Review article
Implication of nuclear EGFR in the development of resistance
to anticancer therapies
Wei-Chien Huang
a,b,c,d,1,*
, Yun-Ju Chen
a,e,f,1, Mien-Chie Hung
a,b,g,**
a
Center for Molecular Medicine, China Medical University Hospital, Taichung 404, Taiwan
bGraduate Institute of Cancer Biology, China Medical University, Taichung 404, Taiwan
cThe PhD Program for Cancer Biology and Drug Discovery, China Medical University, Taichung 404, Taiwan dDepartment of Biotechnology, Asia University, Taichung 413, Taiwan
eDepartment of Medical Research, E-DA Hospital, Kaohsiung 824, Taiwan
fDepartment of Biological Science and Technology, I-Shou University, Kaohsiung 824, Taiwan g
Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
Keywords:
anticancer therapies drug resistance nuclear EGFR
a b s t r a c t
Epidermal growth factor receptor (EGFR) was identified as a major oncogenic factor in various types of cancer, and thereby has been considered as an attractive therapeutic target for cancer therapy. The well-characterized classic function of this plasma membrane-bound receptor is transduction of extracellular mitogenic signals to a variety of intracellular downstream signaling cascades associated with tumorigenesis. Aberrantly expressed EGFR also undergoes direct nuclear translocation to induce transcription of genes associated with cell proliferation, cell cycle regulation, and tumor progression. Emerging evidence suggests the existence of a new role of nuclear EGFR signaling in conferring acquired resistance in response to various anticancer therapies. In this review, we summarize the current understanding of how EGFR translocates into the nucleus in response to ionizing radiation, chemotherapy, and anti-EGFR target agents. The emerging impact of nuclear EGFR in modulating the cellular sensitivity of cancer cells to these anticancer treatments will also be discussed. A better understanding of the nuclear EGFR pathways in response to anticancer therapies will facilitate the development of novel strategies to overcome the acquired resistance.
1.
Introduction
Epidermal growth factor receptor (EGFR; ErbB1), a receptor tyrosine kinase, is frequently overexpressed and widely
involved in the etiology and progression of many types of cancer[1]. Cancer patients whose tumors aberrantly express EGFR tend to have a more aggressive disease and a shorter survival rate, so EGFR not only has been viewed as a predictive * Corresponding author. Center for Molecular Medicine, China Medical University Hospital and Graduate Institute of Cancer Biology, China Medical University, Taichung 404, Taiwan.
** Corresponding author. Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
E-mail addresses:[email protected](W.-C. Huang),[email protected](M.-C. Hung).
marker for poor clinical outcome but also intensely pursued as a therapeutic target[2]. EGFR activation through dimerization and autophosphorylation with ERBB family stimulates multiple intracellular downstream signaling pathways by recruiting effector proteins. Two major pathways initiated by the receptor tyrosine kinase are the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3 K)eAkt pathways[3,4]. Other important growth regulators in cancer in response to EGFR activation are the signal transducer and activator of transcription proteins (STATs), SRC tyrosine kinase, and mammalian target of rapamycin [5e7]. These signaling cascades integrate and transmit the EGFR activation into distinct transcriptional programs associated with prolif-eration, tumorigenesis, metastasis, and survival[3]. In addition to its well-characterized downstream signaling pathways, EGFR and other ErbB family have been found to enter the nucleus of multiple types of cancer cells and possess oncogenic functions, including gene transcription [8e12], DNA repair [13e15], regulation of enzyme activity[16], and translation[17], linking to the aggressiveness of tumors. Interestingly, the nuclear translocation of EGFR was also repeatedly observed in response to the constitutive treatment with different types of anticancer drugs [18e21] and ionizing radiation [14,22,23], suggesting that nuclear EGFR may play a crucial role in the development of therapy resistance. However, the role of nuclear EGFR in the development of resistance to anticancer therapies is still not fully understood. This review will summarize and discuss the current understanding of the nuclear functions of EGFR and its impact on tumor sensitivity to radiation, chemotherapy, and anti-EGFR target therapy.
2.
Biological properties and clinical
implication of nuclear EGFR in cancer
Following the first discovery of nuclear localization of EGFR in liver cancer cells[24], nuclear expression of EGFR and ErbB2 has been continually discovered in a variety of cancer types [8,10,25e31]. The first identified nuclear function of EGFR is regulation of gene transcription[10]. Although the association with AT-rich sequence (ATRS) of its target gene promoters has been proposed to be required for nuclear EGFR-activated gene transcription[10,21], the lack of a DNA-binding domain[10] suggests that nuclear EGFR targets promoters through binding to various transcriptional factors with DNA-binding domains and functions as a transcription cofactor rather than a DNA-binding transcription factor. In response to EGF stimulation, the interaction of nuclear EGFR with signal transducer and activator of transcription-3 (STAT3) has been demonstrated to be required for nuclear EGFR-mediated inducible nitric oxide synthase (iNOS) [8] and cyclo-oxygenase-2 (COX-2)[9]expression. In addition, nuclear EGFR cooperates with STAT5 and E2F1 to enhance aurora A[32]and B-Myb [27]gene expression, respectively. Interestingly, our recent findings further revealed that nuclear EGFR also inter-acts with RNA helicase A (RHA) independent of its ATPase/ helicase activity to associate with the promoter region of its target genes such as cyclin D1 and iNOS[33], indicating that RHA is a DNA-binding partner for nuclear EGFR in regulating its target gene expression. However, overexpression of the
RHA-interaction domain (residues 645e1186) of EGFR is not sufficient and full length of EGFR is required to increase the promoter activity [33], suggesting that other unidentified components may be involved in the EGFReRHA complex.
