Activation of p38 mitogen-activated protein kinase by celecoxib oppositely
regulates survivin and gamma-H2AX in human colorectal cancer cells
Po-Wen Hsiao
a, Chia-Ching Chang
b, Huei-Fang Liu
a,
Chuan-Mei Tsai
b, Ted H. Chiu
a, Jui-I Chao
a,⁎
aInstitute of Pharmacology and Toxicology, College of Life Sciences, Tzu Chi University, 701, Section 3, Chung-Yang Road, Hualien 970, Taiwan bDepartment of Biological Science and Technology, National Chiao Tung University, 75 Bo-Ai Street, Hsin-Chu 300, Taiwan
Received 24 January 2007; revised 17 April 2007; accepted 17 April 2007 Available online 27 April 2007
Abstract
Cancer cells express survivin that facilitates tumorigenesis. Celecoxib has been shown to reduce human colorectal cancers. However, the role and
regulation of survivin by celecoxib in colorectal carcinoma cells remain unclear. Treatment with 40–80 μM celecoxib for 24 h induced cytotoxicity
and proliferation inhibition via a concentration-dependent manner in RKO colorectal carcinoma cells. Celecoxib blocked the survivin protein
expression and increased the phosphorylation of H2AX at serine-193 (
γ-H2AX). The survivin gene knockdown by transfection with a survivin
siRNA revealed that the loss of survivin correlated with the expression of
γ-H2AX. Meanwhile, celecoxib increased caspase-3 activation and
apoptosis. Celecoxib activated the phosphorylation of p38 mitogen-activated protein (MAP) kinase. The phosphorylated proteins of p38 MAP kinase
and
γ-H2AX were observed in the apoptotic cells. SB203580, a specific p38 MAP kinase inhibitor, protected the survivin protein expression and
decreased the levels of
γ-H2AX and apoptosis in the celecoxib-exposed cells. The blockade of survivin expression increased the celecoxib-induced
cytotoxicity; conversely, overexpression of survivin by transfection with a survivin-expressing vector raised the cancer cell proliferation and resisted
the celecoxib-induced cell death. Our results provide for the first time that p38 MAP kinase participates in the down-regulation of survivin and
subsequently induces the activation of
γ-H2AX for mediating apoptosis following treatment with celecoxib in human colorectal cancer cells.
© 2007 Elsevier Inc. All rights reserved.
Keywords: Apoptosis; Celecoxib; Survivin; p38 MAP kinase;γ-H2AX; RKO cells
Introduction
Survivin is expressed in various human cancer cells and is
undetectable in most normal adult cells (
Ambrosini et al., 1997;
Li et al., 1998; Chao et al., 2004
). Survivin inhibits apoptosis and
promotes mitosis (
Li et al., 1998; O'Connor et al., 2000; Kuo
et al., 2004
). It has been shown that the anti-apoptotic effect of
survivin is due to inhibition of the activity of caspases in cancer
cells (
Kawamura et al., 2003; Beltrami et al., 2004
). Moreover,
survivin may serve as a radio- and chemo-resistance factor
(
Rodel et al., 2003; Wall et al., 2003
), and has been correlated
with decreased survival, unfavorable prognosis, and accelerated
rates of recurrences in cancer therapy (
Altieri, 2001
).
Colorectal cancer is the most common cancer and is the
leading cause of cancer-related mortality around the world (
Jemal
et al., 2005
). Non-steroidal anti-inflammatory drugs (NSAIDs)
inhibit COX enzymes and may be employed to reduce colorectal
cancer growth (
Steinbach et al., 2000; Keller and Giardiello,
2003; Sinicrope and Gill, 2004
). Celecoxib exhibits high
selectivity for the COX-2 enzyme and exerts anticarcinogenic
and chemopreventive activities (
Steinbach et al., 2000; Keller and
Giardiello, 2003; Kismet et al., 2004
). Moreover, celecoxib has
been approved by the FDA of USA for adjuvant treatment in
patients with familial adenomatous polyposis. Recently,
cele-coxib has been shown to inhibit the survivin protein expression in
human cancer cells (
Lin et al., 2005; Pyrko et al., 2006
). However,
the regulation of survivin by celecoxib is still unclear.
Anticancer agents exert their activities through the induction of
apoptosis or the inhibition of proliferation in cancer cells (
Tamura
et al., 2000; Brantley-Finley et al., 2003
). It has been proved that
p38 MAP kinase is an important signal molecule to regulate
apoptosis in response to various stimuli (
Schwenger et al., 1997;
Takekawa et al., 2000; Kim et al., 2002
). Anticancer drugs can
⁎ Corresponding author. Fax: +886 3 8570813. E-mail address:[email protected](J.-I. Chao).
0041-008X/$ - see front matter © 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.taap.2007.04.007
induce the activation of p38 MAP kinase leading to the induction
of apoptosis in cancer cells (
Li and Bertino, 2002
). Besides, p38
MAP kinase mediating apoptosis is associated with the activation
of caspases (
Kim et al., 2002; Li and Bertino, 2002
). Nevertheless,
the control of survivin expression by p38 MAP kinase in the
celecoxib-induced apoptosis has not been studied.
