On: 24 December 2014, At: 18:59 Publisher: Taylor & Francis
Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
Click for updates
Liquid Crystals
Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/tlct20
Synthesis of fluorinated terphenyl liquid crystals with
3-propylcyclopentane end group
Tien-Hsin Leea & Chain-Shu Hsua
a
Department of Applied Chemistry, National Chiao-Tung University, Hsinchu, Taiwan Published online: 01 May 2014.
To cite this article: Tien-Hsin Lee & Chain-Shu Hsu (2014) Synthesis of fluorinated terphenyl liquid crystals with
3-propylcyclopentane end group, Liquid Crystals, 41:9, 1235-1245, DOI: 10.1080/02678292.2014.913720
To link to this article: http://dx.doi.org/10.1080/02678292.2014.913720
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) contained in the publications on our platform. However, Taylor & Francis, our agents, and our licensors make no
representations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of the Content. Any opinions and views expressed in this publication are the opinions and views of the authors, and are not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon and should be independently verified with primary sources of information. Taylor and Francis shall not be liable for any losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoever or howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use of the Content.
This article may be used for research, teaching, and private study purposes. Any substantial or systematic reproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in any
form to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http:// www.tandfonline.com/page/terms-and-conditions
Synthesis of fluorinated terphenyl liquid crystals with 3-propylcyclopentane end group
Tien-Hsin Lee and Chain-Shu Hsu*Department of Applied Chemistry, National Chiao-Tung University, Hsinchu, Taiwan (Received 11 February 2014; accepted 7 April 2014)
High performance liquid crystal (LC) displays require nematic mixtures with low rotational viscosity to reduce the response time and large dielectric anisotropy to reduce the threshold voltage. Furthermore, liquid crystals (LCs) with low melting temperature and high clearing temperature are particularly attractive for automobile and outdoor display applications. In this article, we report a series of new fluorinated terphenyl LCs with 3-alkylcyclopentane end group. The terphenyl mesogen is designed for high Δn value, and the lateral fluorine atom is used to reduce the melting temperature. In comparison with the terphenyl analogue containing a 4-alkylcyclohexyl end group, discussed in previous works, the obtained LCs show wider nematic temperature range and smaller rotational viscosity because 3-alkylcyclopentyl moiety is introduced. The effect of alkyl chain length and the lateral fluoro substituent on the mesomorphic properties of the obtained LC is discussed. The results show that the nematic temperature range increases with the alkyl chain length increase. The position of the lateral fluoro substituent also plays an important role; LC with the fluoro substitution on the central benzene ring provides the widest nematic temperature range. In conclusion, we have synthesised a new series of LCs with low melting point, wider nematic temperature range and highΔn value, which is promising in formulating eutectic mixture for display application.
Keywords: terphenyl liquid crystals; cyclopentyl end group; fluoro substituent; birefringence
1. Introduction
Wide mesophase range nematic liquid crystal (LC) with high birefringence (Δn) and low rotational viscosity are attractive materials for applications in reflective LC displays,[1,2] infrared spatial light modulators,[3] polymer-dispersed liquid crystals,[4] cholesteric LCDs,[5] holographic switching devices,[6] polarisers and directional reflectors [7] and laser beam steering.[8] High-Δn LCs can be achieved effectively by extending the conjugated π-bonding length of the molecule [9] by introducing multiple bonds or unsaturated (phenyl) rings,[10] such as diphenyldiacetylene,[11–13] tolane,[14] bis-tolane,[15–20] naphthalene tolanes,[21] thiophenyl-diacetylene [22,23] and thiophenyl-diacetylene systems.[24] However, the LC compounds containing highly conjugated double-bond and triple-bond units suffer from photo and thermal stabilities.[25] Therefore, phenyl ring serves as a more suitable building block for the highly conjugated for LCs for display application. The terphenyl mesogen aids in increas-ing Δn values because of its long molecular conju-gation length. Nevertheless, as the molecular conjugation length increases, the melting point of the material also increases. Moreover, high birefrin-gence LCs often exhibit high viscosity due to the increase of the moment of inertia.[1]
How to lower the melting temperatures and visc-osities of these compounds is a major challenge. One approach to lower the melting point of a highΔn liquid crystal is to introduce a lateral alkyl group [1,15,26–29] or fluorine atom [25,26,28–30] onto its mesogenic core. We chose fluorine atom as the lateral substituent in this series because it not only lowers the melting point but also decreases the smectic tendency of LCs.[31] The other approach to lower the viscosity is to replace the flexible alkyl end group with 4-alkylcyclopentyl group on the mesogen. 4-Alkylcyclohexane end group has been proved to provide various impressive liquid crystalline properties such as low viscosity, good solubility and wide mesophase range.[32,33] Compared to abundant researches concerning cyclo-hexane, cyclopentane relatively remains unexplored.
There are several examples in the literature [33– 36] to replace the cyclohexyl terminal group with cyclopentyl group. The obtained LCs show much lower melting temperatures and viscosities. In this article, we report the synthesis of fluorinated terphe-nyl LCs with 3-alkylcyclopantyl terminal group. The mesomorphic properties as well as optical anisotro-pies of the synthesised LCs are discussed. The general structure of the compounds is given in Figure 1. In this study, n was limited to 2–5 for low viscosity consideration.
*Corresponding author. Email:[email protected]
–1245, http://dx.doi.org/10.1080/02678292.2014.913720
© 2014 Taylor & Francis
2. Experimental section
2.1 General measurement and characterisation All chemicals were purchased from Aldrich or Acros and used as such unless otherwise specified.
1
H NMR spectra were measured using a Varian 300-MHz instrument spectrometer. Thermal
transitions and thermodynamic parameters were measured by a TA Q200 differential scanning calorimeter (DSC), and thermogravimetric analysis was recorded on a Perkin Elmer Pyris under nitro-gen atmosphere at a heating rate of 10°C min−1. A Carl Zeiss Axiophot polarising optical microscope (POM) equipped with a Mettler FP 82 hot stage and a FP 80 central processor was used to observe the thermal transitions and analyse the anisotropic textures.
2.2 Synthetic procedures
Scheme 1outlines the synthesis of 4-(4-propylphenyl)-4′-(3-propylcyclopentyl)-biphenyl (4) and 4-(4- alkylphenyl)-2-fluoro-4′-(3-propylcyclopentyl)-biphe-nyl (5a–5d), and Scheme 2 outlines the synthesis of
n=2–5; H2n+2Cn
A C
B D
A, B, C and D=H, F
Figure 1. Chemical structure of fluorinated terphenyl liquid crystals.
Scheme 1. The synthesis of compounds 4 (X = H, n = 3) and 5a–5d (X = F, n = 2–5).
Scheme 2. The synthesis of compounds 10, 11 and 12.
