國立臺灣大學工學院應用力學研究所 博士論文
Graduate Institute of Applied of Mechanics College of Engineering
National Taiwan University Doctoral Dissertation
四面體雙聚體分子之量子化學計算與分子動力學模擬
Molecular Dynamics Simulations of Tetrahedral Molecule Dimers Properties Using Ab Initio Intermolecular
Interaction Potentials
李皇德
Arvin Huang-Te Li 指導教授:趙聖德 博士 Advisor: Sheng D. Chao, Ph.D.
中華民國 99 年 6 月
June, 2010
國立臺灣大學博士學位論文 口試委員會審定書
四面體雙眾體分子之量子化學計算與分子動 力學模擬-
Molecular Dynamics Simulations of Tetrahedral Molecule Dimers Properties Using Ab Initio
Intermolecular Interaction Potentials
本論文條、李皇德君(學號:
D93543004)
在國立臺灣大 學應用力學研究所完成之博士學位論文,於氏國99 年 6
月3
日承下列考試委員審查通過及口試及格,特此證明
口試委員:
皇室益
莖 2
重室監
奎主 2 至
所
長 莖產盛Acknowledgment
在此要感謝我的爸爸、媽媽以及家人給我的支持與鼓勵,讓我得以無慮的完成 博士班的學業,謝謝您們。再者也要感謝這幾年來讓我在知識與見識上增長的恩師 趙聖德老師與師母,感謝您的諄諄教誨與提攜讓我如沐春風,亦師亦友、同舟共濟 的革命情感相信日後會更加珍惜。也謝謝所長 張建成老師的知遇之恩,讓自己可以 在所內發揮所長。謝謝所上的教過自己的老師讓我獲得許多新知,吳光鐘老師、張 正憲老師、朱錦洲老師、王立昇老師、沈弘俊老師、邵耀華老師、陳國慶老師、陳
瑞琳老師等等,謝謝您們。感謝所有口試委員對自己的肯定,相信自己日後會更
加努力。
感謝多年來實驗室的學弟、研究與行政助理們,謝謝您們的幫忙。謝謝本 實驗室學弟:景政、宏吉、士緯、守正、光惟、宜興、熙葆、俊傑、陳楠、碩 峰、育德、佑儒、思哲,研究助理:奕翔、宛儀、馨瑩、以謝,行政助理:逸璇、
書閔、欣瑩、婷婷、薔涵等等的幫忙。謝謝其他實驗室:在欣、玉清、哲宇、
祺皓、銘儒、義慶、宏志、明峰、冠榮、佳暐、萬霖、大發等等的幫助。所上 翁文斌技士、李孟賢技佐、所辦許文珍小姐、張素端小姐、李瑩蘭小姐的幫忙。
清大生科所的林志侯老師、李寬容老師、張晃猷老師的鼓勵,以及清大的大鈞 學長、昭芬學姐。謝謝當兵時候嘉義縣新港國中的老師們,謝謝您們。
感謝家昕當時候的支持與陪伴,由衷的感謝。謝謝好友瑞苓、瑞玲、Ivo、
育嘉、Amy、Nancy、Yichi、Helen、璽凱、小顏、膺任、彥城、俊甫、珣彣、
啟昌、良玉、慧怡、謝迺岳老師等等的幫忙。
謝謝家人、老師、同儕、朋友,您們都是我的明燈與貴人,萬分感謝。
皇德 謹誌于 國立台灣大學 應用力學所 跨尺度動力學實驗室 R 325A
中華民國 99 年 6 月
摘要
本論文主要探討四面體雙聚體分子之量子化學計算與分子動力學模擬探討熱力 學性質。我們從最簡單的四面體甲烷分子開始研究並推廣到矽烷與四氯化碳分子。
我們首先準確的架構並計算出12 個不同方位的能量曲線,探討方位、中間氫原子數
目對能量關係、吸引力、斥力、剛體與非剛體對能量造成的影響。
在量子計算方面主要使用的方法有 Hartree-Fock (HF)、Density Function Theory (DFT)、Second-order Møller-Plesset Perturbation Theory (MP2) 與 Single-point Coupled Cluster with Single and Double and Perturbative Triple Excitations CCSD(T) 等計算方
法。基底方面使用Pople 的方法從小的基底到 6-311++G (3df, 3pd),並使用 Dunning
的基底 [cc-pVXZ 與 aug-cc-pVXZ,其中 X = D、T、Q、5] 進行一連串完整的計算
並探討能量收斂狀況。計算時中心原子距離從 3.0 Å 到 9.0 Å,並使用四種近似法
(Extrapolation methods) 與完整基底極限展開法 (Complete Basis set limit) 得到鍵結
最低能量極限值,與目前光譜實驗量測所可以測得的容許差值約在 0.03 kcal/mol 之
內。
而在分子動力學模擬方面主要是利用 Lennard-Jones (L-J) potential function 去做
擬合並建構力場,先分別探討 4-site 與 5-site 的影響。4-site 需要決定四個擬合參數
16 條方程式,5-site 需要決定六個擬合參數 25 條方程式。利用得到的擬合參數去模 擬在不同溫度、密度與壓力下Radial Distribution Function (RDF) 的平衡性質並探討
溫度效應與實驗值做比較。而動態性質方面主要是研究沿著汽化線做 Velocity
Autocorrelation Function (VAF) 的模擬,求得擴散係數 (Diffusion Coefficient) 並與文 獻上既有的實驗值做比較。最後做 X-ray 散射 與 Neutron 散射模擬研究皆與真實文
獻上實驗吻合。由上述結果可知,就我們目前所做的量子化學勢能曲線 (Potential Energy Surface) 理論計算結果與所得到的 5-site 理論擬合參數值皆可精準與實驗相 驗證。
關鍵字:Hartree-Fock (HF), Density Function Theory(密度泛函理論),Second-order Møller-Plesset Perturbation Theory (二階 Møller-Plesset 微擾理論) ,Single-point Coupled Cluster with Single and Double and Perturbative Triple Excitations CCSD(T),
Radial Distribution Function (徑向分布函數), Velocity Autocorrelation Function (速度 自相關函數),Diffusion Coefficient (擴散係數),X-ray 散射與 Neutron 散射。
Abstract
In this study, intermolecular interaction energy data for the tetrahedral molecule dimers have been calculated at a spectroscopic accuracy and employed to construct an ab initio potential energy surface (PES) for molecular dynamics (MD) simulations of tetrahedral molecule properties. The full potential curves of the tetrahedral molecule dimers at 12 symmetric conformations were calculated by the supermolecule counterpoise-corrected Hartree-Fock (HF), Density Function Theory (DFT), second-order Møller-Plesset (MP2) perturbation theory and single-point coupled cluster with single and double and perturbative triple excitations [CCSD(T)] calculations were also carried out to calibrate the MP2 potentials. We employed Pople’s medium size basis sets [up to 6-311++G (3df, 3pd)]
and Dunning’s correlation consistent basis sets (cc-pVXZ and aug-cc-pVXZ, X = D, T, Q, 5). For each conformer, the intermolecular carbon–carbon separation was sampled in a step 0.1 Å for a range of 3.0 ~ 9.0 Å. The MP2 binding curves display significant anisotropy with respect to the relative orientations of the dimer. The potential curves at the complete basis set (CBS) limit were estimated using well-established analytical extrapolation schemes and while a large basis set (aug-cc-pVTZ) is required to converge the binding energy at a chemical accuracy (~0.03 kcal/mol). A 4-site and 5-site potential models were used to fit the ab initio potential data. We performed molecular dynamics
simulations using the ab initio force field and compared the simulation results to experiments. Quantitative agreements for the atomwise radial distribution functions, the self-diffusion coefficients, and the X-ray and Neutron diffraction scattering functions over a wide range of experimental conditions can be obtained, thus validating the ab initio force field without using experimental data a priori.
Keyword : Hartree-Fock (HF), Density Function Theory (DFT), Second-order Møller-Plesset Perturbation Theory(MP2), Single-point Coupled Cluster with Single and Double and Perturbative Triple Excitations CCSD(T),Radial Distribution Function (RDF), Velocity Autocorrelation Function (VAF), Diffusion Coefficient, X-ray and Neutron diffraction scattering function.
