrical mirror analyzer to measure kinetic-energy spectrum of photoexcited elec-trons. The overall resolution of the UPS measurement is about 0.15 eV. The absolute energy scale of UPS spectra is referenced to the Fermi level of the system, which is mea-sured on the gold surface before deposition of organic thin films. The ionization energies共IEs兲 of molecular films can be deduced from the energy difference between the HOMO level and the vacuum level 共inferred from the low-energy onset of the UPS spectrum兲.
In quantum chemical calculation of molecular proper-ties, the geometries of molecules were optimized using the semiempirical Austin model 1 共AM1兲 method, which has been providing reliable results for organic molecules.15Yet, it is noteworthy that the calculated results in our work are not markedly sensitive to small geometrical changes. The mo-lecular energy levels of terfluorenes were calculated via AM1
a兲Authors to whom correspondence should be addressed; electronic mail:
[email protected] FIG. 1. The chemical structures of T3, E3, and B3.
APPLIED PHYSICS LETTERS 87, 242107共2005兲
0003-6951/2005/87共24兲/242107/3/$22.50 87, 242107-1 © 2005 American Institute of Physics
based on modified neglect of diatomic overlap Hamiltonian.15The theoretical UPS spectra were then calcu-lated by convoluting these molecular energy levels with Gaussian functions using the full width at half maximum being adapted to the experimental linewidths of 0.4 eV共the broadening of discrete molecular levels is due to the amor-phous nature of terfluorene films and vibronic origins兲.16
The frontier occupied molecular orbitals were calculated using the Hartee–Fock method with 6-13G basis at the optimized conformation.
UPS spectra measured on thin films of three terfluorenes are shown in Fig. 2. The valence-band spectra, representing the DOS of occupied molecular orbitals, are somewhat simi-lar for E3 and T3 for binding energies⬍4 eV. Such a resem-blance suggests that even with ditolyl substitutions, first few HOMOs of T3 still evolve mainly from the terfluorene back-bone as in E3 and both alkyl and aryl substituents might not play significant roles in the frontier occupied molecular or-bitals 共MOs兲. Such results appear consistent with the usual viewpoint that the tetrahedral C9 carbon of fluorene serves as an effective insulating spacer blocking interaction between the backbone and the substitution. The valence-band spec-trum of B3, however, is rather different. There occur various features at low binding energies that are not seen in the spec-tra of E3 and T3. For instance, within the binding energies of 1–5 eV, there are five distinguishable peak/shoulder features 共marked as A, B, C, D, and E兲 in the UPS spectrum of B3, yet there are only three共marked as A, B, and C兲 in those of E3 and T3. The variation is a strong indication of significant differences in electronic structures between terfluorenes with dialkyl/diaryl substitutions and those with spiro-linked con-jugated substitutions and of the subtle role of the spiro-linked conjugated substitution on the electronic structures and prop-erties of fluorenes.
To get further insight into how the spiro-linked substitu-tion influences the electronic structures, the experimental valence-band spectra of three terfluorenes are compared with their calculated energy levels of occupied MOs and the cal-culated valence-band spectra using these energy levels共Fig.
3兲. In all cases, the calculated spectra agree fairly well with the measured ones, particularly for the portion with lower binding energies 共⬍5 eV兲. At higher binding energies, the agreement is compromised by the secondary electron back-ground and different polarization effects on different molecu-lar energy levels.13,16In the calculated molecular energy lev-els, one can see that features A and B in UPS spectra of E3 and T3 both evolve mainly from energy levels of HOMO and HOMO-1. However, from HOMO-2, the molecular energy levels of E3 and T3 start to show differences. While HOMO-2 of E3 is isolated from other MOs, for T3 a bundle of molecular levels close to HOMO-2 result in a broad shoulder on the feature C. The differences are even greater for B3. For the UPS spectrum of B3, the HOMO level gives feature A, HOMO-1, and three nearly degenerate levels group closely together to give feature B, several nearly de-generate MOs around the binding energy of 2.9 eV gives feature C, and so on. The variations in energy levels of fron-tier MOs of the three terfluorenes may be associated with the MO levels of single ethane, toluene, and fluorene units, which are the main components of the substitutions of E3, T3, and B3, respectively. It is found that the HOMO level of ethane is well below HOMO-2 of terfluorene, which thus would not have influences on the first few frontier MOs of terfluorene. The HOMO level of toluene is also below the HOMO-2 of terfluorene, but is close enough to cause inter-action between these two MOs. On the other hand, the HOMO of a single fluorene is energetically close to the HOMO-1 of the terfluorenes, possibly causing interaction of MOs at the top of the valence band.