H2AX is a variant of H2A of histones to maintain genomic
stability (
Paull et al., 2000; Redon et al., 2002
). The
phos-phorylation of H2AX at serine-193, named
γ-H2AX, can be
induced by DNA damage agents (
Paull et al., 2000; Redon et al.,
2002
). Moreover, the presentation of
γ-H2AX phosphorylation
has been shown related to caspase-controlled DNA
fragmenta-tion in the process of apoptosis (
Rogakou et al., 2000
).
In this study, a small interfering RNA (siRNA) and a
survivin-expressed vector were employed to illustrate the role of survivin
in the celecoxib-induced apoptosis. Intriguingly, the blockade of
survivin increased the protein level of
γ-H2AX. Furthermore, p38
MAP kinase participated in the down-regulation of survivin with
subsequent induction of
γ-H2AX and apoptosis after treatment
with celecoxib.
Experimental procedures
Materials. Celecoxib was purchased from Toronto Research Chemical Inc. (North York, Canada) and dissolved in 80% ethanol. The concentration of ethanol wasb0.4% in the control and drug-containing media. Hoechst 33258, propidium iodide (PI), 3-(4,5-dimethyl-thiazol-2-yl) 2,5-diphenyl tetrazolium bromide (MTT), and the Cy3-labeled mouse anti-β-tubulin (c-4585) were purchased from Sigma Chemical Co. (St. Louis, MO). BODIPY FL phallacidin (B-607) was purchased from Invitrogen (Carlsbad, CA). Anti-ATF-2 (F2BR-1), anti-ERK-2 (C-14), anti-GFP (FL), anti-p38 (C-20), anti-survivin (FL-142), and the FITC (fluorescein isothiocyanate)-labeled goat anti-mouse IgG (sc-2010) and donkey anti-goat IgG (sc-2024) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-caspase-3 (3004-100) was purchased from BioVision Research Products (Mountain View, CA). The Cy5-labeled goat anti-rabbit IgG was purchased from Amersham Pharmacia Biotech (Little Chalfont Buckinghamshire, UK). Anti-phospho-ATF-2 (Thr-71) (#9221), anti-phospho-p38 (#9211), SignalSilence™ survivin siRNA (#6351), and SignalSi-lence™ control siRNA (#6201) were purchased from Cell Signaling Technology, Inc. (Beverly, MA). SB203580 was purchased from Calbiochem (San Diego, CA). Anti-phospho-Histone H2A.X (Ser139) (05-636) was purchased from Upstate (Lake Placid, NY).
Cell culture. RKO was a colorectal carcinoma cell line that expressed the wild-type p53 proteins. These cells were maintained in DMEM medium (Invitrogen). The complete medium was supplemented with 10% fetal bovine serum (FBS). The cells were cultured at 37 °C and 5% CO2in a humidified
incubator (310/Thermo, Forma Scientific, Inc., Marietta, OH).
Cytotoxicity assay. The cells were plated in 96-well plates at a density of 1 × 104 cells/well for 16–20 h. Then the cells were treated with 0–100 μM celecoxib for 24 h. At the end of treatment, the cells were washed with phosphate-buffered saline (PBS), and were re-cultured in complete medium for 2 days. Thereafter, the medium was replaced and the cells were incubated with 0.5 mg/ml of MTT in complete medium for 4 h. Finally, the cells were dissolved in dimethyl sulfoxide, and the intensity of formazan was measured at 545 nm using a plate reader (Molecular Dynamics, OPTImax). The relative percentage of cell viability was calculated by dividing the absorbance of treated cells by that of the control.
Cell cycle analysis. The cell cycle progression after treatment with cele-coxib was measured by flow cytometer. The cells were plated at a density of 1 × 106 cells per 60-mm Petri dish in complete medium for 16–20 h. After
drug treatment, the cells were collected and fixed with ice-cold 70% ethanol
overnight at−20 °C. The cell pellets were treated with 4 μg/ml PI solution containing 1% Triton X-100 and 100μg/ml RNase for 30 min. To avoid cell aggregation, the cell solutions were filtrated through nylon membrane (Becton-Dickinson, San Jose, CA). Subsequently, the samples were analyzed by flow cytometer. A minimum of ten thousand cells was analyzed for DNA content, and the percentage of cell cycle phases was quantified by a ModFit LT software (Ver. 2.0, Becton-Dickinson).
Apoptosis analysis. The sub-G1 fractions were determined by flow
cytometer same as above for cell cycle analysis. The percentage of sub-G1
fractions was quantified by using CellQuest software (Becton-Dickinson). To further confirm the level of apoptotic cells, the cells were cultured on coverslip in a 60-mm Petri dish for 16–20 h before treatment. At the end of treatment, the cells were carefully and gently washed with isotonic PBS (pH 7.4), and fixed with 4% paraformaldehyde solution in PBS for 1 h at 37 °C. The nuclei were stained with 2.5μg/ml Hoechst 33258 for 30 min. The cell morphology of apoptosis was confirmed by observation of cell membrane blebbing and formation of apoptotic bodies under a fluorescence microscope. The number of apoptotic nuclei was counted by a hemocytometer. At least 500 cells were examined for the calculation of apoptotic percentage in each treatment.
Cell number assay. To evaluate the effect of celecoxib on cell proliferation, the cells were plated at a density of 1 × 106cells per 60-mm Petri dish in complete medium for 12 h. Then the cells were treated with 0–80 μM celecoxib for 24 h. At the end of treatment, the cells were washed twice with PBS and re-cultured in complete medium for 1–7 days. The total cell number was counted by a hemocytometer.