Contents
口試委員會審定書………..i
Acknowledgment…..………..………...ii
Chinese Abstract……….………..…..…..iii
English Abstract………..………....….. v
Contents ….…….………...vii
Figure Contents ….……...………..………..ix
Table Contents ...……….xiv
Chapter 0 General Introduction ……..……….…..………..….17
Chapter 1 Theoretical Studies on the Methane Dimers……….…..……….22
1.1 Intermolecular Interaction Potentials of Dispersion-Bound Methane Dimer From Coupled Cluster Method at Complete Basis Set Limit…… ………...22
1.2 Intermolecular Potentials of the Methane Dimer Calculated with Møller- Plesset Perturbation Theory and Density Functional Theory and comment on "Intermolecular Interaction Potential of the Methane Dimer From the Local Density Approximation”…………..….…….………42
1.3 Molecular Dynamics Simulations of Fluid Methane Properties Using Ab Initio Intermolecular Interaction Potentials……….……….…….93
Chapter 2 Theoretical Studies on the Silane Dimers……….………..…...131
2.1 Intermolecular Potentials of the Silane Dimer Calculated with Hartree-Fock Theory, Møller-Plesset Perturbation Theory and Density Functional Theory……..131
2.2 Determination of a Silane Intermolecular Force Field Potential Model From an Ab Initio Calculation….……….………..…169
Chapter 3 Theoretical Studies on the Carbon Tetrachloride Dimers………...…187
3.1 Molecular dynamics simulation of liquid carbon tetrachloride using AbInitio
force field.……….………..……….….…187 3.2 Intermolecular Potential of the Carbon Tetrachlorid Dimer Calculated
with Møller-Plesset Perturbation Theory and Density Function Theory………..…213 Chapter 4 Conclusion ………...232
Figure Contents
Fig. 0.0-1... tetrahedral molecule dimers...18
Fig. 0.0-2...Standard operating procedure(SOP)...19
Fig. 0.0-3... 4-site model...20
Fig. 0.0-4... 5-site model...20
Fig. 0.0-5... Radial distribution function...21
Fig. 0.0-6...Velocity autocorrelation function...21
Fig. 1.1-1... The BSSE corrected HF, MP2, and CCSD(T) interaction potentials of the methane dimer using the aug-cc-pVQZ basis set...40
Fig. 1.1-2... The estimated CCSD(T)/CBS potential curves using the direct extrapolation scheme with the four extrapolation methods on the cc-pVXZ potential data..40
Fig. 1.1-3... The estimated CCSD(T)/CBS potential curves using the direct extrapolation scheme with the four extrapolation methods on the aug-cc-pVXZ potential data...41
Fig. 1.2-1... The calculated intermolecular interaction potentials using a series of exchange-correlation functionals...84
Fig. 1.2-2... Comparison of the calculated data with the fittings using the L-J function and the exponential function...85
Fig. 1.2-3... The BSSE corrected HF interaction potentials of the methane dimer using several basis sets...86
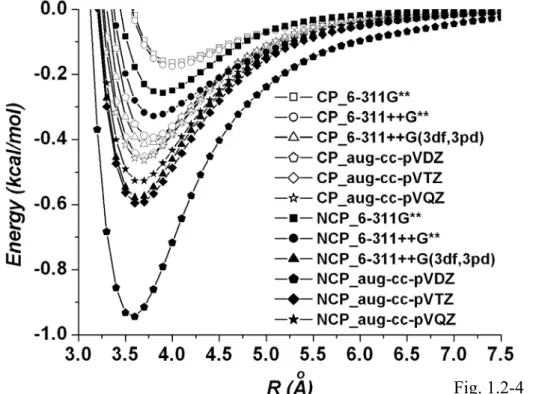
Fig. 1.2-4... The BSSE corrected (CP) and uncorrected (NCP) MP2 potentials of the methane dimer using a series of basis sets...86 Fig. 1.2-5... Comparison of the BSSE corrected MP2 potential curve calculated at the
aug-cc-pVQZ basis set and the sum of the HF potential and the long range
dispersion potentials………87 Fig. 1.2-6... The BSSE corrected DFT intermolecular potential curves using the PW91
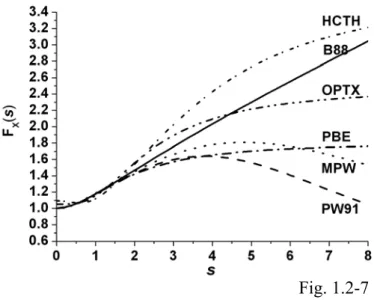
as the exchange or correlation functional...87 Fig. 1.2-7... The GGA exchange enhancement factor as a function of s for the B88,
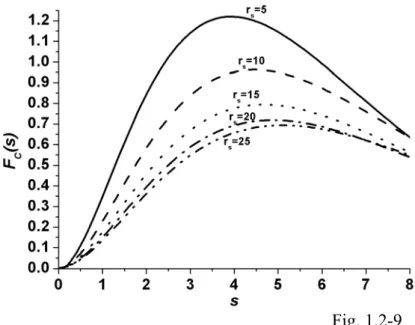
HCTH, OPTX, MPW, PBE, and PW91 exchange functionals...88 Fig. 1.2-8... The GGA correlation enhancement factor as a function of s for the TPSS,
PBE, PW91, P86, and HCTH correlation functionals. Here rs =10... 88 Fig. 1.2-9... The rs dependence of the GGA correlation enhancement factor as a
function of s for the P86 correlation functional...89 Fig. 1.2-10... The BSSE corrected potential curves with varying exchange functionals
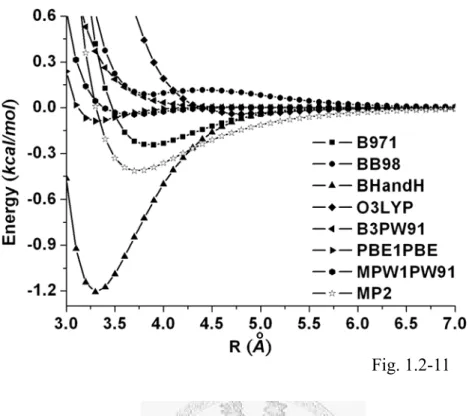
by fixing (a) PBE, (b) PW91, (c) VWN, (d) VP86 and (e) LYP correlation functionals, respectively...89 Fig. 1.2-11... The BSSE corrected potential curves selective using several hybrid
functionals...92 Fig. 1.2-12... The asymptotic behavior of selective DFT potentials versus the MP2
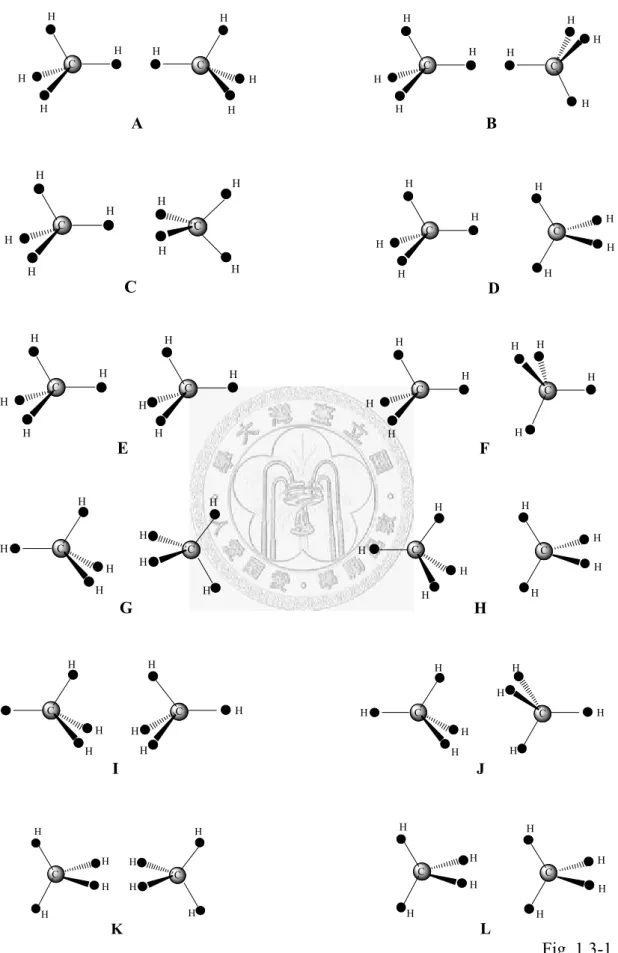
potential via a analysis of the long-range data...92 Fig. 1.3-1... The twelve symmetric conformers of the methane dimer considered in this
paper………118 Fig. 1.3-2... Comparison of the potentials with (rigid) and without (non-rigid) the rigid
monomer assumption for the J conformer...119 Fig. 1.3-3... The MP2/aug-cc-pVQZ potentials of the methane dimer for the 12
conformers...120 Fig. 1.3-4... Comparison of the HF (repulsive) and MP2-HF (attractive) potentials for
the 12 conformers...121 Fig. 1.3-5... The MP2/CBS potential curves using the extrapolation method of
Halgaker et al. …...121
Fig. 1.3-6... A schematic plot of the 4-site model where the sites are located at the
hydrogen positions only...122 Fig. 1.3-7... Comparison of the fitting curves and the calculated potential data...123 Fig. 1.3-8... The six-dimension PES...124 Fig. 1.3-9... Comparison of the experimental (EXP) and the molecular dynamics (MD)
radial distribution functions (RDFs) as a function of R...125 Fig. 1.3-10... Velocity autocorrelation functions as a function of time for several
experimental conditions...127 Fig. 1.3-11... The diffusion coefficient as a function of temperature for different cell
sizes and rcut...128 Fig. 1.3-12... Comparison of the experimental (EXP) and the molecular dynamics (MD)
X-ray scattering as a function of K...129 Fig. 1.3 -13... Comparison of the experimental (EXP) and the molecular dynamics (MD)
thermal effect as a function of K...130 Fig. 2.1-1... The BSSE corrected MP2 potentials using the aug-cc-pVTZ basis set for the
D3d and C3v conformers of the silane dimer...162 Fig. 2.1-2... The BSSE corrected HF interaction potentials of the silane dimer using
several basis sets...162 Fig. 2.1-3... The BSSE corrected (CP) and uncorrected (NCP) MP2 potentials of the
silane dimer using a series of basis sets...163 Fig. 2.1-4... Comparison of the BSSE corrected MP2 potential curve calculated at the
aug-cc-pVQZ basis set and the sum of the HF potential and the long-range dispersion potentials...163 Fig. 2.1-5... The basis set dependence of the DFT potentials calculated with the
PW91PW91 functional (left panel)...164 Fig. 2.1-6... The GGA correlation enhancement factor as a function of s for the TPSS,
PBE, PW91, P86, and HCTH correlation functionals...164 Fig. 2.1-7... The BSSE corrected DFT potential curves with varying exchange
functionals by fixing (a) PBE, (b) PW91, (c) VWN, (d) VP86, and (e) LYP correlation functionals, respectively. (f) The DFT potentials using several hybrid functionals (B3LYP, B3P86, BHandH, MPW1PW91, O3LYP, and PBE1PBE) …...165 Fig. 2.1-8... The asymptotic behaviors of selective DFT potentials versus the MP2