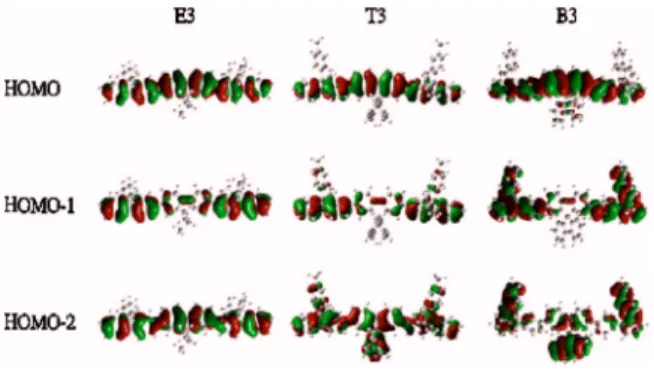
These characteristics can be further understood by in-specting the first few HOMOs共HOMO to HOMO-2兲 of the three terfluorenes共Fig. 4兲. For E3, they are mainly localized
FIG. 2. UPS valence spectra of terfluorenes E3, T3, and B3.
FIG. 3. Calculated molecular energy levels, calculated and experimental UPS valence spectra of E3, T3, and B3. The vertical bars at the bottom of each graph represent the calculated energies of molecular orbitals.
242107-2 Wu et al. Appl. Phys. Lett. 87, 242107共2005兲
on the terfluorene backbone and correlate to frontier MOs of the terfluorene backbone itself, indicating limited interaction between the central chromophore and the peripheral substi-tutions. In T3, although HOMO and HOMO-1 are also mostly on the backbone, there is a relatively significant ex-tension of HOMO-2 onto the substitution due to MO mixing with the energetically close toluene HOMO. For B3, strong backbone-substitution MO mixing observed in HOMO-1 and HOMO-2 is not surprising since the HOMO level of the spiro-linked fluorene substitution is close to HOMO-1 level of terfluorene. On the other hand, although one would expect the HOMO of the terfluorene backbone to be rather intact 共considering energetic difference in HOMO levels of and the perpendicular relationship between the backbone and the substitution兲, one notices a non-negligible portion of B3 HOMO on the substitution. Detailed analysis reveals that such characteristics originate from “spiroconjugation,” an in-teraction that allows for electronic coupling between two mutually perpendicular subsystems linked by a spiro-atom.17–19Overall, C9 substitutions could have signifi-cant electronic coupling with the oligofluorene backbone, de-pending on the characteristics of substitutions共e.g., MO lev-els of the substitution, the way they are linked, etc.兲 Such interaction would lead to backbone-substitution MO mixing and energy splitting, which influences the electronic struc-tures共frontier MOs, MO levels, etc.兲 of oligofluorenes, ex-plaining significant variations in UPS spectra observed for oligofluorenes with different substitutions.
Influences of substitutions on electronic structures of ter-fluorenes have several consequences in electronic properties. The solid-state IEs for B3, T3, and E3, are 5.9, 6.1, and 6.2 eV, respectively. Along with the gas-phase ionization poten-tials calculated with Hartree–Fock method 共7.0 eV for all three terfluorenes兲, the polarization energies, representing electronic screening and intermolecular relaxation in mo-lecular thin films, are estimated to be 1.1, 0.9, and 0.8 eV for B3, T3, and E3, respectively. The solid-state polarization en-ergy of B3 is 0.2–0.3 eV higher than those of T3 and E3, but is still within the typical range reported for organic solids.20–22 Our previous report of larger optical refractive index in B3 films than in T3 films indeed verifies stronger polarization effect in B3,23 since the optical refractive index is also strongly correlated to the polarizability of materials and in general exhibits a similar trend as the polarization energy. The stronger polarization effect of B3, thus giving it a lower solid-state IE, may be attributed to more delocalized frontier MOs and electronic distribution. Finally, it is worthy
of noting that according to previous reports, different types of C9 substitutions and consequent effects on MOs/ electronic structures of oligofluorenes also have strong influ-ences on their carrier-transport properties.19These results in-dicate that the substitutions could considerably change the energy levels, refractive indices, and carrier-transport prop-erties of oligofluorenes that would substantially influence de-vice operation in terms of carrier injection, optical outcou-pling, driving voltages, etc.
In summary, influences of different substitutions on the electronic structures and electronic properties of oligofluo-renes have been systematically studied with photoemission spectroscopy and quantum chemical calculations. Our results show that C9 substitutions of fluorene-based materials could have relatively significant influences on electronic structures. As a consequence, the electronic properties of terfluorenes, which could affect the operation of electronic devices, are also substitution dependent.