4-(4-propylphenyl)-2,5-difluoro-4 ′-(3-propylcyclopen-tyl)biphenyl (10), 4-(4-propylphenyl)-2′-fluoro-4′-(3-propylcyclopentyl)biphenyl (11) and 4-(4-propylphe-nyl)-2′,6′-difluoro-4′-(3-propylcyclopentyl)biphenyl (12). 2.2.1 (3-propylcyclopent-1-enyl)benzene (1a) and (4-propylcyclopent-1-enyl)benzene (1b)
Bromobenzene (3.73 g, 23.7 mmol) was dissolved in dry tetrahydrofuran (THF) and cooled to −78°C. A measure of 2.5 M n-butyl lithium (12.36 mL) was added to the mixture and stirred for 1 h at−78°C. 3-Propylcyclopentanone (3 g, 23.7 mmol) was then added to the reaction mixture and stirred for 1 h. The mixture was allowed to stir at room temperature for another hour. After the solvent was removed, the resi-due was washed twice with brine. The organic layer was dried with anhydrous magnesium sulphate and then the solvent was removed to obtain the crude product. Without further purification, the crude pro-duct was washed twice with brine and twice with 1 M hydrochloric acid (10 mL). The organic layer was dried with anhydrous magnesium sulphate. After the solvent was removed, the crude product was purified by col-umn chromatography (silica gel, hexane as eluent) to yield 3.0 g (67%) of a white solid. The product was a mixture of compounds 1a and 1b. 1H NMR(CDCl3,
400 MHz, ppm): δ 0.94 (t, J = 6.8 Hz, 3H, –CH3),
1.30–1.62 (m, 4H, –CH2CH2–), 2.15–2.88 (m, 5H, C3–
H, C4–H, C5–H), 6.12 (t, J = 2 Hz, –C = CH– of 1b),
6.15 (d, J = 2 Hz,–C = CH– of 1a), 7.21 (t, J = 7.4 Hz, 1H, aromatic proton), 7.29 (t, J = 10 Hz, 2H, aromatic protons), 7.42–7.45 (m. 2H, aromatic protons). MS (EI, C14H18): calcd 186.14; found 186.30. HRMS (EI,
C14H18): calcd 186.1409; found 186.1405.
2.2.2 1-(3-propylcyclopentyl)benzene (2)
A mixture of (3-propylcyclopent-1-enyl)benzene (1a) and (4-propylcyclopent-1-enyl)benzene (1b) (3.00 g, 16.10 mmol) was dissolved in a mixed solvent of methanol (50 mL) and toluene (20 mL). A measure of 10% Pd/C (30 mg) was then added. The mixture was stirred for 16 h at room tempera-ture under H2atmosphere. The mixture was filtered
to remove Pd/C. The solvent was removed under reduced pressure to obtain a colourless liquid (2.80 g) (92%). 1H NMR(CDCl3, 400 MHz, ppm):
δ 0.91 (t, J = 6.8 Hz, 3H, –CH3), 1.28–1.43 (m, 5H,
–CH2CH2CH–), 1.56–2.22 (m, 6H, C2–H, C4–H,
C5–H), 2.98–3.12 (m, 1H, Ph–CH–), 7.14–7.18 (m,
1H, aromatic proton), 7.23–7.30 (m, 4H, aromatic protons). 13C NMR(CDCl3, 100 MHz, ppm): δ
14.35, 21.63, 21.72, 31.93, 33.36, 33.45, 35.12, 38.85, 39.03, 39.28, 39.92, 40.51, 42.26, 44.47,
45.85, 125.63, 127.00, 128.19, 146.49. MS (EI, C14H20): calcd, 188.16; found, 186.2. HRMS (EI,
C14H20): calcd, 188.1565; found, 186.1560.
2.2.3 1-iodo-4-(3-propylcyclopentyl)benzene (3) 1-(3-Propylcyclopentyl)benzene (2) (2.80 g, 14.86 mmol) was dissolved in acetic acid (50 mL). A mix-ture of iodine (1.88 g, 7.40 mmol), iodic acid (0.88 g, 5.00 mmol), sulphuric acid (5 mL) and distilled water (10 mL) was added. The obtained solution was heated to 90°C and stirred for 16 h. After the mixture cooled to room temperature, 1 M sodium thiosulphate was added. Then, the solvent was removed and the residue was washed twice with brine and twice with distilled water. The organic layer was dried with anhydrous magnesium sulphate and then the solvent was removed to obtain the crude product. Further purifi-cation was performed using column chromatography to obtain a colourless liquid (4.50 g) (96%).1H NMR (CDCl3, 300 MHz, ppm):δ 0.87–0.97 (m, 3H, –CH3),
1.09–1.48 (m, 5H, –CH2CH2CH–), 1.56–2.24 (m, 6H,
C2–H, C4–H, C5–H), 2.89–3.02 (m, 1H, Ph–CH–),
6.89 (d, J = 8.1 Hz, 2H, aromatic protons), 7.57 (d, J = 8.1 Hz, 2H, aromatic protons). MS (EI, C14H19I):
calcd, 314.05; found, 314.2. HRMS (EI, C14H19I):
calcd, 314.0531; found, 314.0533.
2.2.4 4-(4-propylphenyl)-4 ′-(3-propylcyclopentyl)-biphenyl (4), 4-(4-ethyllphenyl)-2-fluoro-4 ′-(3-propylcyclopentyl)biphenyl (5a), 4-(4-propylphenyl)-2-fluoro-4′-(3-propylcyclopentyl)biphenyl (5b), 4-(4-butylphenyl)-2-fluoro-4′-(3-propylcyclopentyl) biphenyl (5c) and 4-(4-pentylphenyl)-2-fluoro-4 ′-(3-propylcyclopentyl)biphenyl (5d)
The methods for preparing compounds 4 and 5a–5d are similar. The synthesis of compound 5a is described as follows. To a 250-mL round-bottom flask, compound 3 (1 g, 3.2 mmol), (4 ′-ethyl-3-fluoro-[1,1′-biphenyl]-4-yl)boronic acid (2PGB(OH)2)
(0.8 g, 3.2 mmol), tetrakis(triphenylphospine)palla-dium(0) (0.1 g, 0.1 mmol), sodium hydroxide (0.4 g, 9.6 mmol), Aliquat 336 (0.1 mL), degassed toluene (50 mL) and degassed H2O (10 mL) were added. The
mixture was heated to 90°C under argon gas for 16 h. After cooling to room temperature, the mixture was washed twice with brine. The organic layer was dried with anhydrous magnesium sulphate. After the sol-vent was removed, the crude product was purified by column chromatography (silica gel, hexane as eluent) to yield 0.83 g (66%) of white crystals. 1H NMR (CDCl3, 300 MHz, ppm): δ 0.89–0.94 (m, 3H, –CH3
on 3-propylcyclopentyl), 1.25–1.39 (m, 8H, – CH2CH2CH– on 3-propylcyclopentyl protons and –
CH3on ethyl chain), 1.63–2.25 (m, 6H, cyclopentyl protons), 2.65 (q, J = 7.5 Hz, 2H, –PhCH2–), 3.03– 3.15 (m, 1H,–PhCH–), 7.28–2.54 (m, 11H, aromatic protons). 13C NMR (CDCl3, 100 MHz, ppm): δ 14.37, 15.53, 21.75, 28.53, 31.97, 33.39, 39.09, 39.97, 42.29, 45.65, 114.21, 114.45, 122.65, 122.68, 126.85, 127.19, 128.43, 128.75, 128.78, 130.79, 130.83, 132.92, 141.91, 146.09. MS (EI, C28H31F): calcd,
386.24; found, 386.3. HRMS (EI, C28H31F): calcd
386.2410; found 386.2415.