Immunofluorescence staining and confocal microscopy. To view the localization and expression of proteins after celecoxib treatment, the cells were subjected to immunofluorescence staining and confocal microscopy as described (Chao and Liu, 2006). After fixation with 4% paraformaldehyde solution, the cells were washed three times with PBS, and non-specific binding sites were blocked in PBS containing 10% FBS and 0.3% Triton X-100 for 1 h. Thereafter, the cells were incubated with rabbit anti-survivin (1:50), rabbit anti-phospho-p38 (1:100), or mouse anti-γ-H2AX (1:100) antibodies in PBS containing 10% FBS overnight at 4 °C, and washed three times with 0.3% Triton X-100 in PBS. Then the cells were incubated with goat rabbit Cy5-labeled IgG (1:100), goat anti-mouse FITC-labeled IgG (1:50) in PBS containing 10% FBS for 2.5 h at 37 °C. Theβ-tubulin, F-actin, and nuclei were stained with the Cy3-labeled anti-β-tubulin, BODIPY FL phallacidin, Hoechst 33258, respectively. The samples were immediately examined under a Leica confocal laser scanning microscope (Leica, Wetzlar, Germany).
Western blot. Briefly, proteins were separated on 10–12% sodium dodecyl sulfate–polyacrylamide gels, and transferred electrophoretically onto polyviny-lidene difluoride membranes. The membranes were sequentially hybridized with primary antibody and followed with a horseradish peroxidase-conjugated secon-dary antibody. Thereafter, the protein bands were visualized on the X-ray film using the enhanced chemiluminescence detection system (PerkinElmer Life and Analytical Sciences, Boston, MA). A gel-digitizing software, Un-Scan-It gel (ver. 5.1; Silk Scientific, Inc., Orem, UT), was used to quantify the relative intensity.
Construction of a green fluorescence protein (GFP)–survivin fusion vector. The full-length human survivin cDNA was amplified by polymerase chain reaction by a pair of primer (forward: 5′-GGCCATATGGGTGCCCCGA-3′ and reverse: 5′-GATCCATGGCAGCCAGCT-3′). The survivin cDNA fragment was cloned into a CT–GFP TOPO vector by using a CT–GFP fusion TOPO expression kit (K4820-101, Invitrogen) according to the manufacturer's recommendations. The Escherichia coli strain (BL21 DE3) was transformed with the control or survivin expression vectors for vector amplification. The successful clones of survivin-expressed vectors in E. coli were confirmed by survivin DNA sequencing. One of the successful survivin-expressed vectors was named pCT–GPF–sur8 and the control vector pCT–GFP2.
Transfection. Control siRNA, survivin siRNA, pCT–GFP2 and pCT–GFP– sur8 were employed for transfection using Lipofectamine™ 2000 (Invitrogen)
according to the manufacturer's recommendations. After transfection, the cells were subjected to cytotoxicity, cell number, or Western blot analysis as described above.
Statistical analysis. Data were analyzed by one-way or two-way analysis of variance (ANOVA), and further post-hoc tests using the statistic software of GraphPad Prism 4 (GraphPad software, Inc. San Diego, CA). Differences between control and celecoxib-treated samples were compared by one-way ANOVA with post-hoc Tukey's tests. Differences among control, siRNA, vector, or inhibitor after celecoxib treatments were compared by two-way ANOVA with Bonferroni post-tests. A p value of b0.05 was considered statistically significant.
Results
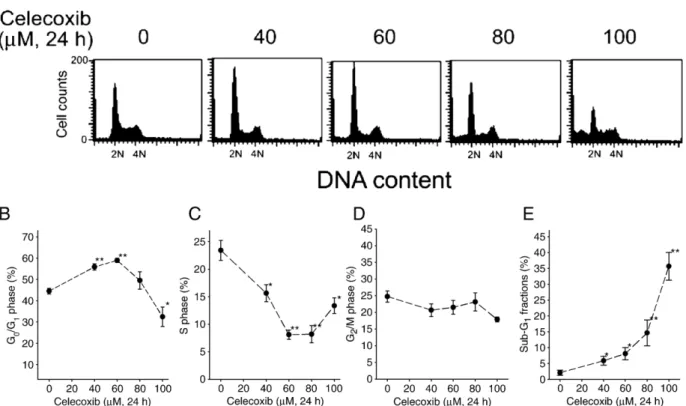
Celecoxib induces both apoptosis and growth arrest in
colorectal carcinoma cells
The effects of celecoxib on the cell cycle progression in
human RKO colorectal cancer cells were examined by flow
cytometer (
Fig. 1
A). The G
1phase of RKO cells was elevated
by 40
–60 μM but was reduced by 100 μM celecoxib; however,
the fraction of S phase was reduced by celecoxib in RKO cells
(
Fig. 1
C). Celecoxib did not significantly alter the G
2/M
frac-tions (
Fig. 1
D). Meanwhile, celecoxib
concentration-depen-dently increased the fraction of sub-G
1indicating apoptotic
induction (
Fig. 1
E). The percentage of apoptotic cells was
counted by nuclear staining under a fluorescence microscope.
Treatment with 60
μM celecoxib for 24 h increased apoptosis
by about 10% in RKO cells (data not shown). Furthermore, the
total cell number was inhibited by celecoxib via a
concentra-tion-dependent manner in RKO cells (
Fig. 2
).