potential via an analysis of the long-range data...168 Fig. 2.2-1... The twelve symmetric conformers of the silane dimer...183 Fig. 2.2-2... The HF potentials for the 12 orientations using the aug-cc-pV5Z basis set.. 184 Fig. 2.2-3... The MP2 potentials for the 12 orientations using the aug-cc-pV5Z basis set.184 Fig. 2.2-4... The MP2-HF potentials for the 12 orientations using the aug-cc-pV5Z basis
set...185 Fig. 2.2-5... The C6 coefficient value has been obtained by fitting to intermolecular
potential energy from ab initio calculation...185 Fig. 2.2-6... Comparison of the fitting curves (line) and the potential data (symbol)...186 Fig. 3.1-1... The twelve symmetric conformers of the carbon tetrachloride dimer...208 Fig. 3.1-2... The HF potentials for the 12 orientations using the aug-cc-pVTZ basis set. .209 Fig. 3.1-3... The MP2 potentials for the 12 orientations using the aug-cc-pVTZ basis
set...209 Fig. 3.1-4... The MP2-HF potentials for the 12 orientations using the aug-cc-pVTZ
basis set…...210 Fig. 3.1-5... Comparison of the fitting curves (line) and the potential data (symbol)...210 Fig. 3.1-6... The calculated C-C, C-Cl and Cl-Cl radial distribution function gCC, gCCl,
gClCl for a range of thermodynamic conditions...211 Fig. 3.1-7... Comparison of theory curve (line) and neutron scattering functions
(symbol)………...212 Fig. 3.1-8... Comparison of theory curve (line) and X-ray scattering functions
(symbol)...212 Fig. 3.2-1... The BSSE corrected MP2 potentials using the aug-cc-pVTZ basis set for
the D3d and C3V conformers of the carbon tetrachloride dimer...229 Fig. 3.2-2... The BSSE corrected HF interaction potentials of the carbon tetrachloride
dimer using several basis sets...229 Fig. 3.2-3... The BSSE corrected (CP) and uncorrected (NCP) MP2 interaction
potentials of the carbon tetrachloride dimer using a series of basis sets...230 Fig. 3.2-4... The BSSE corrected DFT potential curves with varying exchange
functional by fixing VWN5 correlation functionals...230 Fig. 3.2-5... The BSSE corrected DFT potential curves with varying exchange
functional by fixing PL correlation functionals...231
Table Contents
Table 1.1-1 ... The basis set dependence of important potential parameters of the BSSE corrected CCSD(T) intermolecular potentials...37 Table 1.1-2 ... Comparison the estimated CCSD(T) and MP2 binding energies at the
complete basis set limit using the four extrapolation methods……...38 Table 1.1-3 ... Basis set dependence of the estimated potential parameters of the
CCSD(T)/CBS potential...39 Table 1.2-1 ... The basis set dependence of important potential parameters using the
BSSE corrected HF and MP2 intermolecular potentials...80 Table 1.2-2 ... Comparison of the bond lengths and the binding energies calculated
using the several exchange-correlation functionals with the MP2 results using the 6-311++G (3df,3pd) basis set...81 Table 1.2-3 ... Comparison of the bond lengths calculated with the 90 exchange-
correlation functionals using the 6-311++G (3df, 3pd) basis set...82 Table 1.2-4 ... Comparison of the binding energies calculated with the 90 exchange-
correlation functionals using the 6-311++G (3df, 3pd) basis set... 83 Table 1.3-1 ... The basis set dependence of important potential quantities extracted from
the MP2 potentials for the 12 conformers...112 Table 1.3-2 ... The MP2/CBS binding energies using the four extrapolation methods.... 114 Table 1.3-3 ... Comparison of the MP2/aug-cc-pVQZ and MP2/CBS potential data
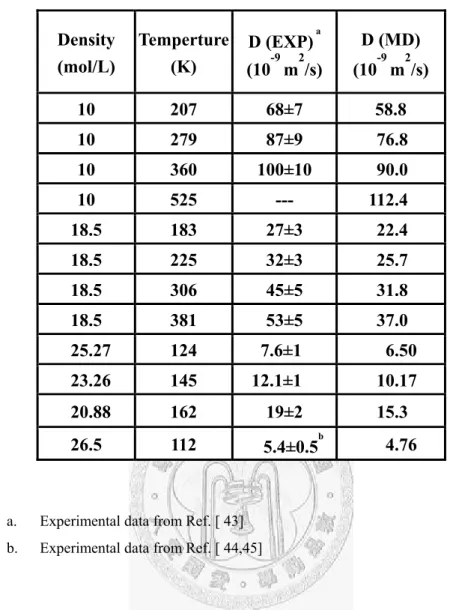
with the CCSD(T)/aug-cc-pVQZ data for the J conformer...115 Table 1.3-4 ... Comparison of the experimental (EXP) and molecular dynamics (MD)
self-diffusion coefficients for a wide range of thermodynamic
conditions………...116
Table 1.3-5 ... The deviation of the fit data from the ab initio data. ABSper and
ABSerr represent the root mean square errors...117 Table 2.1-1 ... The basis set dependence of important potential parameters using the
BSSE corrected HF and MP2 intermolecular potential...158 Table 2.1-2 ... Comparison of the binding energies using the BSSE corrected MP2 and
CCSD(T) intermolecular potentials calculated at several basis sets...159 Table 2.1-3 ... Comparison of the bond lengths calculated with the 108 exchange-
correlation functionals using the 6-311++G (3df,3pd) basis set...160 Table 2.1-4 ... Comparison of the binding energies calculated with the 108 exchange
-correlation functionals using the 6-311++G (3df, 3pd) basis set...161 Table 2.2-1 ... The basis set dependence of MP2 potentials for the G and H conformers.180 Table 2.2-2 ... Predicted C6 coefficient value with literature in twelve conformers...181 Table 2.2-3 ... The estimated MP2/CBS binding energies for the G conformer using the
three extrapolation methods described in the text...182 Table 3.1-1 ... The basis set dependence of some potential parameters of the HF and
MP2 potentials for the J conforme...204 Table 3.1-2 ... The estimated MP2/CBS binding energies for the J conformer. using the
four extrapolation methods described in the text...205 Table 3.1-3 ... Comparison of the calculated peak and valley positions of the RDF with
the experiment …...206 Table 3.1-4... The self-diffusion coefficients using the Green-Kubo formula as
compared to the available experimental data...207 Table 3.2-1 ... The basis set dependence of some potential parameters for the HF and
MP2 potentials...226 Table 3.2-2 ... Comparison of the bond lengths of the interaction potential calculated
with the 80 exchange-correlation functionals using the aug-cc-pVTZ
basis set...227 Table 3.2-3 ... Comparison of the binding energies of the potential calculated with
the 80 exchange-correlation functionals using the aug-cc-pVTZ basis
set...228
Chapter 0 General Introduction
We performed refined quantum chemistry calculations of the intermolecular interactions for dimers of methane, silane, and tetrachloromethane in Fig.0.0-1. For arbitrary molecule in quantum chemistry calculations, the energy can be expansion in Hamiltonian formula,
for total energy (Born-Oppenheimer energy)
Our ab initio calculations focused on the minimum energy curves for a specific relative configuration of the dimers considered. We performed at the HF, DFT, MP2 and CCSD(T) theory
*
2
* 2
1 1
1 1
0 1,
: ˆ
2 (1) (1) (2 )
2 4
slater elec
n n
C
i i ij ij
C i j
e C
HF E H d
Z d J K
m r
DFT: E UNe Ks UeeExc
(0) ' (0) 2
(2) 0
0 (0) (0)
0 0
2 , , ,
| * ˆ | 2 :
| ( | ) ( | ) |
m
HF HF
m m
HF
a b i j i j a b
H d
MP E E E E
E E
ij ab ia jb E
( ) :
Tˆ HFCCSD T e
2 2 2 2 2
2 2 '
, '
0 , 0 , 0 , '
ˆ 1
2 4 4 2 4
C C C
i C
i i C i j C C C
e i C i j C C C
Z Z Z
e e e
H m
r
r
m
R'
total elec c c
E E U
and higher level ab initio calculations for other relative configurations to model the anisotropy of intermolecular interactions of the chosen functional groups. We also perform a nonlinear modeling using force matching and other minimization schemes.