This work is supported by the National Science Council 共Grant No. NSC93-2120-M-002-013兲 and Evervictory Acutech Corporation of the Republic of China.
1C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett. 51, 913共1987兲. 2J. H. Burroughes, D. D. C. Bradley, A. N. Brown, R. N. Marks, K.
Mackay, R. H. Friend, P. L. Burns, and A. B. Holmes, Nature共London兲
347, 539共1990兲.
3Y. Y. Lin, D. J. Gundlach, S. F. Nelson, and T. N. Jackson, IEEE Trans.
Electron Devices 44, 1327共1997兲.
4B. O’Regan and M. Gratzel, Nature共London兲 353, 737 共1991兲. 5D. Neher, Macromol. Rapid Commun. 22, 1365共2001兲.
6U. Scherf and E. J. W. List, Adv. Mater.共Weinheim, Ger.兲 14, 477 共2002兲. 7C. C. Wu, Y. T. Lin, K. T. Wong, R. T. Chen, and Y. Y. Chien, Adv. Mater.
共Weinheim, Ger.兲 16, 61 共2004兲.
8C. C. Wu, T. L. Liu, W. Y Hung, Y. T. Lin, K. T. Wong, R. T. Chen, Y. M.
Chen, and Y. Y. Chien, J. Am. Chem. Soc. 125, 3710共2003兲.
9K. T. Wong, Y. Y. Chien, R. T. Chen, C. F. Wang, Y. T. Lin, H. H. Chiang,
P. Y. Hsieh, C. C. Wu, C. H. Chou, Y. O. Su, G. H. Lee, and S. M. Peng, J. Am. Chem. Soc. 124, 11576共2002兲.
10Y. Geng, A. Trajkovska, D. Katsis, J. J. Ou, S. W. Culligan, and S. H.
Chen, J. Am. Chem. Soc. 124, 8337共2002兲.
11Y. Geng, S. W. Culligan, A. Trajkovska, J. U. Wallace, and S. H. Chen,
Chem. Mater. 15, 542共2003兲.
12N. Johnasson, D. A. dos Santos, S. Guo, J. Cornil, M. Fahlman, J. Salbeck,
H. Schenk, H. Arwin, J. L. Bredas, and W. R. Salanek, J. Chem. Phys.
107, 2542共1997兲.
13G. Greczynski, M. Fahlman, W. R. Salanek, A. Dkhssi, D. A. dos Santos,
and J. L. Bredas, J. Chem. Phys. 116, 1700共2002兲.
14A. Crispin, X. Crispin, D. A. dos Santos, J. Cornil, N. Johnasson, J. Bauer,
J. Salbeck, J. L. Bredas, and W. R. Salanek, J. Chem. Phys. 117, 8195 共2002兲.
15M. J. S. Dewar, E. G. Zoebisch, E. F. Healy, and J. J. P. Stewart, J. Am.
Chem. Soc. 107, 3902共1985兲.
16I. G. Hill, A. Kahn, J. Cornil, D. A. dos Santos, and J. L. Bredas, Chem.
Phys. Lett. 317, 444共2000兲.
17H. E. Simmons and T. Fukunaga, J. Am. Chem. Soc. 89, 5208共1967兲. 18P. Maslak and A. Chopra, J. Am. Chem. Soc. 115, 9331共1993兲. 19C. C. Wu, W. G. Liu, W. Y. Hung, T. L. Liu, K. T. Wong, Y. Y. Chien, R.
T. Chen, T. H. Hung, T. C. Chao, and Y. M. Chen, Appl. Phys. Lett. 87, 052103共2005兲.
20N. Sato, K. Seki, and J. Inokuchi, J. Chem. Soc., Faraday Trans. 277,
1621共1981兲.
21C. I. Wu, Y. Hirose, H. Sirringhaus, and A. Kahn, Chem. Phys. Lett. 272,
43共1997兲.
22C. B. Duke, T. J. Fabish, and A. Paton, Chem. Phys. Lett. 49, 133共1977兲. 23H. W. Lin, C. L. Lin, H. H. Chang, Y. T. Lin, C. C. Wu, Y. M. Chen, R. T.
Chen, Y. Y. Chien, and K. T. Wong, J. Appl. Phys. 95, 881共2004兲. FIG. 4. Frontier occupied MOs of E3, T3, and B3.
242107-3 Wu et al. Appl. Phys. Lett. 87, 242107共2005兲