國立臺灣大學生命科學院分子與細胞生物學研究所 碩士論文
Institute of Molecular and Cellular Biology College of Life Science
National Taiwan University Master Thesis
利用細胞週期及微核糖核酸為基礎的邏輯匣 專一性殺死上皮癌細胞
A cell-cycle and microRNA-based AND logic gate to specifically kill epithelial cancer cells
王敏 Ming Wang
指導教授: 黃筱鈞 博士 Advisor: Hsiao-Chun Huang, Ph.D.
中華民國 107 年 11 月
November 2018
謝 誌
經歷了約兩年半的時間,這段期間全然感謝師長及眾多親友的協助,對於各 方面人事物的支持以及鼓勵。碩士這段期間,必須承認是孤單以及看似永無止盡 的實驗計畫,往往得到的答案又是在台上怯澀地說出:「結果並不如預期,但是 我認為……。」自己口中說出的「原因」像是對台下的直銷話術,又像是對自己 的正反質疑。一開始新鮮人的激昂眼神,如今也成了穩沉於海底而隨時又可拉起 而遠洋的錨。我們都在實驗室裡面對著電腦、實驗桌或是廁所回程後空無一人實 驗室時感受到對於自己選擇的不解以及困惑,但此時此刻卻也是在自己獨自完成 挑戰之後最為恰當的背景,正是因為這個空蕩蕩的時空中被自我意識滿滿地塞滿 了堅持不懈,當我回頭時並不曾疑惑自己是否只是浪費,只是看著黎明映在偶然 經過的窗,我只記得那是如此的美景。
最為感謝的是黃筱鈞老師除了在學術上的指導外(老師的全心全力早就不在
話下),對於我人生更是非常重要而且我畢生感謝的導師。老師不曾因為實驗結
果的不理想或是私人因素導致進度不如預期時放棄我,相反的,老師慷慨地給我 一段空白的時間去整理自己,甚至在我失控提出無理要求時刻,苦口婆心地告訴 我很多從她的人生中得到的體悟,那一字一句對我來說都是人生的轉捩點。我非 常感謝老師在我最脆弱,甚至是影響了整個實驗氣氛時,不斷的鼓勵我,想辦法 站在我的身邊去理解並處理問題,我深深地明白,如果沒有老師拉住我,我是不 可能完成碩士學位的。
另外我想感謝梁安柔學姊,從一開始光是實驗技術就跟著她學習了至少五遍 以上,學姊的專業以及勤奮的態度一直都是讓我佩服不已的地方,更時時刻刻督 促我必須同樣努力。一樣的,在一年的相處時間裡(我是如此地感嘆時間的不 夠)騎著腳踏車去學校周邊吃飯,在實驗室最乏味的時刻鬥鬥嘴或是唱唱歌也 好,與妳在一起的時光永遠是我碩士生活中最為懷念的也最為不捨的。妳不曾吝
得角色。再者,是我的好夥伴鍾佩妤,共同經營相同題目的妳時常配合並協助實 驗上的問題以及瑣事之外,也是我在台大第一個認識的朋友。妳是朋友一樣的攜 手度過許多節日及時光,我記得第一餐是一起吃貴到咋舌的一百元的排骨便當;
妳也是姐姐一樣的體諒並照顧我,謝謝妳往往鼓勵我的給我小紙條以及小餅乾。
非常感謝同期的實驗室同學陳亭均及廖得健,我們四個像是這兩年以來突然被指 定的家人,有過不滿也有過歡樂,在實驗室嘩啦嘩啦的聊天又或著太靠近而爭吵 喧鬧著,對於我來說如今都已成了最為重要的回憶。也謝謝實驗室的碩一碩二學 弟妹在我延畢的這段期間幫忙處理了非常多實驗室的瑣事,非常抱歉我逃了很多 次的值日生工作。
另外還要感謝朱雪萍老師以及吳亘承老師擔任我這次的口試委員,不吝嗇地 給予我非常多的實驗建議以及論文的修改方向,讓我的論文能夠更加完整。也感 謝所辦的魏姊,在我選錯課時以及申請口試時非常用心地協助我。感謝台大生科 館科技共同中心提供各式的儀器以及技術協助。
最後想感謝我的父母在這段時間經濟以及情感上的所有支持,是您們讓我能 夠在台大好好的學習並且完成自己的夢想,是這段時間有您們的鼓勵以及諒解,
我才能夠毫無任何顧慮的埋首於所愛的事物之中,我一直對於讓你們擔心難過感 到非常的抱歉。感謝哥哥在工作繁忙之餘還跑上台北陪我吃飯,雖然你一直以來 都看似沉默不語,卻總是記得寄好吃的東西給我,也私下幫我解決了非常多的困 難而從不邀功。感謝羅仕瑋在碩士的最後期間陪伴在我身邊,每當我情緒以及壓 力緊繃時,堅持不讓我一個人獨處,讓我連普通的便當店都覺得格外的好吃了。
我一直以來都了解自己是如此的幸運才能遇到這麼多的貴人,如今歹戲拖棚 的碩士生活也告一段落了,接下來新的生活,我相信有您們的鼓勵與支持,我一 定可以繼續撐下去的。非常感謝以上提到的所有人以及沒來得及提到的那些人。
中文摘要
在當今的癌細胞治療之中,針對細胞不正常分裂的藥物為相當有潛力的方式 之一。然而,抗有絲分裂的治療方式並沒有精準的專一性,因此可能造成人類體 內也會藉由快速分裂的細胞(例如骨髓幹細胞或皮膚細胞等等)受到影響造成其他 副作用。以骨髓幹細胞為例,當幹細胞受到藥物嚴重的抑制作用後,容易造成嗜 中性白血球低下,進而導致其他疾病產生。因此,本團隊利用合成生物學的方式 去設計足以專一性殺死癌細胞且不會影響造血相關細胞的邏輯匣(genetic circuit)。
此設計利用四環素控制的表現系統 (tetracycline-controllable expression system)作 為開關以及核糖核酸干擾(RNA interference, RNAi)的現象來達到篩選的目的,並 藉由已知骨髓相關細胞株本身含有相較於其他細胞株更為顯著表現的微型核糖核
酸 142(microRNA-142)以及細胞分裂時大量表現的細胞週期素 B1 啟動子(Cyclin
B1 promoter)兩個訊息來偵測胞內環境,最後專一性的對癌細胞表現特定基因促使 其走向凋亡。在本次實驗結果當中,在利用螢光蛋白作為基因表達的指標的情況
下,感染過邏輯匣的細胞確實會在進入細胞循環時啟動細胞週期素 B1 啟動子並