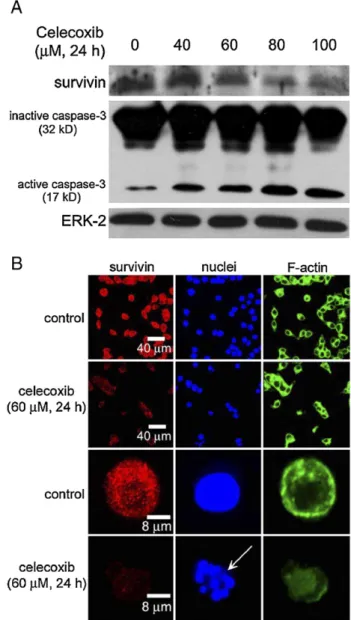
Celecoxib inhibits the survivin protein expression and elevates
the caspase-3 activation
As shown in
Fig. 3
A, treatment with 40–100 μM celecoxib
for 24 h significantly reduced the survivin protein expression in
RKO cells. In contrast, the active forms of caspase-3 (17 kDa)
were induced by celecoxib treatment (
Fig. 3
A). ERK-2 protein
was used as an internal control in this study, which was not
altered by celecoxib. The red fluorescence intensity exhibited
by survivin in RKO cells was significantly decreased when
exposed to celecoxib (
Fig. 3
B). The arrow indicated an
apoptotic cell with the disappearance of survivin proteins
(
Fig. 3
B, arrow). Moreover, the cytoskeleton of F-actin was
disrupted in the celecoxib-induced apoptotic cell.
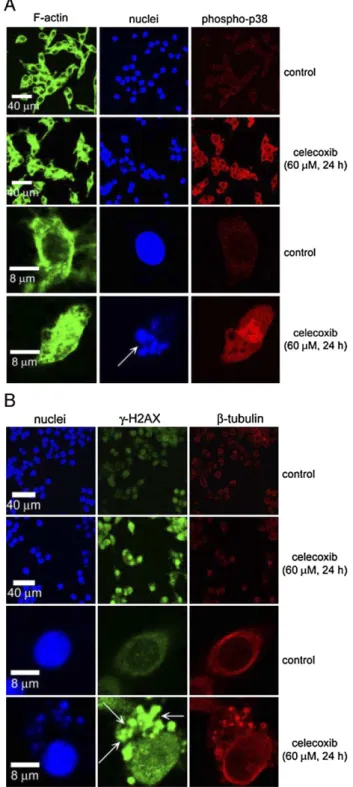
Induction of
γ-H2AX is accompanied by the phosphorylation of
p38 MAP kinase in the celecoxib-exposed cells
We have investigated the phosphorylation of p38 MAP
kinase and
γ-H2AX after treatment with celecoxib in human
colorectal carcinoma cells. Treatment with celecoxib (40–100 μM
for 24 h) increased the phosphorylated proteins of p38 MAP
kinase in RKO cells (
Fig. 4
). The level of
γ-H2AX proteins was
Fig. 1. The effect of celecoxib on the cell cycle progression and apoptosis. (A) RKO cells were treated with 0–100 μM celecoxib for 24 h. At the end of treatment, the cells were trypsinized and then subjected to flow cytometry analyses. (B–E) The populations of G1, S, G2/M, and sub-G1cells were quantified. Results were obtained
increased subsequent to the activation of p38 MAP kinase in the
celecoxib-treated cells (
Fig. 4
).
Inhibition of p38 MAP kinase rescues the survivin protein
expression and reduces cytotoxicity,
γ-H2AX, and active
caspase-3 in the celecoxib-exposed cells
To examine the protein expression and location of
phospho-p38 and
γ-H2AX following celecoxib treatment, the colorectal
carcinoma cells were subjected to immunofluorescence
stain-ing and confocal microscopy. The red fluorescence (Cy5)
intensity exhibited by phospho-p38 proteins was increased and
presented on the apoptotic cells by exposure to 60
μM
celecoxib for 24 h in RKO cells (
Fig. 5
A). The
γ-H2AX proteins
were highly elevated and located on the apoptotic nuclei (
Fig.
5
B, arrows).
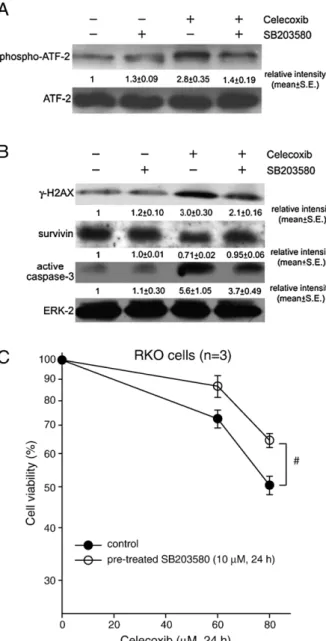
We have further analyzed a well-known substrate of p38
MAP kinase, ATF-2, after treatment with celecoxib in colon
cancer cells. The total protein level of ATF-2 was not altered by
celecoxib (
Fig. 6
A). However, celecoxib increased the
phos-pho-ATF-2 proteins in RKO cells by approximately 3-fold
(
Fig. 6
A). The level of phospho-ATF-2 was reduced after
pre-treatment with the specific p38 MAP kinase inhibitor,
SB203580, in the celecoxib-exposed cells (
Fig. 6
A). Treatment
with SB203580 reversed the survivin protein expression and
simultaneously reduced the protein levels of
γ-H2AX and
active caspase-3 in the celecoxib-treated cells (
Fig. 6
B). The
p values of phospho-ATF-2,
γ-H2AX, survivin, and active
caspase-3 were
b0.05, which indicated significant differences
between celecoxib alone and pre-treatment with SB203580
(
Fig. 6
B). SB203580 alone did not obviously alter the protein
levels of survivin,
γ-H2AX, and active caspase-3 in RKO cells
(
Fig. 6
B). Furthermore, SB203580 significantly reduced the
celecoxib-induced cell death in RKO cells (
Fig. 6
C).