The liquid methane, silane, and tetrachloromethane properties, structural and dynamical, can be well reproduced via molecular dynamics simulations in Fig.0.0-2 ~ Fig. 0.0-6.
Fig. 0.0-1
Standard operating procedure (SOP)
Fig. 0.0-2
Fig. 0.0-3
Fig. 0.0-4
Fig. 0.0-5
Fig. 0.0-6
Chapter 1 Theoretical Studies on the Methane Dimers
1.1 Intermolecular Interaction Potentials of Dispersion-Bound Methane Dimer From Coupled Cluster Method at Complete Basis Set Limit
1. Introduction
The hydrocarbon interactions are very crucial in determining the packing morphology of molecular solids and soft matters such as lipid bi-layer assembly of membranes and active conformation of proteins [1-3]. The potential energy functions are also requested for mesoscale modeling of macromolecules [4] because the hydrocarbon interactions dominate the surface energies, from which many important properties relevant to organic nanostructures can be derived. Determining intermolecular interactions among dispersion-bound complexes from solely experimental measurements is a notoriously challenging task [5-7], mainly due to limited sampling of the potential energy surface. One alternative is to use first-principles electronic structure calculations, at least for small molecular systems [8-10].
Methane molecular interactions can be regarded as a prototype system of any hydrocarbon interactions or of any segment containing hydrocarbons and thus have been intensely studied [11-23]. Jurecka et al. [21] have obtained the CCSD(T) binding
energies of the methane dimer at the complete basis set (CBS) limit using an approximation scheme (see Eq. (1) below). Tsuzuki et al. [22] have estimated the CCSD(T)/CBS binding energies using the method of Halgaker et al. [32] and the method of Feller [34]. Previous CCSD(T) studies on the methane dimer mainly focused on the equilibrium region of the potentials with relatively few discussion on the full potential curve. Nevertheless, to construct a reliable force field model for molecular simulations, the full intermolecular potential surfaces are required.
Recently, Takatani et al. [24] have obtained a CCSD(T)/CBS potential curve for the methane dimer using a specific extrapolation scheme. However, the basis set effects using the CCSD(T) method and the performance of various extrapolation methods to obtain the CBS data have not been systematically investigated. Therefore, in this paper we perform a comprehensive study on interaction potentials of the prototype methane dimer in terms of the CCSD(T) method at the complete basis set limit. The relative performance of several extrapolation methods to obtain the CBS values is thoroughly discussed. The full potential curves are presented to see the overall scope of the interactions.
2. Methods and Calculations
Supermolecular approach has been taken to calculate the interaction energy in which
the intermolecular potential is defined as the total energy difference between the supermolecule and the isolated subsystems. The calculation of electron correlation energies depends on the level of the correlation method, the size of the basis set, and the correction of the BSSE. State-of-the-art choice of the correlation method is the coupled cluster method with iterative single and double substitutions and with noniterative triple excitations [CCSD(T)] method [26]. It has been widely recognized that the CCSD(T) results with large basis sets are close to the results at the complete basis set limit [27]. To study the basis set effects, we have employed a wide range of basis sets from the Slater-type orbitals fitted with Gaussian functions (STO-nG, n=3~6) [28], Pople’s medium size basis sets [up to 6-311++G (3df, 3pd)] [29] to Dunning’s correlation consistent basis sets (cc-pVXZ and aug-cc-pVXZ, X=D, T, Q) [30]. The basis set superposition error (BSSE) was corrected by the counterpoise (CP) method of Boys and Bernardi [31]. The CCSD(T) binding energy at the complete basis set limit has been estimated using the methods of Helgaker et al. [32], Martin [33], Feller [34], and a numerical extrapolation method based on the 3-term Lagrangian formula [35]. The analytic extrapolation formulas are summarized as follows.
Helgaker et al.:
E
X E
bX
3Martin:
4 4
4 4 1 4 4
3 1
( 2 ) ( 2 )
3 1 3 1
( 2 ) ( 2 ) ( 2 ) ( 2 )
X X
X X
E E E
X X X X
Feller:
E
X E
b e
cXAll the calculations are performed using the Gaussian 03 program package [36] on a single node 1-processor IBM 1350 PC cluster with distributed memory. To obtain the most stable intermolecular geometry, the methane molecule was first optimized at the CCSD(T)/aug-cc-pVTZ and the obtained C-H bond length is 1.085 Å. Next we calculated the binding energies of the 12 symmetric dimer conformers such as those considered by Tsuzuki et al. [15] using MP2/aug-cc-pVTZ and found the minimum-energy conformation corresponds to the D3d symmetry conformer. This optimized conformer has been explained in terms of the interplay of the steric stabilization of repulsive hydrogen in opposite monomers [11]. Subsequently the C-C distance was sampled for a quite large range of intermolecular separation (normally 3~9 Å), resulting in a total of 11 configuration points for each basis set. During the scan we first fix the monomer geometry (rigid monomer assumption) and the conformer symmetry. Next, because inclusion of the intramolecular vibrational relaxation could be relevant to molecular dynamics simulations using flexible models [37], validity of the rigid monomer assumption should be checked. We repeat the
above procedure while allowing the C-H bonds to relax during the scan. We found little effect (normally within 0.01 kcal/mol in energy difference) when including the vibrational relaxation. Therefore, in this paper we present the potential data under the rigid monomer assumption in order to compare them with previous studies.
3. Results and Discussions
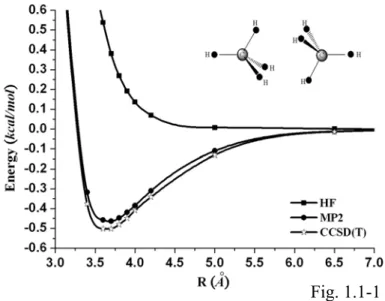
In Fig. 1.1-1 we compare the BSSE corrected HF, MP2, and CCSD(T) potentials of the methane dimer using the aug-cc-pVQZ basis set. The HF and MP2 potentials are obtained from our previous calculations [23]. It is seen that the HF potential is purely repulsive while the MP2 and CCSD(T) potentials display potential wells. The CCSD(T) potential shown in Fig. 1.1-1 displays a clear minimum and a long range attractive potential tail. Because the contributions from the electrostatic and induction energies are small [11], the dispersion energy is mainly responsible for the attractions.
The sharp difference between the HF calculations and the CCSD(T) calculations indicates the importance of including the correlation corrections in the wave function calculations. The MP2 potential is close to the CCSD(T) potential in this case. We also find very strong dependence of the interaction potentials on the BSSE corrections, even using the aug-cc-pVTZ basis set. Only the BSSE corrected potential curves bear the proper trend of systematic convergence. This is similar to our previous studies
using the MP2 method [23].
In Table 1.1-1 we provide a detailed editing of the basis set effects on the equilibrium bond length, the binding energy, and the harmonic frequency. R0 is the distance at which the potential is zero and can be obtained from a two point interpolation of the calculated data. The bond length Rm, the binding energy Eb and the intermolecular vibration frequency can be obtained through a harmonic modeling of the three lowest potential data near the equilibrium regions. It is seen that the interaction energy becomes deeper with more polarization and diffuse functions added. Small cc-pVDZ and cc-pVTZ basis sets lead to underestimation of the binding energy but augmentation of the diffuse functions improves much on the binding energy. The basis set effects presented here are qualitatively similar to previous studies on methane and silane dimers using the MP2 method [23, 38-39]. These observations are expected to apply to other dispersion-bound systems also.