產生有毒基因並造成分裂中的海拉細胞(HeLa cells)以及人類胚胎腎細胞(293T cells)進入細胞凋亡。
另外,為了加強細胞感染的效率,本團隊試圖建立穩定細胞株(Stable cell line) 以達到最適當的效率。首先,我們必須建構出具有持續表現的啟動子(CMV Pro- moter)以及誘導型啟動子(TRE Promoter)的載體。然而,在組合結果後發現兩個啟 動子之間會產生洩漏的效應(leakage),造成在未誘導的情況下皆會造成下游基因 的表現。為了解決該問題,我們添加了絕緣子(Insulator)去阻擋洩漏效應。實驗結 果顯示在兩個啟動子之間加入兩倍的絕緣子中心元素可以有效阻擋下游螢光蛋白 的表現量,進而抑制洩漏效應的發生。我們期許在未來的實驗中,可以進一步的 測試此邏輯匣在懸浮的造血相關細胞內是否能夠同樣保持功能,並作為未來的抗
有絲分裂細胞治療的新方式。
關鍵詞 :癌症、抗有絲分裂、基因治療、合成生物學、核糖酸干擾、細胞週期素、
微型核糖核酸、四環素控制的表現系統、絕緣子。
Abstract
Anti-mitotic chemotherapy is a cancer therapy that kills cells with high division rate. Since abnormal proliferation is one of the most prominent characteristics of cancer cells, antimitotic drugs are expected to have high selectivity and sensitivity. However, non-specific targeting of hematopoietic stem cells (HSCs) that also divide rapidly is one
of the most severe side effects. To overcome this problem, we constructed a synthetic AND genetic logic, utilizing two sensors, cyclin B1 promoter and miR-142-5p binding sites, in mammalian cells. miR-142-5p was chosen as it is present exclusively in hema- topoietic lineages. When cells enter G2 phase of cell cycle, cyclin B1 promoter will be active to express Tet-On 3G transactivator protein. In the presence of doxycycline, the
transactivator will then activate a toxic gene, here hBax-β or truncated caspase-3/7, tagged with miR-142-5p binding sites. In this setting, HSCs, programmed with miR-142 production, toxic gene will be repressed thus cells are protected, whereas in non-hema- topoietic cancer cells, where miR-142 level is significantly lower, apoptosis will be trig- gered. We have successfully cloned and characterized all individual parts in the circuit
and have observed the circuit’s capability to render death in HeLa cell. Moreover, we are currently optimizing the circuit by co-expressing another apoptotic gene and built the stable cell lines for increasing the transfection efficacy. However, when we construct
a vector containing the two different types of promoter, i.e. constitutive and doxycy- cline-inducible, there appears to be interference between promoters. In resolving this, we tested an insulator core element and found that two tandem copies of this sequence can effectively suppress the interference. We envision our circuit will serve as a useful alterative tool for targeted cancer therapy in the near future.
Key words: cancer; anti-mitotic; gene therapy; synthetic biology; RNA interference;
cyclin B1; microRNA; insulator.