Survivin gene knockdown increases the level of
γ-H2AX and
additively elevates the celecoxib-induced cytotoxicity
To examine the blockade of survivin expression on the
regu-lation of celecoxib-induced apoptosis, the cells were transfected
with a survivin siRNA. As shown in
Fig. 7
A, the levels of
survivin proteins were diminished when RKO cells were
trans-fected with 50 nM survivin siRNA. Concomitantly, the
γ-H2AX
proteins were elevated by transfection with survivin siRNA in
Fig. 2. The effect of celecoxib on the total cell number. RKO cells were plated at a density of 1 × 106cells/p100 Petri dish for 12 h. Then the cells were treated
with 0–80 μM celecoxib for 24 h. After drug treatment, the cells were washed with PBS, and incubated for various times before they were counted by a hemocytometer. Results were obtained from 4 experiments and the bar represents the mean ± S.E. pb0.05 (*) and pb0.01 (**), indicate between control and celecoxib treated samples.
Fig. 3. The effect of celecoxib on the survivin protein expression and the activation of caspase-3. (A) RKO cells were treated with 0–100 μM celecoxib for 24 h. The total protein extracts were subjected to Western blot analysis. Representative immunoblot data were shown from 1 of 3 separate experiments with similar findings. (B) RKO cells were treated with or without 60μM celecoxib for 24 h. The cells were incubated with rabbit anti-survivin and then incubated with goat anti-rabbit Cy5. The F-actin, and nuclei were stained with BODIPY FL phallacidin and Hoechst 33258, respectively. The arrow indicated apoptotic nuclei.
RKO cells (
Fig. 7
A). The p values of survivin and
γ-H2AX were
b0.05, which indicated significant difference between
transfec-tion with control and survivin siRNA (
Fig. 7
A). As shown in
Fig. 7
B, celecoxib (60
μM for 24 h) significantly reduced the cell
viability in RKO cells. Transfection of survivin siRNA (50 nM
for 48 h) increased cell death by about 20% when compared to
control siRNA in RKO cells (
Fig. 7
B). Furthermore, transfection
with survivin siRNA additively elevated the celecoxib-induced
cytotoxicity (
Fig. 7
B).
Overexpression of survivin increases the cell proliferation and
resists the celecoxib-induced cell death in colorectal carcinoma
cells
To further determine the role of survivin in regulating the cell
proliferation and the celecoxib-induced apoptosis, we have
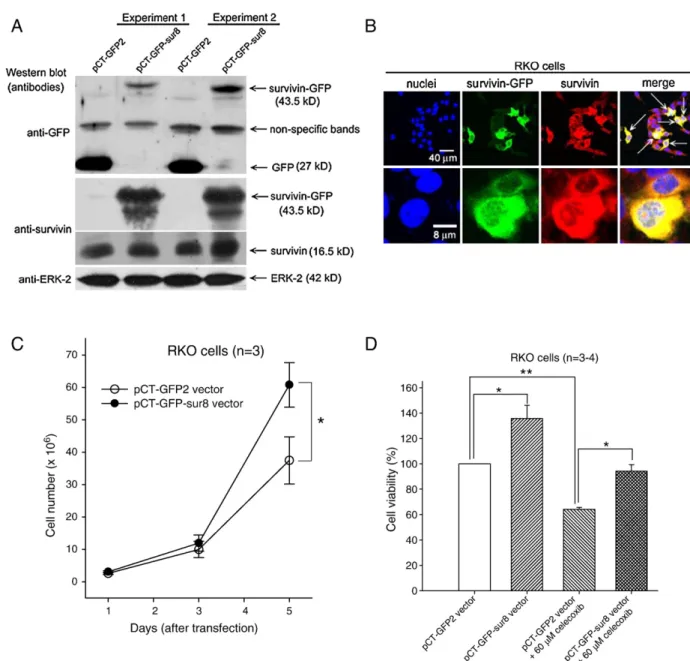
constructed a survivin-expressed vector (pCT–GFP–sur8).
Immunoblot analysis showed that transfection with pCT
–GFP–
sur8 vector expressed a survivin
–GFP–fusion protein (43.5 kDa)
by using anti-GFP or anti-survivin antibodies in RKO cells
(
Fig. 8
A). The endogenous survivin proteins in RKO cells were
recognized as the 16.5 kDa proteins (
Fig. 8
A). The control pCT–
GFP2 vector expressed the GFP proteins (27 kDa) in these cells
(
Fig. 8
A). Unexpectedly, anti-GFP antibody recognized a
non-specific protein band (
Fig. 8
A). Green fluorescence indicated the
expression of survivin–GFP–fusion proteins after transfection
with pCT–GFP–sur8 vector (
Fig. 8
B). The arrows indicated the
overexpressed survivin proteins in RKO cells (
Fig. 8
B).