Compound 4 yielded 36%. 1H NMR (CDCl3,
400 MHz, ppm):δ 0.90–1.03 (m, 6H, –CH3on propyl
terminal chain and–CH3on 3-cyclopentyl), 1.25–1.46
(m, 6H, 3-propylcyclopentyl protons), 1.61–1.79 (m, 3H, 3-propylcyclopentyl proton and–CH2– on propyl
terminal chain), 1.83–2.27 (m, 4H, cyclopentyl pro-tons), 2.64 (t, J = 7.2 Hz, 2H, –PhCH2–), 3.04–3.17
(m, 1H,–PhCH–), 7.26 (d, J = 5.6 Hz, 2H, aromatic protons), 7.32 (d, J = 7.2 Hz, 2H, aromatic protons), 7.55 (d, J = 7.6 Hz, 4H, aromatic protons), 7.64 (s, 4H, aromatic protons). MS (EI, C29H34): calcd,
382.26; found, 382.3.
Compound 5b yielded 66%. 1H NMR (CDCl3,
300 MHz, ppm):δ 0.89–1.00 (m, 6H, –CH3on propyl
terminal chain and –CH3 on 3-propylcyclopentyl),
1.35–1.39 (m, 6H, 3-propylcyclopentyl protons), 1.65–1.72 (m, 3H, 3-propylcyclopentyl protons and –CH2CH3), 1.89–2.25 (m, 4H, 3-propylcyclopentyl protons), 2.64 (t, J = 7.3 Hz, 2H, –PhCH2–), 3.08– 3.11 (m, 1H,–PhCH–), 7.28–7.55 (m, 11H, aromatic protons). 13C NMR (CDCl3, 100 MHz, ppm): δ 13.85, 14.35, 21.73, 24.50, 31.95, 33.37, 37.68, 38.84, 39.95, 42.27, 45.63, 144.19, 114.42, 122.62, 122.65, 126.73, 127.17, 128.74, 128.77, 129.01, 130.76, 130.81, 132.90, 142.50, 146.08. MS (EI, C29H33F):
calcd, 400.26; found, 400.5. HRMS (EI, C29H33F):
calcd 400.2566; found 400.2564.
Compound 5c yielded 36%. 1H NMR (CDCl3,
400 MHz, ppm):δ 0.88–0.97 (m, 6H, –CH3on butyl
terminal chain and –CH3 on 3-propylcyclopentyl),
1.21–1.46 (m, 8H, 3-propylcyclopentyl protons), 1.60–2.25 (m, 7H, 3-propylcyclopentyl protons and – CH2CH2–), 2.66 (t, J = 7.2 Hz, 2H, –PhCH2–), 3.03–
3.15 (m, 1H,–PhCH–), 7.28–2.54 (m, 11H, aromatic protons). MS (EI, C30H35F): calcd, 414.27; found,
414.5. HRMS (EI, C30H35F): calcd, 414.2723;
found, 414.2717.
Compound 5d yielded 40%. 1H NMR (CDCl3,
300 MHz, ppm):δ 0.89–0.94 (m, 6H, –CH3on propyl
terminal chain and –CH3 on 3-propylcyclopentyl),
1.25–1.41 (m, 10H, 3-propylcyclopentyl protons –CH2CH2CH3), 1.63–1.75 (m, 3H,
3-propylcyclopen-tyl protons and –CH2CH2CH2CH3), 1.97–2.25
(m, 4H, 3-propylcyclopentyl protons), 2.65 (t, J = 7.4 Hz, 2H, –PhCH3–), 3.03–3.15 (m, 1H,
–PhCH–), 7.28–2.54 (m, 11H, aromatic protons). MS (EI, C31H37F): calcd, 428.29; found, 428. HRMS (EI,
C31H37F): calcd 428.2879; found 428.2884.
2.2.5 (4-propylphenyl)-1,3,2-dioxaborolane (6a) and 4,4,5,5-tetramethyl-2-(4′-propylbiphenyl-4-yl)-1,3,2-dioxaborolane (6b) The methods for preparing compounds 6a and 6b are similar. The synthesis of compound 6b is described as follows. 4-Bromo-4′-propylbiphenyl (10 g, 36.4 mmol) was dissolved in dry THF (75 mL) and cooled to−78° C. To the mixture, 2.5 M n-butyl lithium (18 mL) was added, and stirred for 1 h at −78°C. 2-Isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (10 mL) is added to the reaction mixture and stirred for 1 h. The mixture was stirred at room temperature for another hour, and then the solvent was removed and the residue was washed twice with brine. The organic layer was dried with anhydrous magnesium sulphate. After the solvent was removed, 9.47 g (80%) of a yellow liquid was obtained. 1H–NMR (CDCl3,
300 MHz, ppm): 0.97 (t, J = 7.3 Hz, 3H, –CH3),
1.36 (s, 12H, –BOC(CH3)2C(CH3)2O), 1.64–1.71 (m,
2H, –CH2CH3), 2.62 (t, J = 7.6 Hz, 2H,
–CH2CH2CH3), 7.24 (d, J = 6.6 Hz, 2H, aromatic
protons), 7.53 (d, J = 8.1 Hz, 2H, aromatic protons), 7.59 (d, J = 8.1 Hz, 2H, aromatic protons), 7.86 (d, J = 6.6 Hz, aromatic protons). Compound 6a yielded 98%. 1H NMR (CDCl3, 300 MHz, ppm): δ 0.93 (t, J = 7.3 Hz, 3H, –CH3), 1.33 (s, 12H, –BOC(CH3)2C(CH3)2O), 1.57–1.70 (m, 2H, –CH2CH3), 2.59 (t, J = 7.3 Hz, 2H, –CH2CH2CH3), 7.14 (d, J = 7.9 Hz, 2H, aromatic
protons), 7.73 (d, J = 7.9 Hz, 2H, aromatic protons). MS (EI, C13H23BO2): calcd, 246.18; found, 246.
2.2.6 2,5-difluoro-4-(4′-propylphenyl)aniline (7a), 3,5-difluoro-4-(4′-propylbiphenyl-4-yl)aniline (7b) and 3-fluoro-4-(4′-propylbiphenyl-4-yl)-a niline (7c) The methods for preparing compounds 7a–7c are similar. The synthesis of compound 7c is described as follows. Compound 6b (9.47 g, 29.4 mmol), 4-bromo-3-fluoroaniline (5.58 g, 29.4 mmol), tetrakis (triphenylphospine)palladium(0) (2.3 g, 1.47 mmol), potassium carbonate (12.2 g, 88 mmol), Aliquat 336 (0.5 mL), degassed toluene (80 mL) and degassed H2O (15 mL) were added to a 250-mL flask. The
mixture was heated to 90°C under argon gas for 16 h. After cooling to room temperature, the mixture was washed twice with brine. The organic layer was
dried with anhydrous magnesium sulphate. After the solvent was removed, the crude product was purified by column chromatography (silica gel, hexane as elu-ent) to obtain 7.0 g (78%) of a yellowish solid. Compound 7a yielded 67%. 1H NMR (CDCl3, 300 MHz, ppm): 0.96 (t, J = 7.3 Hz, 3H, –CH3), 1.63–1.73 (m, 2H, –CH2CH3), 2.60 (t, J = 7.6 Hz, 2H, –CH2CH2CH3), 3.83 (s, 2H, –NH2), 6.55 (dd, J1 = 11.4 Hz, J2 = 7.8 Hz, 1H, aromatic proton), 7.06 (dd, J1 = 11.7 Hz, J2 = 6.9 Hz, 1H, aromatic
proton), 7.22 (d, J = 8.1 Hz, 2H, aromatic protons), 7.39 (d, J = 6.6 Hz, 2H, aromatic protons).