Contents
口試委員審定書 #
謝誌 i
摘要 iii
Abstract v
Contents vii
Figure List x
Table List xii
CHAPTER 1 Introduction 1
1.1 Antimitotic chemotherapy 1
1.2 The design of cell treatment approached by Synthetic biology 1
1.3 Two sensors of the gene circuit 2
1.4 The choice of toxic gene 4
1.5 The insulator to prevent interference 5
CHAPTER 2 Materials and Methods 9
2.1 Bacterial Strain and Culture method 9
2.2.1 Strain 9
2.2.2 Culture 9
2.2 Cloning Methods of Recombinant Constructs 10
2.2.1 Vector 10
2.2.2 Plasmids preparation 11
2.2.3 PCR 12
2.2.4 Enzyme Digestion 13
2.2.5 Gel extraction and PCR Clean Up 14
2.2.6 Oligonucleotides annealing design 15
2.2.7 Ligation 16
2.2.8 Transformation & colony PCR 16
2.3 Cell lines and cell culture 17
2.4 Transfection 18
2.5 Drugs treatment and cell synchronization 19
2.6 Fluorescence microscopy and time-lapse imaging 19
2.7 Flow cytometry 20
2.8 Cell Proliferation Assay 20
2.9 Trypan Blue exclusion assay 21
2.10 primer list 22
CHAPTER 3 Results 23
3.1 Comparison between full-length and truncated cyclin B1 promoter 23
3.2 The degree of apoptosis triggered by hBax-β, cleaved-caspase-3 and cleaved- capsepase-7 25
3.3 Activation of the complete circuit in HeLa cells 26
3.4 Insulator between two promoters 28
CHAPTER 4 Discussion and Future Work 31
CHAPTER 5 Figures 33
CHAPTER 6 Reference 52
Figure List
Fig.1 The diagram of conception and basic design
Fig.2 The flow chat with fusing 4 repeats miR-142 microRNA binding site.
Fig.3 The aligements of procaspase-3 and cleaved caspase-3 ; procasepase- 7 and truncated caspase-7.
Fig.4 The expression level of repoter triggered by full length (949) or truncatred (342) cyclin B1 promoters at G0 and G2 phase.
Fig.5 The transfected cancer cells which constitutively express the toxic gene re- sults in cell apoptosis in HeLa cells and 293T cells.
Fig.6 The inducible promoter triggers the toxic gene results in cell apoptosis in HeLa cell.
Fig.7 Time lapse image of HeLa cells transfected pTRE3G-BI_ hBax-β+EGFP af- ter inducing for 24 hours by doxycycline
Fig.8 Measurement the cell viability and cell death rate by trypan blue exclusion assay in HeLa cells.
Fig.9 Flow chat of cloning the construct containing constitutive and inducible pro- moter.
Fig.10 Designs of vector and two promoters and insulator.
Fig.11 The blocking ability of different copies of core elements of insulator in HeLa cells.
Fig.12 Quantify the expression level of reporter triggered by two promoters con- taining different copies insulator.
Table List
Table 2.1 LB (Luria broth)
Table 2.2 LBA Plate
Table 2.3 Protocol of PCR reaction
Table 2.4 Thermocycling Condition for a Routine PCR
Table 2.5 Time-SaverTM Protocol Table 2.6 Protocol of CIP reaction
Table 2.7 Protocol of T4 PNK reaction
Table 2.8 Protocol of T4 ligation
Table 2.9 Protocol of colony PCR
Table 2.10 Protocol of Lipofectamin 3000 Reagent
Chapter 1 Introduction 1.1 Antimitotic chemotherapy
Mitosis is the process that replicated chromosomes are equally separated to two
nuclei and form the two daughter cells during the cell division.[1] The mitotic check- point is a mechanism for preventing the unequal separate.[2] Anti-mitotics work by dis- rupting the spindle activities, which activate the spindle assembly checkpoint and arrest cells in mitosis, and finally the cells undergo apoptosis after a prolonged mitosis. Based on the unrestricted division in tumors, several antimitotic therapies can effectively insti-
gated the abnormal cancer cells into apoptosis. Moreover, antimitotic drug can selec- tively attack the rapidly and frequently anomalous cell division without targeting the non-cycling cells in normal tissue. [3]
Even thorough the antimitotic drugs are reported as mitosis-selective, they still
have the down side of attacking normal proliferating cells, such as hair follicles, diges-
tive tracts, bone marrow and so on.[4] In other words, an existing problem is how to prevent side effects of antimitotic drugs, that is the equally effective killing of cancer and normal dividing cells. [5] How to specifically target the cancer cells without effect- ing dividing normal cells is an important issue needed to be resolved now.
Synthetic biology is a research field that combines biology and engineering to, in this case, to rationally design novel therapies. The biological pathway is rewritten as electrical circuit analogies and sometimes can be modeled by mathematical equations.