Moreover, the total cell number was augmented after transfection
with pCT–GFP–sur8 vector (
Fig. 8
C). Overexpression of
sur-vivin by pCT–GFP–sur8 vector also increased the cell viability in
RKO cells (
Fig. 8
D). Besides, the transfection of pCT–GFP–sur8
vector was more resistant to the celecoxib-induced cell death than
control vector (
Fig. 8
D).
Discussion
In this study, we provide several lines of evidence on the role of
survivin in regulating the celecoxib-induced apoptosis in
colorectal carcinoma cells. The gene knockdown of survivin
expression increased the celecoxib-induced cell death.
Converse-ly, overexpression of survivin elevated cancer cell proliferation
and resisted the celecoxib-induced apoptosis. These findings
illustrate that the suppression of survivin by celecoxib mediates
Fig. 5. The effect of celecoxib on the expression and location of phosphorylated p38 MAP kinase andγ-H2AX. (A) RKO cells were treated with or without 60μM celecoxib for 24 h. At the end of treatment, the cells were incubated with rabbit anti-phospho-p38 antibody and then incubated with goat anti-rabbit Cy5. (B) At the end of treatment, the cells were incubated with mouse anti-γ-H2AX antibody and then incubated with goat anti-mouse FITC. Theβ-tubulin, F-actin, and nuclei were stained with the Cy3-labeled anti-β-tubulin, BODIPY FL phallacidin, Hoechst 33258, respectively. The arrows indicated apoptotic nuclei orγ-H2AX located on nuclei.
Fig. 4. The effect of celecoxib on the phosphorylation of p38 MAP kinase and the expression ofγ-H2AX. RKO cells were treated with 0–100 μM celecoxib for 24 h. The protein levels of phospho-p38, total p38, andγ-H2AX were analyzed by Western blot. Representative immunoblot data were shown from one of 3 separate experiments with similar findings.
the apoptosis and growth arrest in human colorectal cancers.
Therefore, the blockade of survivin by celecoxib provides an
important strategy for cancer therapy.
The phosphorylation of p38 MAP kinase was elicited by
celecoxib in colorectal cancer cells. The p38 MAP kinase
has been associated with the induction of apoptosis in response
to many different cellular stresses (
Schwenger et al., 1997;
Takekawa et al., 2000; Kim et al., 2002
). Furthermore, p38 MAP
kinase-mediated apoptosis is associated with the activation of
caspases (
Kim et al., 2002; Li and Bertino, 2002
). Celecoxib
increased caspase-3 activation and apoptosis. Indeed, celecoxib
has been shown to induce the activation of 3 and
caspase-9 (
Dandekar et al., 2005; Ding et al., 2005
). Our results showed
that the specific p38 MAP kinase inhibitor, SB203580, attenuated
active caspase-3 and cytotoxicity in the celecoxib-exposed cells;
concomitantly, the survivin protein expression was restored.
Accordingly, these data indicate that p38 MAP kinase may
mediate apoptosis by the down-regulation of survivin.
This is the first report that celecoxib induces the
phosphor-ylation of H2AX, and the gene knockdown of survivin also
elevated the
γ-H2AX induction. H2AX is a variant of H2A of
Fig. 7. The effect of the survivin gene knockdown on the protein levels of survivin andγ-H2AX and the celecoxib-induced cytotoxicity. (A) RKO cells were transfected with 50 nM control or survivin siRNA for 48 h. The relative protein intensity under each treatment was from 3 independent experiments. The results represent the mean ± S.E. (B) The cell survival was measured by MTT assay. Results were obtained from 3 experiments and the bar represents the mean ± S.E. pb0.05 (*) and pb0.01 (**) indicate significant difference between control and survivin siRNA or celecoxib treated samples.
Fig. 6. The effect of SB203580 on the levels ofγ-H2AX, survivin, caspase-3, and cytotoxicity in the celecoxib-exposed cells. (A) RKO cells were pre-treated with 10μM SB203580 for 24 h prior to exposure to 60 μM celecoxib for 24 h. The total protein extracts were prepared for immunoblot analysis using anti-phospho-ATF-2 and anti-anti-phospho-ATF-2 antibodies. (B) RKO cells were pre-treated with 10μM SB203580 for 24 h prior to exposure to 60μM celecoxib for 24 h. The total protein extracts were prepared for immunoblot analysis using anti-γ-H2AX, anti-survivin, anti-caspase-3, and anti-ERK-2 antibodies. The relative protein intensity under each treatment was from 3 independent experiments. The results represent the mean ± S.E. (C) RKO cells were pre-treated with 10μM SB203580 for 24 h before exposure to 60–80 μM celecoxib for 24 h. The cell survival was measured by MTT assay. Results were obtained from 3 experiments and the bar represents the mean ± S.E. #pb0.05 indicates significant difference between
histones that functions to maintain genomic stability (
Paull
et al., 2000; Redon et al., 2002
). Phosphorylation of H2AX at
serine-193, named
γ-H2AX, is induced by DNA damage agents
such as irradiation (
Paull et al., 2000; Redon et al., 2002
).