Compound 7b yielded 65%. 1H NMR (CDCl3,
300 MHz, ppm): 0.97 (t, J = 7.3 Hz, 3H, –CH3),
1.65–1.72 (m, 2H, –CH2CH3), 2.63 (t, J = 7.6 Hz, 2H,
–CH2CH2CH3), 3.9 (s, 2H,–NH2), 6.29 (d, J = 9.6 Hz,
2H, aromatic protons), 7.26 (d, J = 8.0 Hz, 2H, aro-matic protons), 7.48 (d, J = 8.0 Hz, 2H, aroaro-matic protons), 7.54 (d, J = 8.0 Hz, 2H, aromatic protons), 7.63 (d, J = 8.0 Hz, 2H, aromatic protons); MS (EI, C21H19F2N): calcd, 323.39; found, 323.
2.2.7 2,5-difluoro-4-(4′propylphenyl)iodobenzene (8a) 3,5-difluoro-4-(4′-propylbiphenyl-4-yl)
iodobenzene (8b) and 3-fluoro-4-(4′-propylbiphenyl-4-yl)iodobenzene (8c)
The methods for preparing compounds 8a–8c are similar. The synthesis of compound 8c is described as follows. Compound 7c (3 g, 9.8 mmol) was dis-solved in THF (30 mL) and hydrogen chloride (10 mL) was added. After stirring for 1 h at −5°C, 5.5 M sodium nitrite (5 mL) was added slowly. The mixture was stirred for 1 h at −5°C and 5.5 M potassium iodide (5 mL) was then added and then again stirred for 1 h. The mixture was washed twice with brine and twice with 1 M sodium thiosulphate (30 mL), and the organic layer was dried with anhydrous magnesium sulphate. After the solvent was removed, the crude product was purified by column chromatography (silica gel, hex-ane as eluent) to obtain 1.5 g (50%) of white solid.
1H–NMR (CDCl
3, 300 MHz, ppm): 0.98 (t,
J = 7.3 Hz, 3H, –CH3), 1.65–1.73 (m, 2H. –
CH2–), 2.64 (t, J = 7.4 Hz, 2H, –PhCH2–), 7.14–
7.28 (m, 3H, aromatic protons), 7.41–7.68 (m, 8H, aromatic protons). MS (EI, C21H18FI): calcd,
416.04; found, 416. Compound 8b yielded 43%. 1H NMR (CDCl3, 300 MHz, ppm): 0.98 (t, J = 7.3 Hz, 3H, –CH3), 1.65–1.73 (m, 2H, –CH2–), 2.64 (t, J = 7.6 Hz, 2H, PhCH2–), 7.27 (d, J = 4 Hz, 2H, aromatic protons), 7.38 (d, J = 6.8 Hz, 2H, aromatic protons), 7.49 (d, J = 8.1 Hz, 2H, aromatic protons), 7.55 (d, J = 8.1 Hz, 2H, aromatic protons), 7.67 (d, J = 8.4 Hz, 2H, aro-matic protons); MS (EI, C21H17F2I): calcd, 434.27;
found, 434.
2.2.8 2,5-difluoro-4-(4′-propylphenyl)phenylboronic acid (9)
Compound 8a (6 g, 16.7 mmol) was dissolved in dry THF and cooled to−78°C. To the mixture, 2.5 M n-butyl lithium (8.7 mL, 25.0 mmol) was added and stirred for 1 h at −78°C. Trimethyl borate (2 mL, 20.0 mmol) was added to the reaction mixture and stirred for 15 min. To the mixture, 5% hydrochloric acid (0.5 mL) was added. The mixture was stirred at room temperature for another hour, and then the solvent was removed and the residue was washed twice with brine. The organic layer was dried with anhydrous magnesium sulphate. After the solvent was removed, the crude product was purified by column chromatography (silica gel, hexane as eluent) to obtain 2.3 g (50%) of white solid.1H NMR (CDCl3,
300 MHz, ppm): 0.97(t, J = 7.3 Hz, 3H,–CH3), 1.64–
1.72(m, 2H, –CH2CH3), 2.64 (t, J = 7.3 Hz, 2H, –
CH2CH2CH3), 7.14 (dd, J1 = 4.8 Hz, J2 = 5.7 Hz,
1H, aromatic proton), 7.27 (d, J = 6.5 Hz, 2H, aro-matic protons), 7.48 (d, J = 6.5 Hz, 2H, aroaro-matic protons), 7.56 (dd, J1 = 4.9 Hz, J2 = 5.4 Hz, 1H,
aromatic proton).
2.2.9 4-(4-propylphenyl)-2,5-difluoro-4 ′-(3-propylcyclopentyl)biphenyl (10)
Compounds 3 (1.0 g, 8.0 mmol) and 9 (2.2 g, 8.0 mmol), tetrakis(triphenylphospine)palladium(0) (0.1 g, 0.1 mmol), sodium hydroxide (0.9 g, 24.0 mmol), Aliquat 336 (0.1 mL), degassed toluene (50 mL) and degassed H2O (10 mL) were added to a
250-mL flask. The mixture was heated to 90°C under argon gas for 16 h. After cooling to room tempera-ture, the mixture is washed twice with brine. The organic layer was dried with anhydrous magnesium sulphate. After the solvent was removed, the crude product was purified by column chromatography (silica gel, hexane as eluent) to obtain 1.8 g (54%) of white solid. 1H NMR (CDCl3, 300 MHz, ppm): δ
0.89–1.00 (m, 6H, –CH3 on propyl terminal chain
and –CH3 on 3-propylcyclopentyl), 1.35–1.39 (m,
6H, 3-propylcyclopentyl protons), 1.65–1.72 (m, 3H, 3-propylcyclopentyl protons and –CH2– on propyl
terminal chain), 1.89–2.25 (m, 4H, 3-propylcyclopen-tyl protons), 2.64 (t, J = 7.3 Hz, 2H,–PhCH2–), 3.08–
3.11 (m, 1H, –PhCH–), 7.19–7.35 (m, 6H, aromatic protons), 7.50 (d, J = 7.0 Hz, 4H, aromatic protons). MS (EI, C29H32F2): calcd, 418.25; found, 418.4.