Early synthetic biology works include the construction synthetic gene networks, such as toggle switch and repressilators. [6-8] Therefore, we can design novel gene therapy by
the concept of synthetic biology. The engineering-based methods in mammalian cells are similar to these early bacteria works. In the past decades, molecular mechanisms in mammalian cells are getting more and more well understood, and thus the development of gene therapies can use these well-studied mechanisms, such as microRNA and tran- scription factors, as “biosensors” to detect the cellular environment, and functional net-
works that obey the Boolean logic can be readily constructed in mammalian cells.[7, 9]
In a previous report, a multi-input logic circuit is constructed to use the combi- nation of six endogenous miRNA as biosensor to trigger the expression of the toxic gene, human Bcl-2–associated X protein(hBax-β), selectively in HeLa cells without effecting the HEK293.[8] For example, all the “high-expressed” markers must be present or above
the threshold, otherwise the output will be significantly repressed.
Two sensors of the gene circuit
The cancer cell is well known to have high division rate. Hence, the onset of mitosis is a major feature to separate cancer and normal cells. Cell proliferation is controlled by the expression of cyclin genes that activate cyclin-dependent kinase. Most importantly, cyclin B is required for the entering of G2/M phase in somatic cycling mammalian cells.
On the other hand, the G2-enhaced and deregulated expression of cyclin B1 promoter
(CBP) is a hallmark of tumor cells. To sum up, the activation of cyclin B1 promoter is an appropriate feature to detect abnormal division rate in cancer cells. [10, 11]
The microRNAs are a class of non-coding RNA, which are short and single stranded molecules that can regulate gene networks. MicroRNA can bind to the 3’un- translated region (UTR) of the target messenger RNA, which degrade the mRNA or
repress its expression at the translation level.[12] A few microRNAs are enriched in specific tissues. Here, microRNA-142(miR-142) is primarily expressed in hematopoi- etic tissues.[13] Moreover, the pre-miR-142 is the stem-loop structure that encoded two species, miR-142-3p and miR-142-5p, which are conserved in mouse, rat and human.
[12, 14, 15] According to Chung’s thesis, both miR-142-3p and miR-142-5p have higher
expression in hematopoietic cells, such as HL-60 and Jurkat cells [13], than in non- hematopoietic cells, like HeLa and 293T cells. Moreover, the quadruple miR-142-5p multiple binding site (4X miR-142-5p MBS) at 3’UTR can effected render repression in
HL-60 and Jurkat than non-hematopoietic cell lines. [16] Taken together, the miR-142- 5p that is exclusively present in hematopoietic cells is sufficient to inhibit the expression by specifically binding the miR-142-5p MBS.
Based on these, we design an artificial synthetic circuit, here an AND gate, with two sensors to specifically target tumor cells without attacking hematopoietic cells (Fig.
1A). The gate is coupled with a inducible system, that is the Tet system, which was found to negatively regulate the genes in the tetracycline-resistance operon in E.coli.
The core of Tet-On and Tet-off system is the regulatory protein, TetR, which constitutes the hybrid protein, tetracycline-controlled transactivator (tTA). In our case, the Tet-On system, is based on reverse tetracycline-controlled transactivator (rtTA), that binds to
TRE (tetracycline-response element) promoter that contains minimal CMV promoter following with seven repeat of tet operator sequences (TetO), and triggers the expres- sion of the genes we want when doxycycline (Dox) is present. The toxic gene is con- struct after this promoter to kill the cancer cells in our circuit.
1.3 The choice of toxic gene
To achieve effective killing, we sought for previously reported pro-apoptotic genes in the apoptotic network, such as hBax-β and caspases in the caspase family. [8, 13, 17- 19] Caspases are known as endoproteases that regulate the inflammation and cell death.
Scientists have classified caspases into initiator caspases and executioner caspases. They have also found that the procaspases require dimerization and cleavage of the prodomain for activation. In this study, caspase-3 and caspase-7 are tested since they are essential for apoptosis and are of higher expression in cancer than normal cells. According to the previous data, the procaspase-3 is not effective in triggering cell death.[20] Hence, in
this study, the prodomain of both capsease-3 and caspase-7 are removed to directly have the endoproteases ability. (Fig.3) A recent report demonstrated that the truncation mutant at amino acid 57 for caspase-7 can induce apoptosis rapidly.[18]
On the other hand, hBax-β, a member of Bcl-2 family, is upstream of these caspases
and is reported have tumor suppresser activity.[21] hBax-β can affect the permeability
of mitochondria pore to trigger cytochrome c release. Following the induction Apaf-1, apoptosis is triggered by cleavage of procaspase-9. Besides, it has been demonstrated that hBax-β can exert efficient killing in HeLa cells[8, 19].
Taken together, these toxic genes are constructed downstream of pTRE3G-BI, which is only expressed with the conditions of both sensors are satisfied. In this study,
they can cause cell death in non-hematopoietic cancer cells (such as HeLa). (Fig.1C) 1.4 Insulator to prevent interference
The central question is whether hematopoietic cells, like HL-60 and Jurkat [13],
can be protected when the toxic circuit mentioned above is present. Unfortunately, it is too difficult to co-transfect two plasmids into suspension cells by either lipofectamine or electroporation.