The strong basis set dependence and the slow convergence on the dispersion energy call for an estimation of the important potential features at the complete basis set limit in a calculated potential. Complete basis set limit of the binding energy can be estimated using Dunning’s basis sets with an extrapolation method. We consider three
analytical methods [32-34] and a numerical method [35] and the results are shown in Table 1.1-2. The binding energies obtained at the complete basis set limit (using the potential data calculated at Dunning’s basis sets, aug-cc-pVXZ, X=D, T, Q) are -0.509, -0.512, -0.507, and -0.510 kcal/mol using the methods of Helgaker et al. [32], Martin [33], Feller [34] and the numerical method [35], respectively. These values are very close to the results obtained by Tsuzuki et al. [22] (-0.500 kcal/mol) and Zhao et al.
[25] (-0.510 kcal/mol), while a little smaller (in absolute magnitudes) than those obtained by Jurecka et al. [21] (-0.530 kcal/mol) and Takatani et al. [24] (-0.541 kcal/mol). Notice that the estimated binding energies using the data calculated at the
cc-pVXZ basis sets exhibit large deviation among the extrapolation methods. In particular, using the method of Feller with the cc-pVDZ data overestimates much of the energy. This latter observation is at variance with the results of Tsuzuki et al. [22]
while is more in line with other criticisms on using Feller’s method with the cc-pVDZ potential data to obtain the complete basis set limit binding energy [40]. For the other potential parameters, we used the numerical extrapolation based on the vanishing inverse of the number of basis function [35]. We note that the large vibration frequency, or equivalently the zero point energy correction to the binding energy, indicates that the anharmonicity of the dispersion potential well should be considered for comparison with spectroscopic measurements.
Using the above extrapolation methods, the CCSD(T)/CBS binding energies can be estimated. However, our ultimate objective is to obtain the full potential curve.
Because we have obtained the potential curve at each basis set, we can use a direct extrapolation scheme where for each sampled C-C distance the potential data calculated at a series of basis sets are used to extrapolate to the CBS potential energy.
We can then evaluate the effects of the potential data quality and the extrapolation methods on the CCSD(T)/CBS potential curves. In Fig. 1.1-2 and Fig. 1.1-3 we present the CBS potential curves obtained from the above four extrapolation methods [32-35], using the cc-pVXZ and aug-cc-pVXZ potential data, respectively. In Fig.
1.1-2 we see that using the cc-pVXZ data, the estimated CCSD(T)/CBS potential curves exhibit large deviation among the extrapolation methods. On the other hand, as shown in Fig. 3, using the aug-cc-pVXZ data, the CBS potential curves are all close to each other except for that obtained by Feller’s method, which exhibits spurious long-range behavior. It is thus clear that the estimated potential curve depends sensitively on the quality of the calculated potential data which in turn depends on the basis sets used and the precision of the extrapolation methods. By all means one should check the consistency of using the extrapolation methods to obtain the CBS potential curve before any definite conclusion can be made. In the present case, the
CBS potential curves obtained by the methods of Helgaker et al. [32], Martin [33] are more self-consistent than those using the method of Feller [34], in particular if the potential data calculated at small basis sets were used to perform extrapolations. Our potential curve using the method of Helgaker et al. [32] is very close to that of Takatani et al. [24], although they did not compare the results using different extrapolation methods. Therefore, we believe that these data are reliable and serve as an operational standard for further experimental verifications and theoretical calibrations of other lower level ab initio or semi-empirical methods.
In the literature there appears a proposal to obtain the CCSD(T)/CBS potential energy by an approximate formula [39]
( ) 2
( ) 2
CBS CBS
CCSD T MP
small basis small basis
CCSD T MP
E E E
E E E
(1)
Underlying this formula is the assumption that is relatively basis set E independent. We would like to check this assumption more quantitatively by examining the basis set dependence of . Using the CCSD(T) data in Table 1.1-1 E and our previous MP2 data [23] we can estimate the CCSD(T)/CBS binding energy with Eq. (1) for each basis set we employed. In this way we can determine the
applicability of the above approximation more quantitatively. The results are shown in Table III, where our previous MP2 results are also presented as a reference. We see that using the aug-cc-pVDZ is good enough to obtain a binding energy within a 0.01 kcal/mol error with respect to the CBS energy. It is also interesting to study how far
this idea can be applied to estimate the CBS values for other potential quantities;
namely, we propose the formula
( ) 2
( ) 2
CBS CBS
CCSD T MP
small basis small basis
CCSD T MP
Q Q Q
Q Q Q
(2)
where Q= R0, Rm, , etc. To estimate the CBS values for the bond distance and harmonic frequency. The results are shown in Table III. We see that the estimation is surprisingly good. Using even the 6-31G* basis set we have obtained an estimation of the CBS bond distance within a 0.05 Å precision and of the CBS frequency within a 15 cm-1 precision. Using the aug-cc-pVDZ data almost all the potential quantities can be obtained exactly. It remains to be seen whether this kind of good agreement can be extended to other systems.
In passing we would like to comment on comparison of the calculated potential energy with the “empirical” or “semi-empirical” one. Actually the potential energy
itself is not a direct observable in experiments while it depends on auxiliary theoretical modeling. Experimental potentials are often isotropic and depend on thermodynamic conditions so the consistency with the potential retrieved from experiments does not guarantee the correctness and accuracy of the calculated potential. It is highly possible to obtain a potential curve using smaller basis sets (and/or without the BSSE correction) which incidentally is close to the “empirical”
potential. However, this kind of agreement is largely accidental and should not be taken too seriously. Error cancelation can happen from many sources including correlation energy corrections, basis truncation errors, BSSE, and extrapolation approximations. This precaution should also be taken while using lower-level correlation methods or density functional theory using semi-local functionals to obtain the dispersion energies [42-43]. In case of lacking direct experimental verification, a benchmark study like the present one is ultimately required to unambiguously determine the ab initio intermolecular potential.
4. Conclusion
In this paper we have calculated intermolecular potentials of the methane dimer at the most stable D3d conformation using the CCSD(T) method at complete basis set (CBS) limit. With Dunning’s correlation-consistent polarized valence basis sets, we
estimated the CCSD(T)/CBS potential curve using the extrapolation methods of Helgaker et al. [32], Martin [33], Feller [34] and a numerical method [35] on the potential data over the entire potential curve. The relative performance of these extrapolation methods has been carefully evaluated. Depending on the basis set quality, the CBS potential curves obtained by the methods of Helgaker et al. [32] and Martin [33] are more self-consistent than those using the method of Feller [34], in particular if the potential data calculated at small basis sets were used to perform extrapolations. An approximation scheme to obtain the CCSD(T)/CBS binding energy utilizing the MP2 energies has also been studied and extended to estimate other potential quantities. Using our previous MP2 data, we obtained an estimated binding energy within a 0.01 kcal/mol error with the aug-cc-pVDZ basis set.
5. Biblography
[1] J. D. Wright, Molecular Crystals (Cambridge University Press, Cambridge, 1987).
[2] P. Hobza and R. Zahradnik, Intermolecular Complexes : the role of van der Waals systems in physical chemistry and in the biodisciplines (Elsevier, New York,
1988).
[3] G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures (Springer-Verlag, Berlin, 1991).
[4] S. D. Chao, J. D. Kress, and A. Redondo, J. Chem. Phys. 122, 234912 (2005).
[5] A. van der Avoird, P. E. S. Wormer, and R. Moszynski, Chem. Rev. 94, 1931 (1994).
[6] G. Chalasinski and M. M. Szczesniak, Chem. Rev. 100, 4227 (2000).
[7] A. K. Rappe and E. R. Bernstein, J. Phys. Chem. A 104, 6117 (2000).
[8] C. E. Dykstra, G. Frenking, K. S. Kim, and G. E. Scuseria (Eds.), Theory and Applications of Computational Chemistry : The first forty years (Elsevier,
Amsterdam, 2005).
[9] G. C. Groenenboom, E. M. Mas, R. Bukowski, K. Szalewicz, P. E. S. Wormer, and A. van der Avoird, Phys. Rev. Lett. 84, 4072 (2000).
[10] R. Bukowski, K. Szalewicz, G. C. Groenenboom, and A. van der Avoird, Science, 315, 1249 (2007).
[11] M. M. Szczesniak, G. Chalasinski, S. M. Cybulski, and S. Scheiner, J. Chem.
Phys. 93, 4243 (1990).
[12] S. Tsuzuki and K. Tanabe, J. Phys. Chem. 95, 2272 (1991).
[13] J. J. Novoa, M.–H. Whangho, and J. M. Williams, J. Chem. Phys. 94, 4835 (1991).