HRMS (EI, C29H32F2): calcd, 418.2472; found, 418.2470. 2.2.10 4-(4-propylphenyl)-2 ′-fluoro-4′-(3-propylcyclopentyl)biphenyl (11) and 4-(4-propylphenyl)-2′,6′-difluoro-4′-(3-propylcyclopentyl) biphenyl (12)
The methods for preparing compounds 11 and 12 are similar. The synthesis of compound 11 is described as follows. Compound 8c (1.5 g, 3.6 mmol) was dissolved in dry THF (30 mL) and cooled to −78°C. A measure of 2.5 M n-butyl lithium (2.6 mL) was added to the mixture and stirred for 1 h at −78°C. 3-Propylcyclopentan-1-one (0.45 mL 4.6 mmol) was added to the reaction mix-ture and stirred for 1 h. The mixmix-ture was stirred at room temperature for another hour. After the sol-vent was removed and the residue was washed twice with brine, the organic layer was dried with anhy-drous magnesium sulphate. After the solvent was removed, the crude product was transferred to a 150-mL round-bottom flask. Potassium bisulphate (0.5 g, 3.6 mmol) was added to the flask, and the mixture was heated to 100°C and stirred for 5 h. The mixture was purified by column chromatography (silica gel, hexane as eluent) to obtain 0.55 g (38%) of white solid. The white solid was dissolved in methanol (10 mL) and toluene (8 mL). A measure of 10% Pd/C (10 mg) was added to the mixture and stirred for 16 h at room temperature under H2
atmo-sphere. The mixture was filtered to remove Pd/C powder. After the solvent was removed, 0.5 g (99%) of white solid was obtained. 1H NMR (CDCl3, 300 MHz, ppm): 0.87–0.98 (m, 6H, –CH3
on 3-propylcyclopentane and –CH3 on propyl
chain), 1.23–1.39 (m, 6H, 3-propylcyclopentane pro-tons), 1.63–2.19 (m, 7H, 3-propylcyclopentane pro-tons and –CH2– on propyl terminal chain), 2.62 (t,
J = 10.4 Hz, 2H,–PhCH2–), 3.72 (m, 1H, –PhCH–),
7.00–7.08 (m, 2H, aromatic protons), 7.22–7.26 (m, 2H, aromatic protons), 7.37 (t, J = 8.1 Hz, 1H, aromatic protons), 7.52–7.64 (m, 4H, aromatic pro-tons). MS (EI, C29H33F): calcd, 400.26; found, 400.
HRMS (EI, C29H32F2): calcd, 400.2566; found,
400.2566.
Compound 12: 1H NMR (CDCl3, 300 MHz, ppm): 0.88–1.00 (m, 6H, –CH3 on 3-propylcyclopentane
and –CH3 on propyl terminal chain), 1.19–1.28 (m,
2H, 3-propylcyclopentane protons), 1.34–1.40 (m, 4H, 3-propylcyclopentane protons), 1.62–1.73 (m, 3H, 3-propylcyclopentane protons and –CH2– on
propyl terminal chain), 1.96–2.11 (m, 3H, 3-propyl-cyclopentane protons), 2.21–2.24 (m, 1H, 3-propylcyclopentane proton), 2.64 (t, J = 7.2 Hz, 2H, –PhCH2–), 2.99–3.11 (m, 1H, –PhCH–), 6.87 (d, J = 8.9 Hz, 2H, aromatic protons), 7.27 (d, J = 6.4 Hz, 2H, aromatic protons), 7.51–7.57 (m, 4H, aromatic protons), 7.66 (d, J = 8.4 Hz, 2H, aromatic protons). MS (EI, C29H32F2): calcd,
418.25; found, 418. HRMS (EI, C29H32F2): calcd,
418.2472; found, 418.2470.
3. Results and discussions
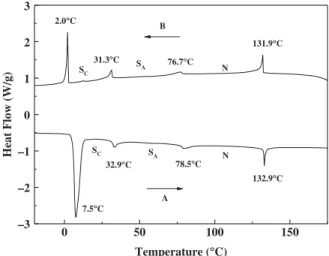
The synthetic procedure for the preparation of compounds 4 and 5a-5d is outlined in Scheme 1. The terphenyl compounds containing 3-propylcy-clopentyl end group were prepared by Suzuki cou-pling of 4-alkyl-2-fluorobiphenyl boronic acid with 1-iodo-4-(4-propylcyclopentyl)benzene. All obtained compounds were purified several times by column chromatography. The thermal and mesomorphic properties of the obtained compounds were char-acterised by DSC and POM. The representative DSC thermograms of compound 5c are presented in Figure 2. Compound 5c exhibits a melting tran-sition at 7.5°C, a smectic C to smectic A trantran-sition at 32.9°C, a smectic A to nematic transition at 79.5°C and a nematic to isotropic phase transition at 132.9°C on the heating scan (curve A). The cooling scan (curve B) looks almost identical to the heating scan, except a very small supercooling (less than 5.5°C) is observed for the first three mesomorphic transitions. The supercooling for the crystallisation transition is larger as expected. Figure 3(a)–3(c) display the typical textures of nematic, smectic A and smectic C exhibited by 5c, respectively. 0 50 100 150 –3 –2 –1 0 1 2 3 SC SC SA SA N 2.0°C 31.3°C 76.7°C 131.9°C 132.9°C 32.9°C 78.5°C A Heat Flow (W/g) Temperature (°C) B 7.5°C N
Figure 2. DSC thermograms of compound 5c: heating scan (curve A) and cooling scan (curve B).
3.1 Effect of terminal alkyl chain
The phase transition temperatures and mesomorphic properties of compounds 5a–5d are listed inTable 1. All of them possess the same mesogenic core except that the left-handed terminal alkyl chains vary from ethyl to pentyl group. Compound 5a, which contains an ethyl end group, reveals an enantiotropic nematic phase. Bearing a propyl terminal alkyl chain, com-pound 5b possesses an enantiotropic nematic phase and two monotropic smectic A and smectic C phases. Compounds 5c and 5d contain a butyl terminal alkyl chain and a pentyl terminal alkyl chain, respectively, and both compounds present enantiotropic nematic, smectic A and smectic C phases. The result demon-strates that the length of terminal alkyl group affects the mesomorphic properties of this series of com-pounds profoundly. Comcom-pounds with longer alkyl chain ends have an obvious tendency to form more ordered smectic phases. In our previous studies, [11,37] we have found that the symmetry of alkyl chain on the both sides of the mesogen has a strong effect in the melting and isotropisation temperatures.
In general, asymmetric compounds generally exhibit much lower melting and isotropisation temperatures than the symmetric analogues. The phenomenon can be observed in these terphenyl compounds as well. Compound 5d which contains symmetric propyl term-inal alkyl chains shows a much higher melting tem-perature and isotropisation temtem-perature than those of compounds 5c and 5d. If one end of the molecule is a considerably short alkyl chain, the phenomenon is not obvious; for instance, the melting temperature of 5a is higher than that of 5b. What is worth noting is the enthalpy change in compounds 5a–5d. Generally, during the transition to isotropic phase, nematic phase exhibits a small enthalpy change that is approximately 1–2 kJ mol−1; however, that of 5b and 5c is obviously larger than expected.[38–40]
3.2 Effect of lateral fluoro substituent
To study the effect of lateral fluoro substituent on the mesomorphic phase behaviour of these terphenyl compounds, compounds 4 and 10–12 were prepared.