To resolve the problem, we aim to generate stable cell lines. mCherry positive signal can be used to select for stably transfected cells using flow cytometer, as it is
under a constitutive CMV promoter. However, EGFP positive signal cannot be used for the similar selection, as it is under the inducible promoter (TRE), which upon induction, the toxic gene will also be expressed to kill the cells. Therefore, we need a vector con- taining constitutive and inducible promoter simultaneously, which means that EGFP should be triggered by CMV promoter (to be detected by sorting), and the toxic genes
should still be triggered by TRE promoter (to be off before the addition of doxycycline).
To achieve this, we digested one of the TRE promoter from pTRE3G-Bi, ligated the element into pBI-CMV1, and use EGFP and mCherry as reporters. However, the interference from CMV promoter causes the inducible promoter to be activated even without the presence of the inducer. One possible reason is that the distance between
two promoters is too short (only 65 bp), which is not far enough to avoid interference.
Thus, we look for insulator elements that can be inserted to shield the promoter form unwanted regulatory elements.
According to previously report, the DNA sequence at the 5’end of the chicken
β-globin locus has the ability for possesses the barrier activity for protecting against
position effects. We use this characteristic to approach the aim blocking the leakage.
The 5’end of the chicken β-globin locus, containing a constitutive DNase I-hypersensi-
tive site (5’HS4)[22], functions as the insulator. The chromatin within the β-globin do- main is DNase I-resistant that may be thought like a “heterochromatic” region. The N-
terminal tail of histone H4 is acetylated within the β-globin domain, transcriptionally, cause the inactive regions. Therefore, the HS4 insulator archives the aim of reducing
enhancing effects on a promoter and preventing the chromosomal position effects. The 250bp of core element of insulator is a CpG domain which is GC-rich sequence and not
to vanish the functions of promoter itself. As the previous data, the insulator activity of two-copy of core element displays greater than the original 1.2-kb fragment one and it
also mentions about that the insulator activity of core element is associated with the numbers of copies, like the three-fold higher insulator activity with six copies of core element than the activity of the two-copy.[23-25] In this thesis, how to design a kind of insulator suitable for our experiment need to satisfied two conditions: efficiency block- ing ability and the certain restriction enzyme inserted at the 3’ and 5’ end which can be
reused and unlimited cloning until the leakage disappeared.
In the results, we successfully triggered the apoptosis happened in HeLa cells trans- fected with our circuit and we also combine the constitutive promoter and inducible
promoter with 2x core element of insulator located between two promoters as the great barrier to avoid the leakage effect. The further experiments will focus on how to resolve the transfected efficiency of hematopoietic cell lines.
Chapter 2 Materials and Methods 2.1 Bacterial Strain and Culture method
2.1.1 Strain.
The E.coli strain DH5α is used for plasmid cloning in this study. The
ECOSTM 101 competent cells was purchased form Yeastern Biotech Co. pre- vented for long-term storage at -80℃. The genotype consist of following:
endA1 recA1 relA1 gyrA96 hsdR17(rk-,mk+) phoA supE44 thi-1Δ(lacZYA- argF) U169Φ80Δ(lacZ)M15 F-
2.1.2 Culture
The bacteria were grown in LB broth (MDbio) or LB agar plates (Fo- cusBio) contained Ampicillin (100μg/ml) as antibiotics and incubated for 14-
16hr.
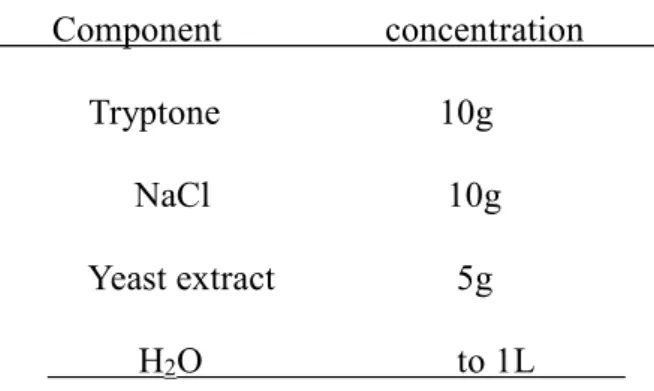
Table 2.1 LB (Luria broth) Component concentration
Tryptone 10g NaCl 10g
Yeast extract 5g
H2O to 1L
Table 2.2 LBA Plate
Component concentration Tryptone 10g
NaCl 10g Yeast extract 5g
Bacto Agar 15g
H2O to 1L
2.2 Cloning Methods of Recombinant Constructs
2.2.1 Vector
pBI-CMV1 (Clontech) is a bidirectional expression vector constitutively ex-
pressing two proteins simultaneously in mammalian cells. Containing with Ampicillin resistance gene (Ampr), it allows selection in E.coli for further ex- periments.
pTRE3G-BI (Clontech) is an inducible vector for mammalian cells and pro-
vide low basal expression and highly expression after induced by Doxycycline.