[14] D. H. Gay, H. Dai, and D. R. Beck, J. Chem. Phys. 95, 9106 (1991).
[15] S. Tsuzuki, T. Uchimaru, K. Tanabe, and S. Kuwajima, J. Phys. Chem. 98, 1830
(1994).
[16] J. Nagy, D. F. Weaver, and V. H. Smith, Mol. Phys. 85, 1179 (1995).
[17] E. Fraschini and A. J. Stone, J. Comput. Chem. 19, 847 (1998).
[18] S. Tsuzuki, T. Uchimaru, and K. Tanabe, Chem. Phys. Lett. 287, 202 (1998).
[19] R. L. Rowley and T. Pakkanen, J. Chem. Phys. 110, 3368 (1999).
[20] S. Tsuzuki and H. P. Luthi, J. Chem. Phys. 114, 3949 (2001).
[21] P. Jurecka, J. Sponer, J. Cerny, and P. Hobza, Phys. Chem. Chem. Phys. 8, 1985 (2006).
[22] S. Tsuzuki, K. Honda and T. Uchimaru, M. Mikami, J. Chem. Phys. 124, 114304 (2006).
[23] A. H.-T. Li and S. D. Chao, J. Chem. Phys. 125, 094312 (2006)..
[24] T. Takatani and C. D. Sherrill, Phys. Chem. Chem. Phys. 9, 6106 (2007).
[25] Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput. 1, 415 (2005).
[26] J. A. Pople, M. Head-Gordon, and K. Raghavachari, J. Chem. Phys. 87, 5968 (1987).
[27] R. J. Bartlett and M. Musial, Rev. Mod. Phys. 79, 291 (2007).
[28] A. Szabo, N. S. Ostlund, Modern Quantum Chemistry. Introduction to Advanced Electronic Structure Theory (Dover, New York, 1996).
[29] R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys. 72, 650
(1980)
[30] T. H. Dunning, Jr., J. Chem. Phys. 90, 1007 (1989).
[31] S. F. Boys and F. Bernardi, Mol. Phys. 19, 553 (1970).
[32] T. Helgaker, W. Klopper, H. Koch, J. Noga, J. Chem. Phys. 106, 9639 (1997).
[33] J. M. L. Martin, Chem. Phys. Lett. 259, 669 (1996).
[34] D. Feller, J. Chem. Phys. 96, 6104 (1992).
[35] W. H. Press, S. A. Teukolsky, W. T. Vetterling, and B. P. flannery, Numerical Recipe in C (Cambridge University Press, Cambridge, 1996).
[36] Gaussian 03, Revision D.01, Frisch, M. J. et al.; Gaussian, Inc., Wallingford CT, 2004.
[37] D. C. Rapaport, The Art of Molecular Dynamics Simulation (Cambridge University Press, New York, 1995).
[38] S. D. Chao and A. H.-T. Li, J. Phys. Chem. A 111, 9586 (2007).
[39] C. C. Pai, A. H.-T. Li and S. D. Chao, J. Phys. Chem. A 111, 11922 (2007).
[40] A. Halkier, W. Klopper, T. Helgaker, P. Jorgensen, P. R. Taylor, J. Chem. Phys.
111, 9157 (1999).
[41] P. Hobza and J. Sponer, J. Am. Chem. Soc. 124, 11802 (2002).
[42] A. H.-T. Li and S. D. Chao, Phys. Rev. A 73, 016701 (2006).
[43] R. J. Meier, Chem. Phys. Lett. 401, 594 (2005).
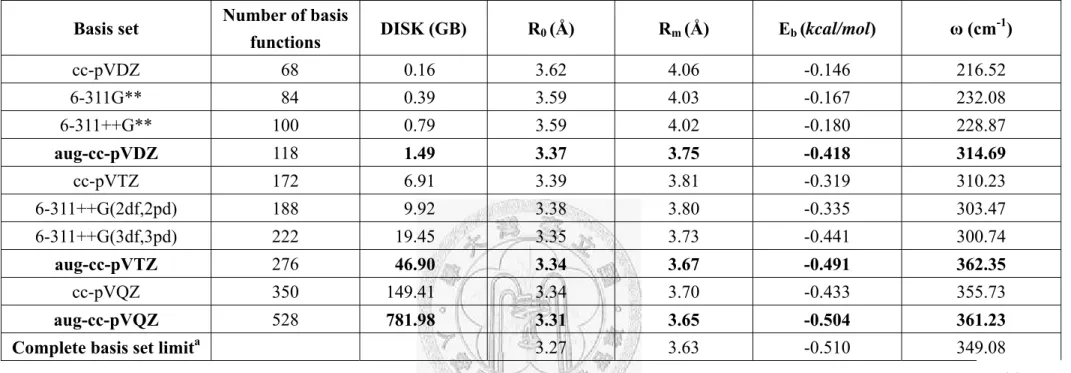
Basis set Number of basis
functions DISK (GB) R0 (Å) Rm (Å) Eb (kcal/mol) ω (cm-1)
cc-pVDZ 68 0.16 3.62 4.06 -0.146 216.52
6-311G** 84 0.39 3.59 4.03 -0.167 232.08
6-311++G** 100 0.79 3.59 4.02 -0.180 228.87
aug-cc-pVDZ 118 1.49 3.37 3.75 -0.418 314.69
cc-pVTZ 172 6.91 3.39 3.81 -0.319 310.23
6-311++G(2df,2pd) 188 9.92 3.38 3.80 -0.335 303.47 6-311++G(3df,3pd) 222 19.45 3.35 3.73 -0.441 300.74
aug-cc-pVTZ 276 46.90 3.34 3.67 -0.491 362.35
cc-pVQZ 350 149.41 3.34 3.70 -0.433 355.73
aug-cc-pVQZ 528 781.98 3.31 3.65 -0.504 361.23 Complete basis set limita 3.27 3.63 -0.510 349.08
a Basis set limit values obtained by the numerical method [40] with the aug-cc-pVXZ (X=D, T, Q) potential data, shown in boldface.
Table 1.1-1. The basis set dependence of important potential parameters of the BSSE corrected CCSD(T) intermolecular potentials. R0 is the
distance at which the potential is zero and is obtained from a two point interpolation of the calculated data. The bond length Rm, the binding
energy Eb and the intermolecular vibration frequency are obtained from an analysis using a quadratic polynomial function form to model
the equilibrium potential well. The disk space requirements (DISK) of the CCSD(T) calculation were recorded on a single node 1-processor
Table 1.1-1
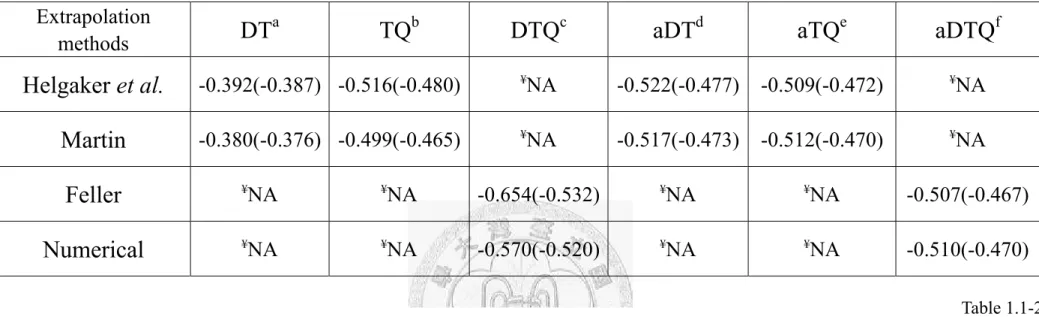
Extrapolation
methods
DT
aTQ
bDTQ
caDT
daTQ
eaDTQ
fHelgaker et al.
-0.392(-0.387) -0.516(-0.480) ¥NA -0.522(-0.477) -0.509(-0.472) ¥NAMartin
-0.380(-0.376) -0.499(-0.465) ¥NA -0.517(-0.473) -0.512(-0.470) ¥NAFeller
¥NA ¥NA -0.654(-0.532) ¥NA ¥NA -0.507(-0.467)Numerical
¥NA ¥NA -0.570(-0.520) ¥NA ¥NA -0.510(-0.470)aBasis set limit estimation with the cc-pVXZ(X=D and T).
bBasis set limit estimation with the cc-pVXZ(X=T and Q).
cBasis set limit estimation with the cc-pVXZ(X=D, T, and Q).
dBasis set limit estimation with the aug-cc-pVXZ(X=D and T).
eBasis set limit estimation with the aug-cc-pVXZ(X=T and Q).
fBasis set limit estimation with the aug-cc-pVXZ(X=D, T, and Q).
¥ Not available.