(a) (b) (c)
Figure 3. Optical micrographs of (a) nematic texture obtained at 110°C, (b) smectic A texture obtained at 54°C and (c) smectic C texture obtained at 25°C for compound 5c.
Table 1. The phase transitions and corresponding enthalpy changes of compounds 4, 5a–5d and 10–12.
Compound Phase transition temperature (°C) and enthalpy in parenthsis (kJ mol−1)
4 Cr 164.3 (37.3) I I 160.9 (36.8) Cr 5a Cr 50.9 (92.4) N 71.3 (1.1) I I 68.7 (0.8) N 47.4 (1.4) Cr 5b Cr 36.1 (72.9) N 144.3 (4.0) I I 143.0 (7.0) N 18.4 (3.7) SA−2.3 (1.5) SC−13.2 (15.5) Cr 5c Cr 7.5 (45.8) SC32.9 (2.8) SA79.5 (2.4) N 132.9 (4.1) I I 131.9 (5.2) N 76.7 (1.6) SA31.3 (2.4) SC2.0 (8.6) Cr 5d Cr 1.8 (39.7) SC36.6 (4.9) SA68.3 (2.1) N 89.8 (1.2) I I 83.9 (1.9) N 68.9 (3.0) SA32.4 (3.5) SC1.0 (7.9) Cr 10 Cr 52.5 (119.4) N 76.9 (3.2) I I 74.1 (4.4) N 2 (35.6) Cr 11 Cr 61.8 (109.7) N 142.5 (5.4) I I 141.1 (5.2) N 42.0 (4.81) S 29.0 (8.4) S 12.3 (52.1) Cr 12 Cr 76.4 (139.2) I I 74.4 (4.3) N 36.9 (45.7) Cr
Compound 4 containing no lateral fluoro substituent exhibits only crystalline phase and its melting point is much higher than the melting point compounds 5a–5d. The result indicates that the lateral fluoro substituent has a significant effect on lowering the melting point and formation of mesophases. To study the different positions of fluoro substituents and the fluoro-substituent number on the meso-morphic properties of the obtained compounds, we also synthesised compounds 10–12 (Scheme 2). Compounds 10–12 bear symmetry propyl groups on both terminal ends. Compound 10 contains two fluoro substituents on 2 and 5 positions of central phenyl ring of the terphenyl compounds, whereas compounds 11 and 12 contain, respectively, a fluoro substituent and two fluoro substituents on the phenyl ring close to cyclopentyl end group. Comparing mesomorphic properties of compound 10 with those of 5b, compound 10 merely exhibits an enantiotropic nematic phase, whereas compound 5b displays an enantiotropic nematic phase and two monotropic smectic A and C phases. The results indicate that addition of a second fluoro substituent onto the 5 position of central phenyl ring inhibits the formation of smectic phases. Nevertheless, its nematic tempera-ture range also decreases. The phenomena can be attributed to the fact that the stability of mesophase is interrupted by the inter-annular twisting caused by lateral fluorine atom.[41] Both compounds 11 and 5b contain only one fluorine atom, but the fluorine sub-stituent for compound 11 locates on the phenyl ring close to cyclophenyl group. Their mesomorphic beha-viours are similar except that compound 11 exhibits a narrower mesomorphic temperature range. Compound 12 contains two fluoro substituents on the phenyl ring close to cyclophenyl group, and it reveals only a monotropic nematic phase. By compar-ing the mesomorphic properties of compound 12 with that of compound 10, it seems that phenyl ring next to the cyclopentyl end group bearing two fluoro substi-tuents will decrease the stability of nematic phase. The slightly large enthalpy changes on the transition of nematic and isotropy phase for compounds 10 and 12 are unusual and noteworthy.[38–40]
3.3 Effect of 3-propylcyclopentane end group In the literature, the cyclohexyl group is more pop-ular than cyclopentyl group to be used as a building block for LCs. However, there are still several exam-ples [33–36] that demonstrate compounds replacing cyclohexyl terminal group with cyclopentyl group. Those compounds with cyclopentyl end group show much lower melting temperatures than those com-pounds with cyclohexyl end group. The phenomenon
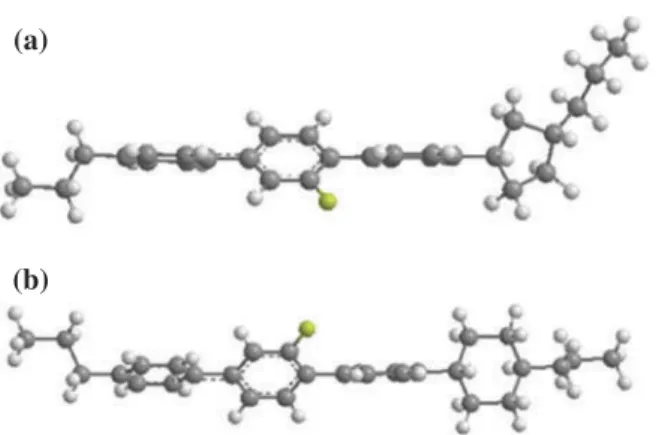
is also verified in the terphenyl system we have synthesised in this article.Figure 4shows the chemi-cal structure and mesomorphic property of com-pound 13.[42] Both comcom-pounds 13 and 5d have identical mesogenic core except the terminal group. Compound 13 that has a cyclohexyl terminal group shows only an enantiotropic nematic phase, whereas compound 5d that possesses a cyclopentyl end group shows three enantiotropic nematic, smectic A and smectic C phases. The melting temperature of 5d is lower than that of 13 by approximately 90°C. The decrease in melting temperature can be attributed to the mixture of cis- and trans-forms of 1,3-substitued cyclopentyl end group and the non-linear molecular structure of both cis- and trans-forms.[43,44] We use proton NMR and nuclear Overhauser effect spectro-scopy (NOESY) to determine the cis/trans isomers ratio. The proton NMR peak of C1 proton (con-necting to the phenyl ring) on cis-cyclopentyl group presents at δ 2.98–3.02 ppm and that of C1 proton on trans-cyclopentyl group presents at δ 3.04–3.11 ppm. The ratio of cis/trans-forms of the synthesised compounds is 1:0.31. It depicts that there are more cis-forms than trans-forms.Figure 5 shows the simu-lated molecular structures of cis-form of 5b, which presents a much curved molecular skeleton and cyclohexyl analogue that shows a more linear structure.
Figure 4. Chemical structure of compound 13.[42]
(a)
(b)
Figure 5. (colour online) The molecular structures of (a) cis-form of 5b and (b) analogue with a cyclohexane end group.