Based on Tet-On Advanced system, the transcription regulator, Tet-On 3G transactivator protein with high sensitive relation with Doxycyclin. It also contain the Ampicillin resistance gene for selection in in E.coli.
plVX-MetLuc
2.2.2 Plasmids preparation
Plasmids used in this study are first purified using Mini PlusTM Plasmid DNA Extraction System (Viogene). Most of steps followed as the protocol for Spin Method in manufacturer’s recommended protocols, and some parts are modified as follows: (i) 5 ml the plasmid-containing bacterial cells in LB me-
dium are all-pelleted centrifuge at full-speed. (ii) After centrifuge at full-speed for 3 min to remove the ethanol, the columns change into a new 1.5ml centri- fuge tube and place to on dry bath at 60℃ for 5 min to make sure the ethanol
actually removed. (iii) Add 55μl elution buffer to elute.
Due to the low copy numbers of vectors we used, plasmids further prep- aration by GeneaidTM Midi Plasmid Kit & GeneaidTM Midi Plasmid Kit (En- dotoxin free) (Geneaid). Culturing the 150ml bacterial cells, the cells transfer to 50 ml centrifuge tube and centrifuge at 4000 rpm for 10 min. At step 8.
(Elution), elute plasmids are precipitated by adding the same volumes of iso-
propanol and concentrated to 400μl by PrestoTM Plasmid DNA Concentration
Kit (Geneaid) started from step 3 of Plamid DNA Concentration Vacuum Pro- tocol. All the plasmid are long-term storage at
-20℃.
2.2.3 PCR
To amplify the gene of interest, it is Q5 High-Fidelity DNA polymerase
(NEB) that we used to produce the insert fragments from plasmid DNA or genomic DNA.
Table 2.3 protocol of PCR reaction
Component Final concentration 5X Q5 Reaction Buffer 1X 10mM dNTPs 200μM 10μM Forward Primer 0.5μM /1μM 10μM Reverse Primer 0.5μM /1μM Template DNA 1-5 ng Q5 High-Fidelity DNA polymerase 0.02U/μl 5XQ5 High GC Enhancer (optional) (1X) Nuclease-Free Water to 50μl
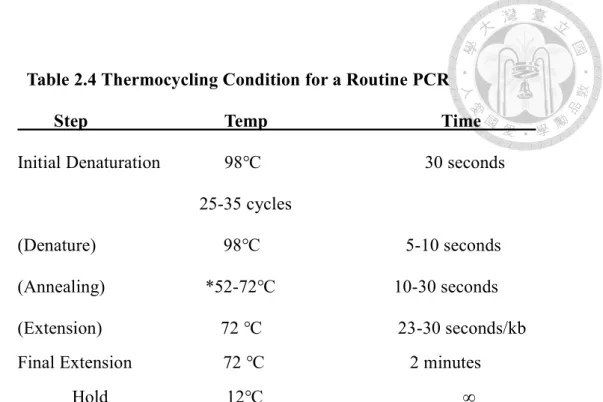
Table 2.4 Thermocycling Condition for a Routine PCR
Step Temp Time Initial Denaturation 98℃ 30 seconds
25-35 cycles
(Denature) 98℃ 5-10 seconds (Annealing) *52-72℃ 10-30 seconds (Extension) 72 ℃ 23-30 seconds/kb Final Extension 72 ℃ 2 minutes Hold 12℃ ∞
*Using the online tool of Tm calculator on the NEB website.
2.2.4 Enzyme Digestion
All the restricted enzymes are purchased form New England Biolabs (NEB). The reaction time is about 45 min to 2 hours at 37℃ for reaching the best efficiency.
Table 2.5 Time-SaverTM Protocol
Component Amount per reaction Restriction Enzyme 1μl
DNA 1μg 10X NEBuffer 5μl
Preventing from self-ligation, we used the Alkaline Phosphatase, Calf Intesti- nal (CIP) to nonspecifically catalyze the dephosphorylation of the 5’-end of
DNA. Incubated for 30 min at37℃, the CIP was used during the enzyme di-
gestion reaction.
Table 2.6 Protocol of CIP reaction
Component Amount per reaction DNA 1 pmol of DNA ends NEBuffer (10X) 2μl
CIP 1 unit
H2O to 20μl
2.2.5 Gel extraction and PCR Clean Up
To purify the DNA from agarose, Gel AdvancedTM Gel Extraction System (Vi- ogene) is used after agarose gel electrophoresis. 1%(w/v) agarose (Infinigen)/ 1X TAE Buffer is used for this experiment and run in 1X TAE buffer under 100V, stained by 0.5 μg/ml Ethidium Bromide Solution. TAE buffer (50X) is prepared by 242g Tris-base (J.T.Baker), 100 ml 0.5X EDTA (Calbiochem) and 57.1 mL glacial
acetic acid(Sigma-Aldrich) in deionized water to 1 liter. PCR AdvancedTM PCR
Clean Up System is used for PCR products or DNA fragment of enzymatic reaction, purified from 100-bp to 10-kb.