Table 1.1-2. Comparison the estimated CCSD(T) and MP2 (numbers in parenthesis) binding energies at the complete basis set
limit using the four extrapolation methods. The unit is kcal/mol.
Table 1.1-2
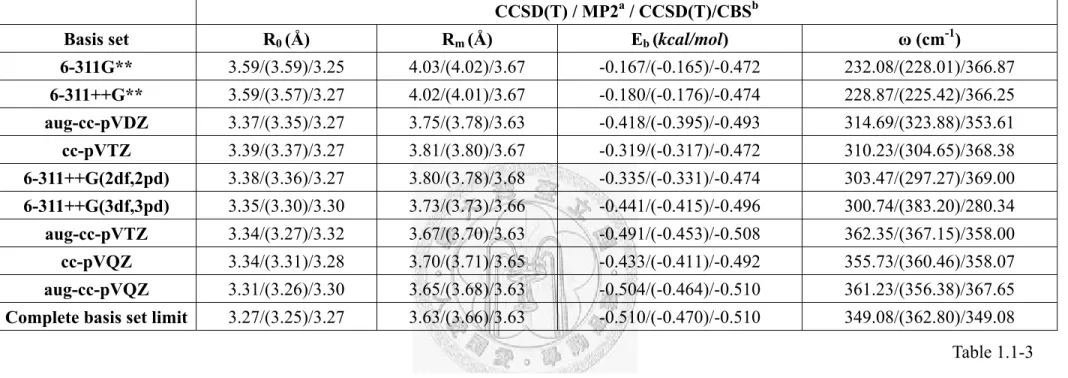
CCSD(T) / MP2a / CCSD(T)/CBSb
Basis set R0 (Å) Rm (Å) Eb (kcal/mol) ω (cm-1)
6-311G** 3.59/(3.59)/3.25 4.03/(4.02)/3.67 -0.167/(-0.165)/-0.472 232.08/(228.01)/366.87 6-311++G** 3.59/(3.57)/3.27 4.02/(4.01)/3.67 -0.180/(-0.176)/-0.474 228.87/(225.42)/366.25 aug-cc-pVDZ 3.37/(3.35)/3.27 3.75/(3.78)/3.63 -0.418/(-0.395)/-0.493 314.69/(323.88)/353.61 cc-pVTZ 3.39/(3.37)/3.27 3.81/(3.80)/3.67 -0.319/(-0.317)/-0.472 310.23/(304.65)/368.38 6-311++G(2df,2pd) 3.38/(3.36)/3.27 3.80/(3.78)/3.68 -0.335/(-0.331)/-0.474 303.47/(297.27)/369.00 6-311++G(3df,3pd) 3.35/(3.30)/3.30 3.73/(3.73)/3.66 -0.441/(-0.415)/-0.496 300.74/(383.20)/280.34 aug-cc-pVTZ 3.34/(3.27)/3.32 3.67/(3.70)/3.63 -0.491/(-0.453)/-0.508 362.35/(367.15)/358.00 cc-pVQZ 3.34/(3.31)/3.28 3.70/(3.71)/3.65 -0.433/(-0.411)/-0.492 355.73/(360.46)/358.07 aug-cc-pVQZ 3.31/(3.26)/3.30 3.65/(3.68)/3.63 -0.504/(-0.464)/-0.510 361.23/(356.38)/367.65 Complete basis set limit 3.27/(3.25)/3.27 3.63/(3.66)/3.63 -0.510/(-0.470)/-0.510 349.08/(362.80)/349.08
a The MP2 binding energy from Ref. [26].
b The estimated CCSD(T) basis set limit binding energy using the Eq.(1).
c A missing factor 2 to account for the reduced mass in calculating the frequency from our previous MP2 calculations [26] is included.
Table 1.1-3. Basis set dependence of the estimated potential parameters of the CCSD(T)/CBS potential using Eqs. (1) and (2).
Table 1.1-3
FIG. 1.1-1. The BSSE corrected HF, MP2, and CCSD(T) interaction potentials of the methane dimer using the aug-cc-pVQZ basis set.
FIG. 1.1-2. The estimated CCSD(T)/CBS potential curves using the direct extrapolation scheme with the four extrapolation methods on the cc-pVXZ potential data.
Fig. 1.1-1
Fig. 1.1-2
FIG. 1.1-3. The estimated CCSD(T)/CBS potential curves using the direct extrapolation scheme with the four extrapolation methods on the aug-cc-pVXZ potential data.
Fig. 1.1-3
1.2
Intermolecular Potentials of the Methane Dimer Calculated with Møller-Plesset Perturbation Theory and Density Functional Theory and Comment on "Intermolecular Interaction Potential of the Methane Dimer From the Local Density Approximation”1. Introduction
To verify the recently calculated intermolecular interaction potentials of the methane dimer within the density functional theory using the (Perdew) local density approximation (LDA) [Chen et al., Phys. Rev. A 69, 034701 (2004)], we have performed a parallel series of calculations using the LDA/6-311++G (3df, 3pd) level of theory with selected exchange functionals (B, G96, MPW, O, PBE, PW91, S, and XA). None of the above calculated intermolecular interaction potentials from the local density approximation reproduce the results reported in the commented paper. In addition, we point out the inappropriateness of using the Lennard-Jones function to model the long-range parts of the calculated intermolecular interaction potentials, as suggested positively by Chen et al.
Chen et al. [1] recently reported the intermolecular interaction potentials of the methane dimer (CH4)2 within the standard density functional theory (DFT) scheme [2]
using the (Perdew) local density approximation (LDA) [3], the pseudopotential [4,5], and the plane-wave expansion [6]. Their results agreed surprisingly well with those
obtained by the correlation-corrected Møller-Plesset (MP2, MP3) [7] and coupled cluster (CCSD(T)) [8] methods using a large basis set [9]. Because it has been known for some time that the usual DFT based approaches, using either the LDA or the generalized gradient approximation (GGA), can not calculate the intermolecular interaction potentials of molecular dimers to such a high level of accuracy [10-12], it is important to perform a parallel series of calculations using the available implementations of commonly used exchange-correlation functionals to verify the proposed results by Chen et al.
Intermolecular interaction potentials, or van der Waals interactions, or non-covalent-bonded interactions, play an essential role in condensed matter physics, materials chemistry and structural biology. While these interactions are normally one or two orders of magnitude weaker than typical covalent bonds, they are crucial in determining the thermodynamic properties of molecular liquids and solids [13], the energy transfers among molecular complexes [14], and the conformational tertiary structures of macromolecules such as protein and DNA [15]. Unlike intramolecular covalent bonds, intermolecular bonds do not originate from sharing of electrons but rather arise from simultaneous electron correlation of the separated subsystems [16].
Different from stiff covalent bonds, they are relatively soft and non-rigid. Early studies of intermolecular interactions can be traced back to one century ago [17],
while measurements of these interactions are still challenging in the present time [18].
The main difficulty in determining intermolecular interactions experimentally resides at limited samplings of the potential energy surface. For example, experiments using the X-ray crystallography or the laser spectroscopy mainly explore the equilibrium regions of the potential, while thermodynamic measurements in the gas or liquid phase often yield isotropic potential data without the desired stereo-chemical responses. In addition, the measured potentials sensitively depend on the thermodynamic conditions. Usually two measurements carried out in different conditions cannot be compared directly but rely on auxiliary theoretical modeling.
Alternatively intermolecular potentials can be calculated in terms of correlation-corrected quantum chemistry methods [19-21] or density functional theory (DFT) [22-23]. These quantum mechanics based potentials are requested by ab initio molecular dynamics simulations [24] and by classical molecular simulations using force field constructions [25]. Among the components of an intermolecular interaction, the London dispersion force is the most difficult to calculate. The reason is that dispersion interactions arise from the non-local “dynamic” correlations [26]. This non-locality demands full exploration of the time-dependent perspective of quantum mechanics. Often an electron correlation-corrected method and a large basis set are
required to obtain accurate dispersion forces [27]. Also a technical note is in order.
Most present implementations of quantum chemistry programs utilize Gaussian type functions to fasten the calculations of Coulomb repulsion integrals. Because Gaussian type functions are local functions, a large basis set is indispensable to perform a correlation energy calculation. Moreover, these functions do not have the correct asymptotic behavior as the intermolecular separation becomes large. Therefore, the basis set limit of the calculated potential must be estimated so as to be consistent with the conventional perturbation theory.
For general polar molecular systems, the relatively weak dispersion energy is masked by the competing electrostatic energies and hydrogen bond interactions. Nonpolar atomic and molecular dimers are usually taken as a prototype case to study the dispersion energy. Many previous studies on dispersion forces have focused on atomic inert gas dimers and several important conclusions have been drawn from the calculations [28]. There are, however, comparatively fewer detailed studies on whether similar conclusions can be applied to molecular dimers. Because of the extra degrees of freedom and the stereo-chemical responses, the conclusions about atomic dimers may need to be extended or modified in dealing with molecular dimers.