Given the fact that nuclear EGFR retains its tyrosine kinase activity, regulation of protein stability and enzymatic activity of its nuclear target proteins via tyrosine phosphorylation has been explored as another important nuclear function of EGFR. Proliferating cell nuclear antigen (PCNA) was the first identi-fied nuclear substrate of EGFR tyrosine kinase[13]. Nuclear EGFR stabilized chromatin-bound PCNA protein via phos-phorylating at its Tyr211 and preventing its polyubiquitination and proteasomal degradation. The increased PCNA Tyr211 phosphorylation by nuclear EGFR promotes cell proliferation and DNA repair, and is closely correlated with poor survival of breast cancer patients. In addition to targeting PCNA, nuclear EGFR was also found to enhance DNA repair via regulating DNA-dependent protein kinase (DNAePK), an enzyme involved in repairing double-strand breaks and V(D)J recom-bination[34]. A substantial amount of DNAePK was found to be colocalized with EGFR in anti-EGFR mAb-treated cells in the confocal microscope analysis[34]. The physical interac-tion between nuclear EGFR and DNAePK was also observed in cancer cells treated with radiation [14]or anti-EGFR mono-clonal antibody[34]. Furthermore, the nuclear level of EGFR is associated with phosphorylation of DNAePK at residue T2609, an indicator of DNAePK activity during nonhomolo-gous end-joining DNA repair[14,22], and inhibition of EGFR signaling was accompanied by a reduction in the level and activity of DNAePK in the nuclear fraction[34,35]. Although there is no evidence revealing that DNAePK is directly phosphorylated by nuclear EGFR, these findings support that nuclear EGFR modulates DNA repair in response to DNA damage through regulating the kinase activity of the DNAePK complex[36].
Although not all functions of nuclear EGFR have been elucidated, several studies suggest that nuclear EGFR may serve as a prognostic marker for poor clinical outcome. In a population of 130 breast cancer patients, tumor tissues from 37.7% of this cohort were immunostained positively for nuclear EGFR, and a significant inverse correlation existed between the high nuclear EGFR expression and overall survival[28]. Hadzisejdic et al also reported nuclear EGFR as an independent prognostic factor for mortality in another cohort of breast cancer patients[37]. The correlation between poor survival rate and high level of nuclear EGFR in the cancer cells was also observed in several cohorts of cancer patients with oral squamous carcinomas [28], oropharyngeal carcinomas [29], ovarian cancer [30], and esophageal squamous carci-nomas [38]. These observations suggest that nuclear EGFR may be considered as a prognostic indicator for poor clinical outcome and also revealed a crucial role of nuclear EGFR in mediating tumor progression.
3.
Nuclear EGFR facilitates the development
of resistance to a variety of anticancer therapies
In most studies, nuclear localization of EGFR was observed in EGF-treated cancer cells or in the human primary tumor
tissues. EGF induces EGFR nuclear localization rapidly and transiently within 2 h of treatment[39]. Coat protein complex I-mediated retrograde trafficking from the Golgi to the endoplasmic reticulum (ER) has been shown to regulate EGF-induced EGFR nuclear transport [40]. In addition to these physiological situations, our and others’ recent studies uncovered that some anticancer therapies also drive EGFR import into the nucleus of various cancer cells, adding a role of nuclear EGFR in the development of drug resistance. Unlike the transient nuclear localization by EGF stimulation, EGFR is steadily present in the nucleus in response to these anticancer treatments[14,18e22,36].
Ionizing radiation (IR) was found to stimulate EGFR nuclear transport in human bronchial and squamous carcinoma cells [14,18,41]. Other DNA-damaging stimuli, such as cisplatin and H2O2 also initiated EGFR internalization and subsequent
nuclear import[14,42]. In the nucleus, EGFR has been demon-strated to play important roles in unhooking cisplatin-induced interstrand crosslinks and in repairing IR-induced strand breaks, indicating the involvement of nuclear EGFR in confer-ring chemoresistance and radioresistance[36]. Supporting this notion, our data further showed that after reconstruction of a functional nuclear localization sequence in its nuclear localization signal (NLS)-deleted mutant, EGFR is able to restore the DNA repair activity and consequently reduced the sensitivity of cancer cells to cisplatin[19].
In addition to IR and cisplatin, two EGFR-targeted thera-peutic agents, cetuximab [41] and gefitinib[21], were also found to elicit the accumulation of EGFR in the nucleus. Cetuximab (Erbitux) is an EGFR-blocking antibody that has been approved for the treatment of patients with head and neck squamous cell carcinoma (HNSCC) and metastatic colorectal cancer. Ectopic expression of the NLS-tagged EGFR can reduce the sensitivity of NCI-H226 non-small cell lung cancer (NSCLC) cells to cetuximab both in vitro and in mouse xenografts[20], supporting the association of nuclear EGFR with tumor resistance to cetuximab. Gefitinib (ZD1839, Ire-ssa), a small molecular weight EGFR kinase inhibitor, has been used for advanced and metastatic NSCLC with expres-sion of activating EGFR mutants, such as EGFR L858R mutant [43], and most of NSCLC cancer patients bearing wild-type (wt) EGFR frequently are insensitive to this drug [43]. Our recent study reported that, in a wt EGFR-expressing cancer cell line, nuclear translocation of EGFR was increased in response to chronic treatment with gefitinib, and mediated the gene expression of breast cancer resistant protein (BCRP) to cause the development of drug resistance through efflux of gefitinib[21]. However, the nuclear translocation of EGFR and its mediated BCRP expression were only observed in cancer cells expressing wt EGFR but not its activating mutant, suggesting a possible mechanism explaining why gefitinib is not beneficial to most wt EGFR-expressing NSCLC patients [21,44] The reason why the mutant EGFR lacks nuclear translocation ability is not clear. One possibility could be that the recently identified tracking mechanism of cell surface EGFR to the nucleus is impaired[40,45]. Similar to this observation, cells bearing EGFR L858R, which do not show nuclear expression, also possess less ability to repair cisplatin- and IR-induced DNA damage [36]. These studies imply a nuclear-specific role of wt EGFR in conferring the
resistance in response to EGFR target therapy, chemo-therapy, and IR.