2.2.6 Oligonucleotides annealing design
To construct the 4X miR-142-5p MBS for microRNA-142 bind and repressed the gene expression (Fig.2), the sequences are separate to two paired single strand DNA
(about 50 bp per strand)with the cleavaged cutting sites, BglII and EcoRV, ordered form Genomics BioSci & Tech.
First, to anneal the two paired DNA, which are designed with part of overlapping sequence, we mix the 20 μM DNA into in a microtube incubated in 95℃ for 5 min and slowly cool down to room temperature (-1℃ per 1 min) . Second, to phosphorylate the
5’end for subsequent ligation, the DNA are incubated ing 37℃ for 30 min by following the T4 polynucleotide kinase protocol in NEB. The last step is ligation into the down- stream of the output gene.
Table 2.7 Protocol of T4 PNK reaction
Component Amount per reaction T4 ligase Buffer 5μl
DNA up to 300 pmol of 5´ termini T4 PNK 1μl
2.2.7 Ligation
To produce the final construct, ligation is the most traditional way that using the T4 ligase to ligate the enzyme digested DNA incubated in 16℃ overnight or in room
temperature for 15 min. The protocol is following as below.
Table 2.8 Protocol of T4 ligation
Component Amount per reaction T4 ligase Buffer(10X) 2μl
Molar ratio of vector DNA to insert DNA 3:1 T4 ligase 1μl
H2O to 20μl 2.2.8 Transformation & colony PCR
ECOSTM competent cells, DH5α, is used for our study. The cells mixed with the plasmid or ligation product are thawed on the ice for 5 min, heat shocked in 42℃ for 45 seconds, and spreading the dilution onto the selection plate. The colony growth
after incubated in 37℃ overnight.
To identify whether the constructs are successfully transform into the competent cells, the colony PCR is a quick way to used. Mixing the forward and reverse primers with the 2X Ready-to-load PCR Taq Master Mix (MDBio), the single colony is picked
up into the reaction for routine PCR.
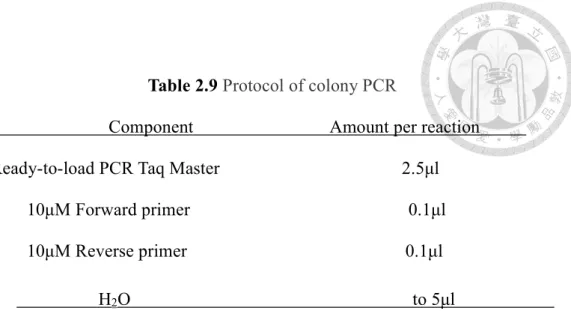
Table 2.9 Protocol of colony PCR
Component Amount per reaction Ready-to-load PCR Taq Master 2.5μl
10μM Forward primer 0.1μl 10μM Reverse primer 0.1μl
H2O to 5μl 2.3 Cell lines and cell culture
HeLa (human cervical cancer cell line) was acquired from Dr. Fon-Chun Ke's
lab and 293T (human embryonic kidney cell line) was acquired from Dr. Jean Lu’s lab. Both of HeLa and 293T cells are growth in high-glucose DMEM medium sup- plemented with 10% FBS, 100 units/mL of penicillin and 100 μg/mL streptomycin in the present of 5%CO2 at 37℃.
When cells growth to 80% confluence, the cells are subculture to the new plate
for continual growth. First, we remove the medium and wash once by 1X DPBS.
Second, the cells are incubated with the trypsin/EDTA for 5 min in 37℃ until the cells are dispersed and inactive the action of trypsin/EDTA by adding the same volume medium. The cells solution is transfer into the new tube for centrifuge at
750 rpm for 5 min to collect the cell pellet and replace the fresh medium after
throwing the subtenant. Eventually, the cells are seeded into the new plate and con-
tinued incubation.
2.4 Transfection
According to the different transfection efficiency, the Liposome-mediated
transfected mainly used in adhesion cells, like HeLa cells and 293T cells, and the electroporation used for the low transfection efficiency cells. In this study, it only display the Lipofectamin 3000 Reagent transfection. The cells are seeded into the wells of 12 or 24-well plate (Corning) and incubated for 24 hours. Importantly, the Lipofectamine 3000 Reagent is mixed with the Opti-MEM (Gibco) for a while.
Then, preparing the solution that the protocol is follow as below, the well-inverted
tube incubated for at least 10 min. Finally, the reagents are adding to the each well.