Methane is a non-polar molecule with vanishing dipole and quadrupole moments and
the first nonvanishing electrostatic interaction is the octopole-octopole interaction.
The higher order electrostatic interactions are rather weak and decay fast at large intermolecular separation. The dominant long-range attraction for the methane dimer is thus due to the London dispersion force. On the other hand, the strong repulsive force almost comes from the exchange-repulsion interaction due to the overlapping of electron clouds. Because the exchange-repulsion interactions have been incorporated in the Hatree-Fock (HF) self-consistent theory, post-HF methods such as the Møller-Plesset (MP) perturbation theory and the coupled cluster (CC) theory are often used to calculate the correlation effect. Contrasting both sets of calculation helps to delineate the relative importance of the dispersion energy in the overall intermolecular interaction. In addition, the interaction potentials of alkanes, among them methane being the smallest model, is very crucial in determining the packing morphology in solids and liquids and in lipid bilayers [29-31]. The potentials are also requested for mesoscale simulations for macromolecules [32] because many polymers contain alkyl groups as their backbone units or side chains. Therefore, the calculation of intermolecular interactions of the methane dimer is a “must” and serves as a prototype example to start to investigate the various factors affecting the calculations of these interactions.
There have been many quantum chemistry studies of intermolecular potentials of the methane dimer using correlation-corrected methods [33-44]. Szczesniak et al. [33]
have used the MP2 method with the Sadlej basis to calculate the interaction energies for the six conformers of the methane dimer. They found that the minimum-energy conformation is the D3d conformer and the dimer structure is determined by minimizing the steric repulsion between hydrogen atoms belonging to opposite subsystems. Tsuzuki et al. [36] have studied the basis set effects using basis sets from 6-31G* to 6-311++G (2d,2p) to calculate the interaction energies for two conformers of the methane dimer (but none of them is the D3d conformer). They observed little basis set effect on the HF calculations as long as one uses a basis set larger than the 6-31G* one. The basis set effect is significant for the MP2 interaction energies and the basis set superposition error (BSSE). The dispersion energy can be seriously underestimated if a smaller (than 6-31G*) basis set has been used. They found that the BSSE uncorrected interaction energies do not systematically converge to a destined value, in contrast to those with BSSE corrections. Tsuzuki et al. [39, 43] studied twelve conformers and verified that the D3d conformer corresponds to the minimum-energy geometry. They observed that a large basis set with multiple polarization functions is necessary to evaluate the dispersion energy accurately. They found that augmentation of the diffuse d and p functions to the 6-311G** basis set
more efficiently yields the dispersion energy. Tsuzuki et al. [40, 43] explored the effect of the choice of the correlation-correction methods on the calculations of intermolecular interactions. They demonstrated that the MP2 and MP3 energies are not too far away from the higher level MP4 (SDTQ) calculations, while the latter is not less expensive than the CCSD(T) calculation. They tested the DFT using the BLYP, BPW91 and B3LYP functionals but found unbound interactions while the PW91 functional underestimates only 8% of the potential well depth. They suggest that DFT with the PW91 functional could be an alternative to the ab initio methods.
Recently, Tsuzuki et al. [44] have estimated the MP2 and CCSD(T) interaction energies of the n-alkane dimers at the basis set limit using Dunning’s correlation consistent basis sets.
Many previous studies mainly focused on the equilibrium region of the potentials with relatively few discussions of the full potential curves. Nevertheless, to construct a reliable force field model for molecular simulations, the full intermolecular potential surfaces are required. In this paper we perform a comprehensive up-to-date study on interaction potentials of the prototype methane dimer in terms of the HF, MP2 and DFT methods to gain more understanding of this system. With current computational powers, a detailed editing of the potential data base can be obtained for small size
molecular clusters. It is thus so important to obtain general features of the calculations that we can follow to explore large scale molecular simulations via similar procedures.
The purpose of this paper is twofold. First, we use the state-of-the-art methodology to obtain accurate potential energies for the methane dimer. We would like to verify or modify the previous conclusions about the basis set effects and the effect of including the BSSE on the calculation details of the intermolecular interactions. The basis set effects on repulsion exponents, equilibrium bond lengths, binding energies, and asymptotic coefficients of the calculated intermolecular potentials are thoroughly studied. This is achieved using basis sets from STO-3G [45] to aug-cc-pVQZ [46].
The full potential curves are presented in order to see the overall scope of the potential. In particular, both the BSSE corrected and uncorrected results are presented to emphasize the importance of these corrections. Second, this paper attempts to re-assess the utilities of using the available implementation of the density functional theory in determining the intermolecular interactions. From the studies of atomic dimers, it has been found that conventional DFT based on the local density approximation (LDA) and generalized gradient approximation (GGA) cannot calculate the intermolecular interactions to a satisfying level of accuracy [47]. To address the title issue, we carry out a systematic DFT study of the equilibrium binding energies and bond lengths of the methane dimer using 90 functionals. Methane is a
non-polar molecule with vanishing dipole and quadrupole moments. The first nonvanishing electrostatic interaction is the octopole-octopole interaction and all higher order interactions are weak and decay fast at large intermolecular separation.
The dominant attraction for the methane dimer is thus due to the van der Waals force.
Therefore, the calculation of van der Waals interactions of the methane dimer serves as a prototype study to investigate the various factors affecting the calculations of these interactions.
2. Methods and Calculations
All the calculations were performed using the Gaussian03 package suite [48] and followed a theoretical procedure similar to that employed by Tsuzuki et al. [9] Fig.
1.1-1 shows the calculated interaction potentials of (CH4)2 with a set of selected exchange functionals (B, G96, MPW, O, PBE, PW91, S, and XA) [49], together with the Perdew correlation functional dubbed as PL (Perdew local) [3]. Although we have used a pretty large basis set, 6-311++G (3df, 3pd), which has been shown to lead to convergent results for (CH4)2 at a chemical accuracy [50], none of the calculated intermolecular interaction potentials reproduce the results of Chen et. al. A puzzling point in the commented paper is that there are two sets of data, one reported in their Fig. 1.2-1 and the other as numerical data in the text [1], while the latter is twice the former. Because there was no further clarification on this apparent inconsistency in
the commented paper, we present both data for comparison in Fig. 1.2-1 (open symbol-lines). Restated, neither of them can be reproduced in the present calculations.
In the case of methane dimers, a large part of the exchange-repulsion interactions can be calculated by the HF method. The calculation of electron correlation energies depends on the level of the correlation-corrected method, the size of the basis set, and the correction of the BSSE. The state-of-the-art choice of the correlation-corrected method is either the Møller-Plesset (MPx, x=2, 3, 4) perturbation method [51] or the coupled cluster method with iterative single and double substitutions and with noniterative triple excitations [CCSD(T)] method [52]. It has been found that the MP2 results for the methane dimer are not too much different from those calculated by the much more expensive CCSD(T) as long as a large basis set has been used [43]. To study the basis set effects, we have employed comprehensive basis sets from the Slater-type orbitals fitted with Gaussian functions (STO-nG, n=3~6) [45], Pople’s medium size basis sets [up to 6-311++G (3df, 3pd)] [53] to Dunning’s correlation consistent basis sets (aug-cc-pVXZ, X=D, T, Q) [46]. The basis set superposition error (BSSE) was corrected by the counterpoise (CP) method of Boys and Bernardi [54]. The MP2 interaction potentials at the basis set limit have been estimated using the methods of Helgaker et al. [55] and Feller [56] and a numerical extrapolation scheme based on the Lagrangian formula [57].
All the HF, MP2 and DFT calculations are performed using the Gaussian 03 program package [58] on a single node 2-processor AMD 250 PC cluster with distributed memory. The equilibrium geometry of a single methane molecule was first optimized at the MP2/6-31G* level of theory. To obtain the most stable intermolecular geometry, the methane dimer has been modeled by first fixing the carbon-carbon (C-C) distance while letting the two monomers to rotate freely. By approaching the monomers from the far side with several initial choices of mutual orientation, we found the minimum-energy conformation corresponds to the D3d symmetry conformer. This optimized conformer has been reached through the interplay of the steric stabilization of repulsive hydrogens in opposite monomers [33]. Subsequently the C-C distance was sampled in step 0.1 Å for a quite large range of intermolecular separation (normally 3~9 Å). During the scan we allow the individual methane molecule to be fully relaxed. This means that we do not fix the monomer geometry and the methane molecule is not assumed to be rigid. Although it is not expected to see much deviation from the rigid molecule approximation, in the real condensed phase environment, stretching, bending and torsional relaxations could be important for many subtle thermodynamic properties. The inclusion of intramolecular relaxation is especially relevant to the construction of force fields for use in molecular dynamics simulations