EGFR tyrosine kinase activation has been reported to be required for the nuclear translocation of EGFR in response to EGF stimulation[10]. In addition to the wt form, EGFR is also present as truncated mutant (EGFRvIII) with constitutive kinase activity [46], and is associated with the aggressive biology of glioma [47]. The constitutively activated EGFR variant, EGFRvIII, is present in the nuclei of glial cells[48]and glioblastoma [48,49] revealing the crucial role of tyrosine kinase activation in the EGFR nuclear import. In response to irradiation, however, EGFR has been found to enter the nucleoplasm in a ligand-independent process that involves free radicals[14,50,51]. Furthermore, the L858R and exon 19 deletion mutants of EGFR, which exhibit constitutive kinase activity, did not show nuclear import after irradiation or cisplatin treatment[36], suggesting that other mechanisms, in addition to its kinase activity, may regulate the nuclear import of EGFR in response to these anticancer therapies. We have recently discussed the detailed mechanism by which the full-length receptors embedded in the endosomal membrane travel all the way from the cell surface to the early endosomes and pass through the nuclear pore complexes [40,45,52]. The specific regulations of EGFR nuclear import in response to different anticancer treatments will be described below.
4.
Nuclear trafficking of EGFR in response to
anticancer therapies
4.1. Ionizing radiation-induced EGFR nuclear
localization involves karyopherin alpha and protein kinase C epsilon
Karyopherin proteins, also named importin, are nuclear transport factors and mediate the majority of nucleocytoplas-mic transport[53]. We have demonstrated that interaction with importin beta is involved in the translocation of EGFR[39,45,54] and ErbB2 [55] into the nucleus through the nuclear port complex. Mutation of its NLS disrupts the interaction of EGFR with importins, indicating that EGFR may interact with importins through its NLS motif[39,54]. Dittmann et al showed that ionizing radiation also enhanced nuclear EGFR to form a complex with karyopherin alpha and Ran protein, which are essential for formation of a nuclear localization sequence-dependent nuclear import complex[14]. Their work suggested that IR triggers EGFR import into the nucleus in a karyopherin alpha-linked manner. In parallel with the role of nuclear EGFR in repairing DNA damage, karyopherin has also been consid-ered as a marker for global chemoresistance and been reported as an important factor of tumorigenesis and progression of breast cancer[56]. Interestingly, the interaction between EGFR and the alpha form of karyopherin was only observed in response to IR but not EGF stimulation [14], suggesting that interactions with karyopherin alpha and beta forms may be responsible for radiation- and EGF-induced EGFR nuclear translocation, respectively. It would also be of great interest to know what the different regulations for the EGFR interactions with various karyopherins are in response to these stimuli.
Phosphorylation of EGFR Thr654 by protein kinase C epsilon (PKCε) is another regulation for EGFR nuclear translocation following irradiation[23]. EGFR Thr654 phosphorylation has been reported earlier to block Cbl induced ubiquitination and lysosomal degradation of EGFR, leading to EGFR stabilization [57]. PKCε has been identified as the kinase responsible for this modification following irradiation[18]. Furthermore, deletion of Thr654 blocked nuclear transport of EGFR, whereas muta-tion to Glu increased this shuttling, demonstrating that phos-phorylation of this residue is essential for nuclear EGFR shuttling following irradiation[23]. Because the Thr654 phos-phorylation is located within the NLS motif of EGFR, it raises the possibility that this phosphorylation by PKCε may regulate EGFR interaction with karyopherins.
4.2. Phosphorylation by Src family kinases drives EGFR nuclear import to reduce the sensitivity to cetuximab
Cetuximab (C225, Erbitux), a humanized monoclonal anti-body, recognizes the extracellular domain of both wt EGFR and EGFRvIII has been approved as the second-line treatment after failure to chemotherapy or as the first-line treatment with
radiation therapy for advanced HNSCC. Cetuximab is also used in combination with irinotecan for treating metastatic colorectal cancer after failure to chemotherapy. Reduction of c-Cbl-mediated internalization and degradation of EGFR under the chronic exposure to cetuximab leads to steady-state expression of EGFR, and the increased EGFR confers cetux-imab resistance through binding and activating HER2 or HER3 to maintain signaling to MAPK and Akt pathways [58]. In addition to activating other receptor tyrosine kinases, the increased EGFR expression also caused the accumulation of EGFR in the nucleus[20]. In contrast to the ligand-independent manner in the IR-treated cells, the nuclear translocation of EGFR in cetuximab-resistant cells seems to rely on over-expression of several ErbB family ligands, including EGF, amphiregulin, heparin-binding (HB) EGF and beta-cellulin[20]. Overexpression of these ligands enhances the nuclear trans-location of EGFR through Src family kinases (SFKs), and treatment of cetuximab-resistant cells with SFK inhibitor dasatinib (BMS354825) can resensitize cells to cetuximab[20]. Because inhibition of SFK activity and EGFR Y845 phosphory-lation by dasatinib resulted in loss of cetuximab-induced nuclear EGFR expression (Fig. 1) and increase in membrane
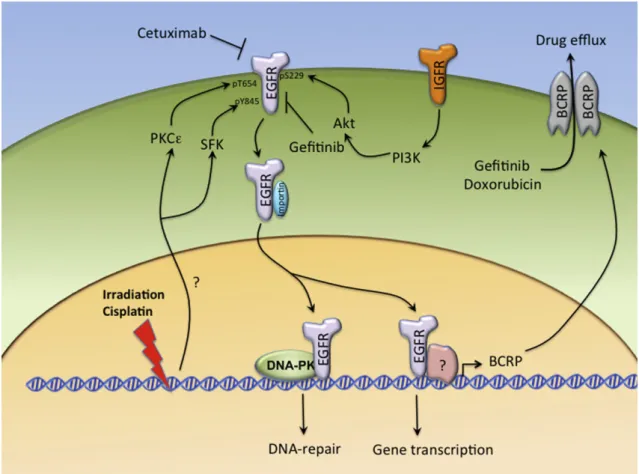
Fig. 1e The nuclear roles of EGFR in conferring the resistance to irradiation, cisplatin, and anti-EGFR agents. In response to chronic treatment with irradiation and cisplatin, the nuclear translocation of EGFR is enhanced by PKCε and SFK-dependent phosphorylation at Thr654 and Tyr845, respectively, and by interaction with importins. The nuclear EGFR can interact with and activate DNAePK to promote DNA repair, and thereby confers radioresistance and chemoresistance. In gefitinib-resistant cells, the compensatory Akt activation by IGFR signaling also facilitates EGFR nuclear import through
phosphorylation of its Ser229. EGFR in the nucleus functions as a transcriptional regulator to mediate BCRP expression, which recognizes gefitinib and doxorubicin as substrates to result in the efflux of these drugs.