Besides, the
γ-H2AX formation is due to caspase-controlled
DNA fragmentation in the course of apoptosis which facilitates
the packaging of fragmented DNA into apoptotic bodies
(
Rogakou et al., 2000
). We found that the phosphorylated
proteins of p38 MAP kinase and
γ-H2AX were located on the
apoptotic bodies. Moreover, the inhibition of p38 MAP kinase
by SB203580 reduced the level of
γ-H2AX in the
celecoxib-exposed cells. These results suggest that the down-regulation
of survivin by p38 MAP kinase can induce the activation of
γ-H2AX and caspase-3 for apoptotic induction.
It has been shown that survivin protein can be stabilized by
the COX-2 overexpression leading to the reduction of apoptosis
in cancer cells (
Krysan et al., 2004
). However,
Pyrko et al.
(2006)
recently reported that the suppression of survivin by
celecoxib was via a COX-independent pathway. In addition,
celecoxib has been shown to mediate cell cycle arrest and
Fig. 8. The effect of the overexpression of survivin on the cell proliferation and the celecoxib-induced cytotoxicity. (A) RKO cells were transfected with 10μg control vector (pCT–GFP-2) or survivin-expressing vector (pCT–GFP–sur8). The total protein extracts were subjected to Western blot analysis. Representative immunoblot data were shown from duplicate experiments. (B) After transfection, RKO cells were incubated with rabbit anti-survivin and then incubated with goat anti-rabbit Cy5. The survivin protein displayed the red color. The nuclei were stained with Hoechst 33258 that displayed the blue color. The GFP proteins displayed the green fluorescence. The arrows indicated the yellow color for the merged picture presenting with the survivin and GFP at the same location. (C) After transfection, the cells were washed with PBS, and incubated for various times before they were counted by a hemocytometer. (D) After transfection and treatment with celecoxib, the cell viability was measured by MTT assay. Results were obtained from 3 to 4 experiments and the bar represents the mean ± S.E. *pb0.05 indicates significant difference between control and survivin vector or celecoxib treated samples. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
apoptosis via COX-2-independent pathways in human
cholan-giocarcinoma and prostate cancer cells (
Han et al., 2004; Kulp
et al., 2004
). Indeed, the gene knockdown of COX-2 did not
significantly induce the phosphorylation of p38 MAP kinase in
colorectal cancer cells (unpublished data). Therefore, we
sug-gest that the activation of p38 MAP kinase by celecoxib results
from a COX-2-independent pathway. Nevertheless, the
pre-cise mechanisms of COX-2-dependent and -independent
path-ways on apoptosis after treatment with celecoxib need further
investigation.
In conclusion, we propose that survivin plays an important
role in the celecoxib-mediated apoptosis, and p38 MAP kinase
may down-regulate the survivin expression. Understanding the
mechanisms by which these signal molecules mediate the
celecoxib-induced growth arrest and apoptosis in human cancer
cells may provide a novel strategy in cancer therapy.
References
Altieri, D.C., 2001. The molecular basis and potential role of survivin in cancer diagnosis and therapy. Trends Mol. Med. 7, 542–547.
Ambrosini, G., Adida, C., Altieri, D.C., 1997. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 3, 917–921. Beltrami, E., Plescia, J., Wilkinson, J.C., Duckett, C.S., Altieri, D.C., 2004.
Acute ablation of survivin uncovers p53-dependent mitotic checkpoint functions and control of mitochondrial apoptosis. J. Biol. Chem. 279, 2077–2084.
Brantley-Finley, C., Lyle, C.S., Du, L., Goodwin, M.E., Hall, T., Szwedo, D., Kaushal, G.P., Chambers, T.C., 2003. The JNK, ERK and p53 pathways play distinct roles in apoptosis mediated by the antitumor agents vinblastine, doxorubicin, and etoposide. Biochem. Pharmacol. 66, 459–469.
Chao, J.I., Liu, H.F., 2006. The blockage of survivin and securin expression increases the cytochalasin B-induced cell death and growth inhibition in human cancer cells. Mol. Pharmacol. 69, 154–164.
Chao, J.I., Kuo, P.C., Hsu, T.S., 2004. Down-regulation of survivin in nitric oxide-induced cell growth inhibition and apoptosis of the human lung carcinoma cells. J. Biol. Chem. 279, 20267–20276.
Dandekar, D.S., Lopez, M., Carey, R.I., Lokeshwar, B.L., 2005. Cycloox-ygenase-2 inhibitor celecoxib augments chemotherapeutic drug-induced apoptosis by enhancing activation of caspase-3 and -9 in prostate cancer cells. Int. J. Cancer 115, 484–492.
Ding, H., Han, C., Zhu, J., Chen, C.S., D'Ambrosio, S.M., 2005. Celecoxib derivatives induce apoptosis via the disruption of mitochondrial membrane potential and activation of caspase 9. Int. J. Cancer 113, 803–810. Han, C., Leng, J., Demetris, A.J., Wu, T., 2004. Cyclooxygenase-2 promotes
human cholangiocarcinoma growth: evidence for cyclooxygenase-2-inde-pendent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 64, 1369–1376.
Jemal, A., Murray, T., Ward, E., Samuels, A., Tiwari, R.C., Ghafoor, A., Feuer, E.J., Thun, M.J., 2005. Cancer statistics, 2005. CA Cancer J. Clin. 55, 10–30. Kawamura, K., Sato, N., Fukuda, J., Kodama, H., Kumagai, J., Tanikawa, H.,
Shimizu, Y., Tanaka, T., 2003. Survivin acts as an antiapoptotic factor during the development of mouse preimplantation embryos. Dev. Biol. 256, 331–341.