3.4 Electric and optical properties
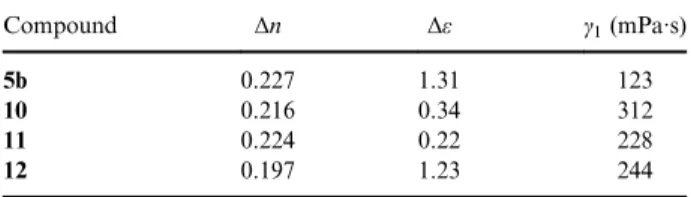
Table 2 lists the birefringence (Δn), dielectric ani-sotropy (Δε) and rotational viscosity (γ1) of
com-pounds 5b and 10–12. The Δn, Δε and γ1 are
measured using guest–host method.[45] The bire-fringence is measured by Abbe refractometer. The dielectric anisotropy and rotational viscosity are measured by Instec automatic liquid crystal tester ALCT4. All four compounds show moderate values of Δn around 0.2. Both compounds 5b and 11 containing only one fluoro substituent reveal a higher value of Δn than those of com-pounds 10 and 12, which contain two fluoro sub-stituents. It can be attributed to the fact that fluoro substituent is an electron-withdrawing group.[9,46,47] The Δε value is strongly related to the molecular dipole moment of the molecule. All four compounds show low Δε value because they contain no strong dipole substituents besides fluoro substituent. Compound 5b contains a lateral fluoro substituent, and the Δε is 1.31, which is the highest among four compounds. Compound 10 contains two fluoro substituents. Both fluoro sub-stituents are located at para position of central phenyl ring, and their dipoles cancel each other. Therefore, it shows a smaller Δε value of 0.34. Compound 11 contains only one fluoro substituent on the phenyl ring close to the cyclopentyl ring. We speculate that its fluoro dipole is cancelled by the terminal cyclopentyl group, the reason for the smallest Δε value of 0.224. Compound 12 also contains two fluoro substituents. Both fluoro sub-stituents are located at meta position of phenyl ring close to cyclopentyl group, and their dipoles do not cancel each other, the reason for the med-ium value of Δε of 1.23. Finally, all four com-pounds possess rather large rotational viscosities. The rotational viscosity is related to the linearity of molecular structure. Those compounds contain-ing cyclopentyl group exhibit bend molecular structures. This is the reason why these com-pounds show large γ1 values. Compound 10 with
two kink fluoro substituents shows the largest γ1
value of 312.
4. Conclusion
We have synthesised a series of fluorinated terphenyl LCs containing cyclopentyl end group. All com-pounds in this series show wide mesomorphic tem-perature range. As anticipated, introduction of fluoro substituent and cyclopentyl end group decreases the melting point of these terphenyl compounds. Most of the terphenyl compounds discussed in previous litera-ture always show very high melting points. Our study provides an efficient way in molecular design to achieve the moderate birefringence LCs with low melting points. The obtained LC, such as compound 5b, that possess moderate birefringence, wide nematic temperature range, low melting point and moderateγ1
value is suitable to formulate eutectic mixture for LC applications.
References
[1] Parisha A, Gauzaa S, Wu ST, Dziaduszekb J, Dabrowskib R. New fluorinated terphenyl isothiocya-nate liquid crystals. Liq Cryst. 2008;35:79–86. doi:10.1080/02678290701749917
[2] Fergason JL. Polymer encapsulated liquid crystals for display and light control applications. SID Symp Dig. 1985;16:68.
[3] Wu ST, Efron U, Grinberg J, Hess LD. Visible-to-infrared dynamic image converter (VIDIC): present status. SID Symp Dig. 1985;16:262–265.
[4] Doane JW, Vaz NA, Wu BG, Žumer S. Field con-trolled light scattering from nematic microdroplets. Appl Phys Lett. 1986;48:269–271. doi:10.1063/1.96577 [5] Li J, Hoke C, Fredly DS, Bos PJ. A bistable LCD using polymer stabilization. SID Symp Dig. 1996;96:265.
[6] Sutherland RL, Natarajan LV, Tondiglia VP, Bunning T. Bragg gratings in an acrylate polymer consisting of periodic polymer-dispersed liquid-crystal planes. Chem Mater. 1993;5:1533–1538. doi:10.1021/ cm00034a025
[7] Bowley CC, Yuan H, Crawford GP. Morphology of holographically-formed polymer dispersed liquid crys-tals (H-PDLC). Mol Cryst Liq Cryst. 1999;331:209– 216. doi:10.1080/10587259908047518
[8] Mcmanamon PF, Watson EA, Dorschner TA, Barnes LJ. Applications look at the use of liquid crystal wri-table gratings for steering passive radiation. Opt Eng. 1993;32:2657–2664. doi:10.1117/12.148094
[9] Reiffenrath V, Finkenzeller U, Poetsch E, Rieger BA, Coates D, Doane JW, Yaniv Z. Synthesis and proper-ties of liquid crystalline materials with high optical anisotropy. Liq Cryst Disp Appl. 1990;1257:84–94. doi:10.1117/12.19930
[10] Kula P, Herman J, Chojnowska O. Synthesis and properties of terphenyl- and quaterphenyl-based chiral diesters. Liq Cryst. 2013;40:83–90. doi:10.1080/ 02678292.2012.733033
[11] Wu ST, Hsu CS, Shyu KF. High birefringence and wide nematic range bis-tolane liquid crystals. Appl Phys Lett. 1999;74:344–346. doi:10.1063/1.123066 Table 2. The electro-optical properties of compounds 5b
and 10–12. Compound Δn Δε γ1(mPa·s) 5b 0.227 1.31 123 10 0.216 0.34 312 11 0.224 0.22 228 12 0.197 1.23 244
[12] Wu ST, Hsu CS, Chuang YY, Cheng HB. Physical properties of polar bis-tolane liquid crystals. Jpn J Appl Phys. 2000;39:L38–L41. doi:10.1143/JJAP.39. L38
[13] Miao Z-C, Wang D, Zhang Y-M, Jin Z, Liu F, Wang F-F, Yang H. Asymmetrical phenyldiacety-lenes liquid crystalline compounds with high bire-fringence and characteristics of selective reflection. Liq Cryst. 2012;39:1291–1296. doi:10.1080/02678292. 2012.714801
[14] Dziaduszekb J, Kula P, Dąbrowski R, Drzewiński W, Garbat K, Urban S, Gauza S. General synthesis method of alkyl–alkoxy multifluorotolanes for negative high birefringence nematic mixtures. Liq Cryst. 2012;39:239–247. doi:10.1080/02678292.2011.636457 [15] Hsu CS, Shyu KF, Chuang YY, Wu ST. Synthesis of
laterally substituted bistolane liquid crystals. Liq Cryst. 2000;27:283–287. doi:10.1080/026782900203100 [16] Seed AJ, Toyne KJ, Goodby JW, Hird MJ. Synthesis,
transition temperatures, and optical properties of var-ious 2,6-disubstituted naphthalenes and related 1-ben-zothiophenes with butylsulfanyl and cyano or isothiocyanato terminal groups. J Mater Chem. 2000;10:2069–2080. doi:10.1039/b003818k