expression of EGF, Src may regulate the nuclear import of EGFR through phosphorylation of its Y845 and thereby contribute to cetuximab resistance[41].
4.3. Akt enhances wt EGFR nuclear import via phosphorylating its Ser229 response to gefitinib resistance
Gefitinib (ZD1839, Iressa) is the first small molecular inhibitor of EGFR tyrosine kinase. Although EGFR is overexpressed in many cancer types, targeting EGFR tyrosine kinase activity by gefitinib showed more dramatic efficacy and clinical benefits for NSCLC patients, particularly those characterized as East Asian, nonsmoker, adenocarcinoma histological type, and female gender, but only modest effects on many other cancer types[43]. The encouraging responses in these selected NSCLC patients to gefitinib showed strong association with specific gain-of-function mutations within the EGFRetyrosine kinase domain [43,59]. Several mechanisms, including activation of c-MET and insulin growth factor receptor (IGFR) to raise the compensatory survival signals[60,61]and the loss of phosphatase and tensin homolog (PTEN) [62,63], have been shown to play a role in determining the sensitivity of wt EGFR-expressing cancer cell to gefitinib. Interestingly, these mechanisms commonly elevate the PI3KeAkt signaling pathway to maintain the cell survival in the presence of gefitinib[64,65]. In addition to providing the survival signaling, our recent study further disclosed that the elevated Akt activity was also associated the nuclear trans-location of wt EGFR to mediate gefitinib resistance[21].
As in cetuximab-resistant cells[20]and irradiation-resistant cells[14,66], nuclear accumulation of EGFR in response to gefi-tinib resistance is observed in wt EGFR-expressing cell lines but not in EGFR mutant-expressing cell lines. We identified EGFR Ser229 as a novel Akt substrate and Ser229 phosphorylation of EGFR was detected in both nuclear and cytoplasmic fraction of gefitinib-resistant cells. Overexpression of Akt can dramatically increase the nuclear level of EGFR, and the Akt-mediated EGFR nuclear accumulation was attenuated by substitution of Ser229 to Ala, demonstrating that this phosphorylation is required for EGFR nuclear translocation[21]. Because elevated or contin-uous activation of Akt survival signaling is commonly observed in cancer cells with the characteristic of chemoresistance[19], radioresistance [14], or cetuximab insensitivity [20,58], Akt-dependent phosphorylation might be an general regulation for the nuclear translocation of EGFR in response to various anticancer therapies (as illustrated in Fig. 1). Currently, we further reported that sequential Akt-dependent phosphoryla-tion and polyubiquitinaphosphoryla-tion are required for IkB kinase (IKKa) nuclear transportation in response to hepatitis B virus X protein overexpression[67]. Because ubiquitination has been widely found to be involved in protein nucleocytoplasmic shuttling [68e71], it raises the possibility that polyubiquitination of EGFR also occurs following Akt-dependent phosphorylation and mediates EGFR nuclear import. These observations suggest that phosphorylation by Akt may function as a common signal to drive the nuclear trafficking of target proteins, including EGFR. Other regulations, such as polyubiquitination, involving inter-actions with nuclear importer or exporter might be required to decide the destination of these cargo proteins. However, to elucidate the detail mechanism further studies are needed.
5.
Molecular actions of nuclear EGFR in the
development of resistance to anticancer
therapies
5.1. Nuclear EGFR regulates DNA repair involves DNAePK activation
The exploration of physical interaction between EGFR and DNAePK[34], which is a major enzyme of nonhomologous end-joining DNAedouble-strand break repair, initiated the extended studies to understand the roles of nuclear EGFR in DNA repair and resistance to DNA-damaging radiotherapy and alkylators (Fig. 1). After treatment with cisplatin and irradiation, the interaction of EGFR with the catalytic subunit of DNAePK (DNAePKcs) and its regulatory heterodimeric complex Ku70/80 was observed in the nucleus in vivo and in vitro[34]. Because the EGFR NLS mutation interrupts the association of EGFR with DNAePKcs and reduces the nuclear localization of DNAePKcs, EGFR has been suggested to cotranslocate with DNAePKcs into the nucleus and regulate the formation and activation of the DNAePK complex after cisplatin treatment and IR[36]. Indeed, nuclear EGFR is asso-ciated with phosphorylation of DNAePK at residue T2609, an indicator of DNAePK activity during nonhomologous end-joining DNA repair[14,22]. Nuclear EGFR, in association with DNAePK or Ku70/80, retains its intrinsic kinase activity[34]. Blockage of EGFR activation by its antibody cetuximab resul-ted in the decrease of DNAePK activity, the increase of residual DNA damage, and the subsequent enhancement of the radiosensitivity of human A549 lung carcinoma cell line [22], suggesting that the tyrosine kinase activity of nuclear EGFR is required for the activation of the DNAePK complex. However, the evidence revealing DNAePK as a direct substrate of nuclear EGFR is lacking. In addition, EGFR tyrosine kinase activity may be essential but not sufficient for EGFR-dependent DNAePKcs activation as overexpression of EGFR LNLS mutant, which contains both a constitutive activating mutation at L858 and an NLS mutation, cannot activate DNAePKcs activity[36], further bolstering the crucial role of nuclear existence of EGFR in contributing to radioresistance and chemoresistance.