Keller, J.J., Giardiello, F.M., 2003. Chemoprevention strategies using NSAIDs and COX-2 inhibitors. Cancer Biol. Ther. 2, S140–S149.
Kim, S.J., Ju, J.W., Oh, C.D., Yoon, Y.M., Song, W.K., Kim, J.H., Yoo, Y.J., Bang, O.S., Kang, S.S., Chun, J.S., 2002. ERK-1/2 and p38 kinase oppositely regulate nitric oxide-induced apoptosis of chondrocytes in association with p53, caspase-3, and differentiation status. J. Biol. Chem. 277, 1332–1339.
Kismet, K., Akay, M.T., Abbasoglu, O., Ercan, A., 2004. Celecoxib: a potent cyclooxygenase-2 inhibitor in cancer prevention. Cancer Detec. Prev. 28, 127–142.
Krysan, K., Dalwadi, H., Sharma, S., Pold, M., Dubinett, S., 2004. Cyclooxygenase 2-dependent expression of survivin is critical for apoptosis resistance in non-small cell lung cancer. Cancer Res. 64, 6359–6362. Kulp, S.K., Yang, Y.T., Hung, C.C., Chen, K.F., Lai, J.P., Tseng, P.H., Fowble,
J.W., Ward, P.J., Chen, C.S., 2004. 3-Phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Res. 64, 1444–1451. Kuo, P.C., Liu, H.F., Chao, J.I., 2004. Survivin and p53 modulate
quercetin-induced cell growth inhibition and apoptosis in human lung carcinoma cells. J. Biol. Chem. 279, 55875–55885.
Li, W., Bertino, J.R., 2002. Fas-mediated signaling enhances sensitivity of human soft tissue sarcoma cells to anticancer drugs by activation of p38 kinase. Mol. Cancer Ther. 1, 1343–1348.
Li, F., Ambrosini, G., Chu, E.Y., Plescia, J., Tognin, S., Marchisio, P.C., Altieri, D.C., 1998. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 396, 580–584.
Lin, J., Hsiao, P.W., Chiu, T.H., Chao, J.I., 2005. Combination of cyclooxygenase-2 inhibitors and oxaliplatin increases the growth inhibition and death in human colon cancer cells. Biochem. Pharmacol. 70, 658–667. O'Connor, D.S., Grossman, D., Plescia, J., Li, F., Zhang, H., Villa, A., Tognin, S., Marchisio, P.C., Altieri, D.C., 2000. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc. Natl. Acad. Sci. U. S. A. 97, 13103–13107.
Paull, T.T., Rogakou, E.P., Yamazaki, V., Kirchgessner, C.U., Gellert, M., Bonner, W.M., 2000. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895. Pyrko, P., Soriano, N., Kardosh, A., Liu, Y.T., Uddin, J., Petasis, N.A., Hofman,
F.M., Chen, C.S., Chen, T.C., Schonthal, A.H., 2006. Downregulation of survivin expression and concomitant induction of apoptosis by celecoxib and its non-cyclooxygenase-2-inhibitory analog, dimethyl-celecoxib (DMC), in tumor cells in vitro and in vivo. Mol. Cancer 5, 19.
Redon, C., Pilch, D., Rogakou, E., Sedelnikova, O., Newrock, K., Bonner, W., 2002. Histone H2A variants H2AX and H2AZ. Curr. Opin. Genet. Dev. 12, 162–169.
Rodel, C., Haas, J., Groth, A., Grabenbauer, G.G., Sauer, R., Rodel, F., 2003. Spontaneous and radiation-induced apoptosis in colorectal carcinoma cells with different intrinsic radiosensitivities: survivin as a radioresistance factor. Int. J. Radiat. Oncol. Biol. Phys. 55, 1341–1347.
Rogakou, E.P., Nieves-Neira, W., Boon, C., Pommier, Y., Bonner, W.M., 2000. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 275, 9390–9395. Schwenger, P., Bellosta, P., Vietor, I., Basilico, C., Skolnik, E.Y., Vilcek, J., 1997.
Sodium salicylate induces apoptosis via p38 mitogen-activated protein kinase but inhibits tumor necrosis factor-induced c-Jun N-terminal kinase/ stress-activated protein kinase activation. Proc. Natl. Acad. Sci. U. S. A. 94, 2869–2873.
Sinicrope, F.A., Gill, S., 2004. Role of cyclooxygenase-2 in colorectal cancer. Cancer Metastasis Rev. 23, 63–75.
Steinbach, G., Lynch, P.M., Phillips, R.K., Wallace, M.H., Hawk, E., Gordon, G.B., Wakabayashi, N., Saunders, B., Shen, Y., Fujimura, T., Su, L.K., Levin, B., 2000. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N. Engl. J. Med. 342, 1946–1952.
Takekawa, M., Adachi, M., Nakahata, A., Nakayama, I., Itoh, F., Tsukuda, H., Taya, Y., Imai, K., 2000. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 19, 6517–6526.
Tamura, K., Southwick, E.C., Kerns, J., Rosi, K., Carr, B.I., Wilcox, C., Lazo, J.S., 2000. Cdc25 inhibition and cell cycle arrest by a synthetic thioalkyl vitamin K analogue. Cancer Res. 60, 1317–1325.
Wall, N.R., O'Connor, D.S., Plescia, J., Pommier, Y., Altieri, D.C., 2003. Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 63, 230–235.