[17] Wu ST, Hsu CS, Chuang YY. Room temperature bis-tolane liquid crystals. Jpn J Appl Phys. 1999;38:L286– L288. doi:10.1143/JJAP.38.L286
[18] Gauza S, Du F, Wu JR, Wu ST, Spadło A, Dabrowsk R, Janarthanan N, Hsu CS. 32.2: high birefringence and low viscosity LC mixtures. SID Symp Dig Tech Pap. 2003;34:1054–1057. doi:10.1889/1.1832469 [19] Jia D, Yang C, Peng Z, Li X, Liu Y, Yao L, Cao Z,
Mu Q, Hu L, Lu X, Xuan L. Wide-angle switchable negative refraction in high birefringence nematic liquid crystals. Liq Cryst. 2013;40:599–604. doi:10.1080/ 02678292.2013.774065
[20] Zhang Y-M, Wang D, Miao Z-C, Jin S-K, Yang H. Novel high birefringence bistolane liquid crystals with lateral fluorosubstituent. Liq Cryst. 2012;39:1330– 1339. doi:10.1080/02678292.2012.725871
[21] Hird M, Toyne KJ, Goodby JW, Gray GW, Minter V, Tuffin RP, McDonnell DG. Synthesis, mesomorphic behaviour and optical anisotropy of some novel mate-rials for nematic mixtures of high birefringence. J Mater Chem. 2004;14:1731–1743. doi:10.1039/ b400630e
[22] Sekine C, Konya N, Minai M, Fujisawa K. Synthesis and properties of high birefringence liquid crystals: thiophenylacetylene and benzothiazolylacetylene deri-vatives. Liq Cryst. 2001;28:1361–1367. doi:10.1080/ 02678290110061386
[23] Sekine C, Ishitobi M, Iwakura K, Minai M, Fujisawa K. Novel high birefringence dibenzothiophenylacety-lene liquid crystals. Liq Cryst. 2002;29:355–367. doi:10.1080/02678290110102434
[24] Wu ST, Margerum JD, Meng HB, Dalton LR, Hsu C, Lung S. Room-temperature diphenyl-diacetylene liquid crystals. Appl Phys Lett. 1992;61:630–632. doi:10.1063/ 1.107829
[25] Spadlo A, Dabrowski R, Ziolek A, Kula P, Urban S, Gauza S, Wu ST, Wolinski TR, Warenghem M, Wu S. Comparison of the mesogenic and physical properties of polar tolanes, biphenyls, and terphenyls. Liq Cryst: Opt Appl. 2005;5947:594711–594716. doi:10.1117/ 12.622878
[26] Catanescu O, Chien LC. High birefringence difluoroi-sothiocyanate biphenyl tolane liquid crystals. Liq Cryst. 2006;33:115–120. doi:10.1080/02678290500241835 [27] Yao YH, Kung LR, Chang SW, Hsu CS. Synthesis of
UV‐curable liquid crystalline diacrylates for the appli-cation of polarized electroluminescence. Liq Cryst. 2006;33:33–39. doi:10.1080/02678290500450931 [28] Herman J, Dziaduszek J, Dąbrowski R, Kędzierski J,
Kowiorski K, Dasari VS, Dhara S, Kula P. Novel high birefringent isothiocyanates based on quaterphenyl and phenylethynyltolane molecular cores. Liq Cryst. 2013;40:1174–1182. doi:10.1080/02678292.2013.808768 [29] Kula P, Herman J, Pluczyk S, Harmata P, Mangelinckx G, Beeckman J. Synthesis and meso-morphic properties of laterally substituted 4,4 ′′′-dia-lkyl-p-quaterphenyls. Liq Cryst. 2014;41:503–513. doi:10.1080/02678292.2013.859755
[30] Dąbrowski DR. New liquid crystalline materials for photonic applications. Mol Cryst Liq Cryst. 2004;421:1–21. doi:10.1080/15421400490501112 [31] Gray GW, Hogg C, Lacey D. The synthesis and
liquid crystal properties of some laterally fluorinated trans-cyclohexane-1-carboxylate and benzoate esters. Mol Cryst Liq Cryst. 1981;67:1–23. doi:10.1080/ 00268948108070871
[32] Karamysheva LA, Geyvandova TA, Agafonova IF, Roitman KV, Torgova SI, Geivandov RKH, Petrov VF, Rabinovich AZ, Grebyonkin MF, Crystalline L. 1,3-disubstituted cyclopentanes. Mol Cryst Liq Cryst. 1990;191:237–246.
[33] Eidenschink R. Low viscous compounds of highly nematic character. Mol Cryst Liq Crys. 1983;94:119– 125. doi:10.1080/00268948308084251
[34] Carr N, Gray GW. The preparation and liquid crystal properties of materials incorporating a mono-substi-tuted ring system. Liq Cryst. 1989;6:467–480. doi:10.1080/02678298908034191
[35] Petrov VF. Cyclopentane as a structural fragment in liquid crystals. Mol Cryst Liq Cryst. 2010;517:10–26. doi:10.1080/15421400903507755
[36] Coates D, Jenner JA, Relffenrath V, Finkenzeller U Fluorinated terphenyls. German Patent GB2240778A. 1991 Aug 14.
[37] Chang CY, Chien SY, Hsu CS, Gauza S, Wu ST. Synthesis of laterally substituted α‐methylstilbene‐ tolane liquid crystals. Liq Cryst. 2008;35:1–9. doi:10.1080/02678290701743027
[38] Imrie CT. Laterally substituted dimeric liquid crystals. Liq Cryst. 1989;6:391–396. doi:10.1080/0267829890 8034184
[39] Imrie CT, Taylor L. The preparation and properties of low molar mass liquid-crystals possessing lateral alkyl chains. Liq Cryst. 1989;6:1–10. doi:10.1080/ 02678298908027317
[40] Collings PJ, Hird M. Introduction to liquid crystals: chemistry and physics. Boca Raton (FL): CRC Press; 1997.
[41] Hird M. Fluorinated liquid crystals – properties and applications. Chem Soc Rev. 2007;36:2070–2095. doi:10.1039/b610738a
[42] Yamada S, Ikukawa S, Nakayama S, Yudasaka Y. Seiko Epson Corp. Cyclohexyl terphenyl derivative and liquid crystal composition containing the same and liquid crystal display element using the same. Japanese patent. JP04356432. 1992 Dec 10.
[43] Ruiz de Ballesteros O, Cavallo L, Auriemma F, Guerra G. Conformational analysis of poly(methy-lene-1,3-cyclopentylene) and chain conformation in the crystalline phase. Macromolecules. 1995;28:7355– 7362. doi:10.1021/ma00126a012
[44] Jacquemet A, Mériadec C, Lemiègre L, Artzner F, Benvegnu BT. Stereochemical effect revealed in self-assemblies based on archaeal lipid analogues bearing a central five-membered carbocycle: a SAXS study. Langmuir. 2012;28:7591–7597. doi:10.1021/la2045948
[45] Khoo IC, Wu TS. Optics and nonlinear optics of liquid crystals. Singapore: World Scientific; 1993.
[46] Schad H, Kelly SM. A novel liquid crystal compound with very large dielectric constants. J Chem Phys. 1984;81:1514. doi:10.1063/1.447757
[47] Kelly SM. The synthesis and transition tempera-tures of benzoate ester derivatives of 2-fluoro-4-hydroxy-and 3-fluoro-4-hydroxybenzonitriles. Helv Chim Acta. 1984;67:1572–1579. doi:10.1002/hlca. 19840670623