Although EGFR nuclear localization has been demon-strated to be required for modulation of cisplatin and IR-induced repair of DNA damage, the interaction between EGFR and DNAePKcs was induced by cisplatin or IR but not by EGF stimulation or EGFR nuclear translocation per se [36]. Other mechanisms specifically elicited by DNA damage may be involved in the regulation of nuclear EGFR binding with DNAePKcs. Interestingly, treatment with celecoxib, a COX-2 specific inhibitor, has been shown to obviously increase the radiosensitivity of multiple cancer cell lines via attenuating the radiation-induced EGFR nuclear transport and DNAePK activation [72]. However, the radiosensitization by celecoxib seems to be independent of COX-2 activity[72]. Because cel-ecoxib and its analogs possess an off-target effect on dis-rupting Akt signaling[73], which has been demonstrated to regulate EGFR nuclear import [21], it is worthy to further pursue whether radiation induces the interaction between nuclear EGFR and DNAePK in an Akt-dependent manner.
5.2. Nuclear EGFR functions as a transcription regulator to increase expression of gefitinib efflux pump
Despite that the effects of nuclear EGFR on the sensitivity to gefitinib are not well understood, nuclear presence of EGFR seems to be a general event in different gefitinib-treated cells [21]. As illustrated in Fig. 1, we have reported that nuclear EGFR functions as a transcription regulator to mediate BCRP/ ABCG2[21]and thereby confers gefitinib resistance[44]. The expression of BCRP/ABCG2, a well-known transporter of ATP-binding cassette (ABC) family involved in chemoresistance to doxorubicin as well as many other chemotherapies[74,75], was found in 46% of advanced NSCLC patients[76]. Several studies have demonstrated that gefitinib is also a substrate of BCRP/ABCG2[44e46]and can be pumped out of the cells, resulting in development of gefitinib resistance[44]. Aberrant expression of this transporter was not only correlated with the intrinsic insensitivity of wt EGFR-expressing patients to gefitinib[44] but also increased in the wt EGFR-expressing NSCLC patient with acquired gefitinib resistance [77e79], revealing BCRP as a valuable marker to predict the clinical outcome of gefitinib-treated patients without EGFR activating mutations and as a potential target to overcome the acquired resistance to gefitinib [44]. The BCRP promoter contains ATRSs and has been found to be targeted by nuclear EGFR in an Akt-dependent manner [21]. Mutation of EGFR NLS or silence of importin, which mediates the nuclear EGFR translocation, can abolish EGFR-dependent BCRP expression [21]. Although the promoter region of multiple drug resis-tance 1 (MDR-1/ABCB1), another ABC transporter, also contains ATRSs putative EGFR binding sites, the increase in MDR1 expression was not detected in the gefitinib-resistant cells[21]. Nuclear EGFR has been suggested to form a het-eromeric transcription complex with the signal transducer and activator of transcription (Stat) proteins to mediate c-Myc expression in pancreatic cancer cells [80]. The over-lapped binding site on BCRP promoter for Stat5 and ATRSs might account for the specific regulation of BCRP but not MDR-1 expression by nuclear EGFR[21].
6.
Perspectives and future directions
The studies in nuclear functions of EGFR conducted in the past decade have disclosed several important pathological roles of the nuclear EGFR pathway in tumorigenesis. Since Dittmann and colleagues reported the involvement of nuclear EGFR in the activation of the DNAePK complex to mediate the DNA repair in response to irradiation [14], a novel aspect of nuclear EGFR in the development of acquired resistance to anticancer therapies has emerged. This finding has hence evoked many other studies on the mode of action of nuclear EGFR in conferring resistance to chemotherapy and EGFR target therapy. These studies have had profound implications for the development of a novel strategy to overcome drug resistance by targeting nuclear EGFR signaling. Although researchers gain new insights into the therapeutic impact of nuclear EGFR on human cancers, many questions remain to be addressed:
(1) Are there additional mechanisms underlying nuclear EGFR-mediated resistance? Although the regulations of DNAePK activation and BCRP expression by nuclear EGFR have been demonstrated to mediate the resistance to DNA-damaging treatment and gefitinib, respectively, the detail mechanisms remain unclear. Novel nuclear proteins phosphorylated and functionally modulated by nuclear EGFR in response to anticancer treatments await to be discovered. In addition, several target genes or proteins of nuclear EGFR implicated in tumorigenesis have been identified. It is worthy to further pursue whether these known targets of nuclear EGFR also contribute to the formation of drug resistance.
(2) Are there common mechanistic regulations that drive EGFR nuclear transport in response to different anticancer therapies? As described above, Akt activation has been viewed as a common signaling pathway to mediate the acquired resistance to multiple drugs and also plays a role in regulating gefitinib-induced EGFR nuclear translocation. Therefore, regulation by Akt may be a common mecha-nism for the EGFR import into the nucleus. However, other regulations, such as phosphorylation by SFK and PKCε, may compensate for this process.
(3) Does nuclear EGFR function as a key regulator in cross-resistance among irradiation, chemotherapy, and EGFR target therapy? The nuclear EGFR-mediated BCRP expres-sion in gefitinib-resistance cells has been found to cause the cross-resistance to doxorubicin. It would be of great interest to examine whether chemoresistant cells also exhibit nuclear EGFR and BCRP expression to reduce their sensitivity to gefitinib. In addition, the nuclear import of EGFR and the subsequent DNAePK activation have been commonly observed in response to various treatments including irradiation and cisplatin. Further studies are needed to demonstrate whether the nuclear EGFR-mediated DNAePK activation can result in the cross-resistance between these DNA-damaging therapies. Addressing this question will provide very important information to determine the use and priority of anti-cancer therapies.
(4) Is there any efficient strategy for targeting nuclear EGFR signaling and thereby overcoming drug resistance? Once the nuclear role of EGFR in developing the resistance to anticancer therapies is extensively understood, blockage of nuclear EGFR signaling may be a new strategy to fight treatment resistance[23]. Indeed, targeting nuclear EGFR-dependent tyrosine phosphorylation of PCNA by blocking peptides has been shown to inhibit prostate cancer growth [81]. This finding revealed a promising strategy to over-come the nuclear EGFR-dependent resistance.
(5) Do other plasma membrane receptors also function in the nucleus to confer the resistance to cancer therapies? In addition to EGFR, nuclear translocations of other plasma membrane-bound receptors, such as HER2[17,55], ErbB4 [82e86], and fibroblast growth factor receptor (FGFR)[87], have also been observed in various cancer types and are associated with etiology and tumor progression of these cancers. It would be of interest to understand whether these receptors in the nucleus also play a crucial role
in determining the cellular sensitivity to anticancer therapies.
Collectively, elucidation of these aspects of nuclear EGFR will help us evaluate the possibility of using nuclear EGFR as a biomarker to predict the sensitivity to various anticancer treatments and develop novel strategies to prevent or over-come the acquired resistance. If this is the case, nuclear EGFR may serve as a biomarker to help us stratify patients for personalized cancer therapy.
Acknowledgments
We are grateful to Dr Stephanie Sellers for editing the manu-script. This work was supported by grants from the National Science Council of Taiwan (2320-B-039-021, NSC-100-2320-B-039-022-, NSC-97-2320-B-039-033-MY3, and NSC-99-3112-B-039-002 to W.C.H, and NSC-2632-B-001-MY3 to M.C.H.), the National Health Research Institutes of Taiwan (NHRI-EX-98-9812BC to W.C.H), Cancer Center Research of Excellence (DOH-TD-C-111-005 to M.C.H.), and the University of Texas MD Anderson Cancer Center/China Medical University Hospital Sister Institution Fund. All authors declare no financial interests.
r e f e r e n c e s
[1] Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005;5: 341e54.
[2] Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol 2004; 44:195e217.
[3] Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127e37.
[4] Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science 2004;306: 1506e7.
[5] Yu H, Jove R. The STATs of cancere new molecular targets come of age. Nat Rev Cancer 2004;4:97e105.
[6] Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell 2004;6:209e14.
[7] Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335e48.
[8] Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005;7:575e89.
[9] Lo HW, Cao X, Zhu H, Ali-Osman F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol Cancer Res 2010;8: 232e45.
[10] Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 2001;3:802e8. [11] Xie Y, Hung MC. Nuclear localization of p185neu tyrosine
kinase and its association with transcriptional
transactivation. Biochem Biophys Res Commun 1994;203: 1589e98.
[12] Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell 2004;6: 251e61.
[13] Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol 2006;8:1359e68.
[14] Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, et al. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem 2005;280: 31182e9.
[15] Chen DJ, Nirodi CS. The epidermal growth factor receptor: a role in repair of radiation-induced DNA damage. Clin Cancer Res 2007;13:6555e60.
[16] Dittmann K, Mayer C, Fehrenbacher B, Schaller M,
Kehlbach R, Rodemann HP. Nuclear epidermal growth factor receptor modulates cellular radio-sensitivity by regulation of chromatin access. Radiother Oncol 2011;99:317e22. [17] Li LY, Chen H, Hsieh YH, Wang YN, Chu HJ, Chen YH, et al.
Nuclear ErbB2 enhances translation and cell growth by activating transcription of ribosomal RNA genes. Cancer Res 2011;71:4269e79.
[18] Wanner G, Mayer C, Kehlbach R, Rodemann HP, Dittmann K. Activation of protein kinase C epsilon stimulates DNA-repair via epidermal growth factor receptor nuclear accumulation. Radiother Oncol 2008;86:383e90.
[19] Hsu SC, Miller SA, Wang Y, Hung MC. Nuclear EGFR is required for cisplatin resistance and DNA repair. Am J Transl Res 2009;1:249e58.
[20] Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009;28:3801e13.
[21] Huang WC, Chen YJ, Li LY, Wei YL, Hsu SC, Tsai SL, et al. Nuclear translocation of EGFR by AKT-dependent phosphorylation enhances BCRP/ABCG2 expression in gefitinib-resistant cells. J Biol Chem 2011;286:20558e68. [22] Dittmann K, Mayer C, Kehlbach R, Rothmund MC, Peter
Rodemann H. Radiation-induced lipid peroxidation activates src kinase and triggers nuclear EGFR transport. Radiother Oncol 2009;92:379e82.
[23] Dittmann K, Mayer C, Fehrenbacher B, Schaller M,
Kehlbach R, Rodemann HP. Nuclear EGFR shuttling induced by ionizing radiation is regulated by phosphorylation at residue Thr654. FEBS Lett 2010;584:3878e84.
[24] Marti U, Burwen SJ, Wells A, Barker ME, Huling S, Feren AM, et al. Localization of epidermal growth factor receptor in hepatocyte nuclei. Hepatology 1991;13:15e20.
[25] Marti U, Ruchti C, Kampf J, Thomas GA, Williams ED, Peter HJ, et al. Nuclear localization of epidermal growth factor and epidermal growth factor receptors in human thyroid tissues. Thyroid 2001;11:137e45.
[26] Cordero JB, Cozzolino M, Lu Y, Vidal M, Slatopolsky E, Stahl PD, et al. 1,25-Dihydroxyvitamin D down-regulates cell membrane growth- and nuclear growth-promoting signals by the epidermal growth factor receptor. J Biol Chem 2002; 277:38965e71.
[27] Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog 2006;45:10e7.
[28] Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res 2005;65:338e48. [29] Psyrri A, Yu Z, Weinberger PM, Sasaki C, Haffty B, Camp R,
et al. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in
oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin Cancer Res 2005;11:5856e62. [30] Xia W, Wei Y, Du Y, Liu J, Chang B, Yu YL, et al. Nuclear
expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinog 2009;48:610e7.
[31] Xu Y, Shao Y, Zhou J, Voorhees JJ, Fisher GJ. Ultraviolet irradiation-induces epidermal growth factor receptor (EGFR) nuclear translocation in human keratinocytes. J Cell Biochem 2009;107:873e80.
[32] Hung LY, Tseng JT, Lee YC, Xia W, Wang YN, Wu ML, et al. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res 2008;36:4337e51.
[33] Huo L, Wang YN, Xia W, Hsu SC, Lai CC, Li LY, et al. RNA helicase A is a DNA-binding partner for EGFR-mediated transcriptional activation in the nucleus. Proc Natl Acad Sci USA 2010;107:16125e30.
[34] Bandyopadhyay D, Mandal M, Adam L, Mendelsohn J, Kumar R. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J Biol Chem 1998;273:1568e73. [35] Um JH, Kwon JK, Kang CD, Kim MJ, Ju DS, Bae JH, et al.
Relationship between antiapoptotic molecules and
metastatic potency and the involvement of DNA-dependent protein kinase in the chemosensitization of metastatic human cancer cells by epidermal growth factor receptor blockade. J Pharmacol Exp Ther 2004;311:1062e70. [36] Liccardi G, Hartley JA, Hochhauser D. EGFR nuclear
translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res 2011;71:1103e14. [37] Hadzisejdic I, Mustac E, Jonjic N, Petkovic M, Grahovac B.
Nuclear EGFR in ductal invasive breast cancer: correlation with cyclin-D1 and prognosis. Mod Pathol 2010;23: 392e403.
[38] Hoshino M, Fukui H, Ono Y, Sekikawa A, Ichikawa K, Tomita S, et al. Nuclear expression of phosphorylated EGFR is associated with poor prognosis of patients with esophageal squamous cell carcinoma. Pathobiology 2007;74:15e21.
[39] Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem 2007;282:10432e40.
[40] Wang YN, Wang H, Yamaguchi H, Lee HJ, Lee HH, Hung MC. COPI-mediated retrograde trafficking from the Golgi to the ER regulates EGFR nuclear transport. Biochem Biophys Res Commun 2010;399:498e504.
[41] Li C, Iida M, Dunn EF, Wheeler DL. Dasatinib blocks cetuximab- and radiation-induced nuclear translocation of the epidermal growth factor receptor in head and neck squamous cell carcinoma. Radiother Oncol 2010;97:330e7. [42] Li F, Yang W, Guo D, Hu Z, Xu H, Ye Z. LRIG1 combined with
cisplatin enhances bladder cancer lesions via a novel pathway. Oncol Rep 2011;25:1629e37.
[43] Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying
responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129e39.
[44] Chen YJ, Huang WC, Wei YL, Hsu SC, Yuan P, Lin HY, et al. Elevated BCRP/ABCG2 expression confers acquired resistance to gefitinib in wild-type EGFR-expressing cells. PLoS One 2011;6:e21428.
[45] Wang YN, Yamaguchi H, Huo L, Du Y, Lee HJ, Lee HH, et al. The translocon Sec61beta localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus. J Biol Chem 2010;285:38720e9.
[46] Batra SK, Castelino-Prabhu S, Wikstrand CJ, Zhu X, Humphrey PA, Friedman HS, et al. Epidermal growth factor ligand-independent, unregulated, cell-transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ 1995;6:1251e9.
[47] Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor
receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res 2008;14:6042e54.
[48] de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, et al. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev 2008;22:449e62. [49] Lo HW. EGFR-targeted therapy in malignant glioma: novel
aspects and mechanisms of drug resistance. Curr Mol Pharmacol 2010;3:37e52.
[50] Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res 2000;6:2166e74. [51] Harari PM, Huang SM. Modulation of molecular targets to
enhance radiation. Clin Cancer Res 2000;6:323e5. [52] Wang YN, Yamaguchi H, Hsu JM, Hung MC. Nuclear
trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene 2010;29:3997e4006.
[53] Xu D, Farmer A, Chook YM. Recognition of nuclear targeting signals by karyopherin-beta proteins. Curr Opin Struct Biol 2010;20:782e90.
[54] Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem 2006;98:1570e83.
[55] Giri DK, Ali-Seyed M, Li LY, Lee DF, Ling P, Bartholomeusz G, et al. Endosomal transport of ErbB-2: mechanism for nuclear entry of the cell surface receptor. Mol Cell Biol 2005;25:11005e18. [56] Gluz O, Wild P, Meiler R, Diallo-Danebrock R, Ting E,
Mohrmann S, et al. Nuclear karyopherin alpha2 expression predicts poor survival in patients with advanced breast cancer irrespective of treatment intensity. Int J Cancer 2008; 123:1433e8.
[57] Bao J, Alroy I, Waterman H, Schejter ED, Brodie C, Gruenberg J, et al. Threonine phosphorylation diverts internalized epidermal growth factor receptors from a degradative pathway to the recycling endosome. J Biol Chem 2000;275:26178e86.
[58] Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008;27:3944e56.
[59] Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497e500. [60] Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C,
Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316:1039e43.
[61] Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2008;14:2895e9.
[62] Guillamo JS, de Bouard S, Valable S, Marteau L, Leuraud P, Marie Y, et al. Molecular mechanisms underlying effects of epidermal growth factor receptor inhibition on invasion, proliferation, and angiogenesis in experimental glioma. Clin Cancer Res 2009;15:3697e704.
[63] Mellinghoff IK, Cloughesy TF, Mischel PS. PTEN-mediated resistance to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res 2007;13:378e81.
[64] Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest 2008;118:2609e19.
[65] Cappuzzo F, Toschi L, Tallini G, Ceresoli GL, Domenichini I, Bartolini S, et al. Insulin-like growth factor receptor 1 (IGFR-1) is significantly associated with longer survival in
non-small-cell lung cancer patients treated with gefitinib. Ann Oncol 2006;17:1120e7.
[66] Das AK, Chen BP, Story MD, Sato M, Minna JD, Chen DJ, et al. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR-mediated radioprotection in non-small cell lung carcinoma. Cancer Res 2007;67:5267e74.
[67] Huang WC, Chen WS, Chen YJ, Wang LY, Hsu SC. Hepatitis B virus X protein induces IKKalpha nuclear translocation via Akt-dependent phosphorylation to promote the motility of hepatocarcinoma cells. J Cell Physiol; 2011. doi:10.1002/jcp. 22860.
[68] Geetha T, Kenchappa RS, Wooten MW, Carter BD. TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor 1. EMBO J 2005;24:3859e68. [69] Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH. C-terminal ubiquitination of p53 contributes to nuclear export 2. Mol Cell Biol 2001;21:8521e32.
[70] Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling 2. Cell 2006;125:665e77. [71] Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J,
Yang H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression 1. Cell 2007;128:141e56.
[72] Dittmann KH, Mayer C, Ohneseit PA, Raju U,
Andratschke NH, Milas L, et al. Celecoxib induced tumor cell radiosensitization by inhibiting radiation induced nuclear EGFR transport and DNA-repair: a COX-2 independent mechanism. Int J Radiat Oncol Biol Phys 2008;70:203e12. [73] Kucab JE, Lee C, Chen CS, Zhu J, Gilks CB, Cheang M, et al.
Celecoxib analogues disrupt Akt signaling, which is commonly activated in primary breast tumours. Breast Cancer Res 2005;7:R796e807.
[74] Kuo MT. Roles of multidrug resistance genes in breast cancer chemoresistance. Adv Exp Med Biol 2007;608:23e30. [75] Takara K, Sakaeda T, Okumura K. An update on overcoming
MDR1-mediated multidrug resistance in cancer chemotherapy. Curr Pharm Des 2006;12:273e86.
[76] Yoh K, Ishii G, Yokose T, Minegishi Y, Tsuta K, Goto K, et al. Breast cancer resistance protein impacts clinical outcome in platinum-based chemotherapy for advanced non-small cell lung cancer. Clin Cancer Res 2004;10:1691e7.
[77] Usuda J, Ohira T, Suga Y, Oikawa T, Ichinose S, Inoue T, et al. Breast cancer resistance protein (BCRP) affected acquired resistance to gefitinib in a "never-smoked" female patient with advanced non-small cell lung cancer. Lung Cancer 2007; 58:296e9.
[78] Sugimoto Y, Tsukahara S, Ishikawa E, Mitsuhashi J. Breast cancer resistance protein: molecular target for anticancer drug resistance and pharmacokinetics/pharmacodynamics. Cancer Sci 2005;96:457e65.
[79] Elkind NB, Szentpetery Z, Apati A, Ozvegy-Laczka C, Varady G, Ujhelly O, et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib). Cancer Res 2005;65:1770e7.
[80] Jaganathan S, Yue P, Paladino DC, Bogdanovic J, Huo Q, Turkson J. A functional nuclear epidermal growth factor receptor, SRC and Stat3 heteromeric complex in pancreatic cancer cells. PLoS One 2011;6:e19605.
[81] Zhao H, Lo YH, Ma L, Waltz SE, Gray JK, Hung MC, et al. Targeting tyrosine phosphorylation of PCNA inhibits prostate cancer growth. Mol Cancer Ther 2011; 10:29e36.
[82] Xu S, Kitayama J, Yamashita H, Souma D, Nagawa H. Nuclear translocation of HER-4/c-erbB-4 is significantly correlated with prognosis of esophageal squamous cell carcinoma. J Surg Oncol 2008;97:44e50.
[83] Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G.
Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell 2006; 127:185e97.
[84] Carpenter G. Nuclear localization and possible functions of receptor tyrosine kinases. Curr Opin Cell Biol 2003;15: 143e8.
[85] Ni CY, Murphy MP, Golde TE, Carpenter G. g-Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 2001;294:2179e81.
[86] Srinivasan R, Gillett CE, Barnes DM, Gullick WJ. Nuclear expression of the c-erbB-4/HER-4 growth factor receptor in invasive breast cancers. Cancer Res 2000;60:1483e7. [87] Maher PA. Nuclear translocation of fibroblast growth factor
(FGF) receptors in response to FGF-2. J Cell Biol 1996;134: 529e36.