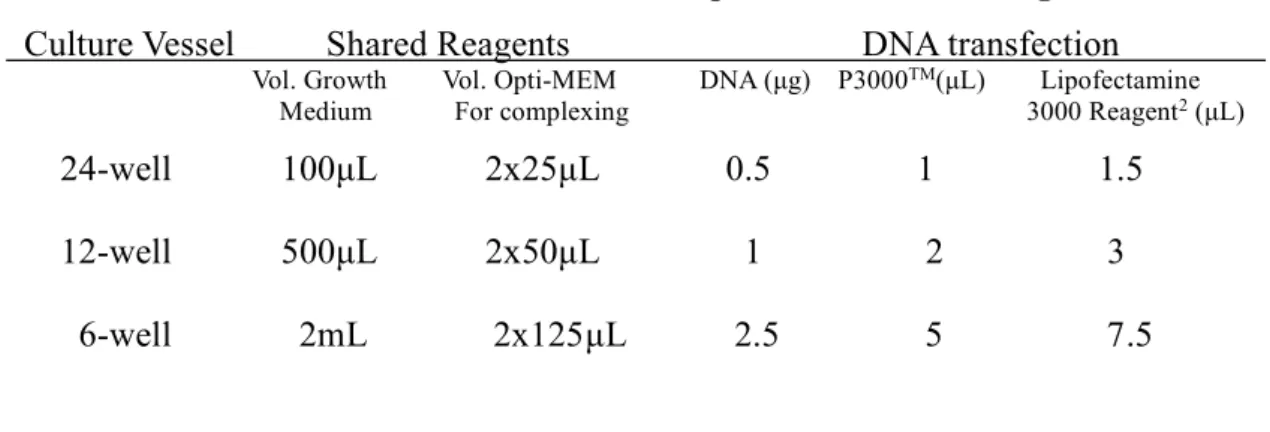
Table 2.10 Protocol of Lipofectamin 3000 Reagent
Culture Vessel Shared Reagents DNA transfection
Vol. Growth Vol. Opti-MEM DNA (μg) P3000TM(μL) Lipofectamine Medium For complexing 3000 Reagent2 (μL)
24-well 100μL 2x25μL 0.5 1 1.5 12-well 500μL 2x50μL 1 2 3 6-well 2mL 2x125μL 2.5 5 7.5
2.5 Drugs treatment and cell synchronization
Doxycycline hydrochloride (Dox, Sigma-Aldrich) is used for activate the Tet-On
system and trigger the output expression, incubating the cell in medium with 1,000 ng/ml Dox. STLC (S-trityl-L-cysteine, Merck Millipore) can block the cell cyclin in M phase with the medium containing 5 μM STLC, and it is also used as positive
control of apoptosis test treated for 48 hours. To observe the different expression level of each phase in cell cycle, the cells are culture in the medium containing 2 mM thymidine (Sigma-Aldrich) for 19 hours, washed by DPBS and incubated in fresh medium for 9 hours. Then, the cells are replaced with 2 mM thymidine for 16 hours, blocking cells in G1/S phase. To make the cell entry the G2 phase, the cells
which are already blocked in G1/S phase are replaced in fresh medium again for 2 hours. By culturing the cells in medium containing 0.5% FBS for a period of 72
hours to G0 phase.
2.6 Fluorescence microscopy and time-lapse imaging
All the images are taken by Inverted fluorescence microscopy Axio Ob-
server Z1 (Carl Zeiss) with ZEN software (Carl Zeiss). The filters we chose is the most adept for different fluorescence, like EGFP used the channel of 470/40 nm (excitation) 525/50 nm (emission) and mCherry used the channel of 560/40 nm
are seed in 24-well plate and covered by a layer of mineral oil (Sigma-Aldrich) on medium avoiding the evaporation. The pictures are automatically captured once every 15 minutes in an incubation chamber at 37°C with 5% CO2 continually pro-
vided for 24 hours.
2.7 Flow cytometry
To count the ratio of the fluoresces in the adhesion cells transfected with different circuit, we use the FACSCanto IITM flow cytometer (BD Biosciences) to gather these data. The cells are resuspended in the 1mL 1xDPBS and collected 105 cells that specific gated above the certain threshold defined by non-transfected cells.
We use the Coherent Sapphire 20 mW 488 nm solid state blue laser as our excitation.
Moreover, the measurement of experiment used different bandpass filter, 530/30 nm one with a PMT 250 V of EGFP and 585/42 nm one with a PMT 350V.
2.8 Cell Proliferation Assay
To measure the killing ability of genetic circuit, we chose the CellTiter 96®
AQueous One Solution Cell Proliferation Assay (MTS, Promega) as our tool to quantify. The transfected cells are seed into the 96-well plate (5000 cells/well) in 100μl of culture medium. After adding 20μl CellTiter 96® AQueous One Solution
Reagent into each well and incubating for 1-4 hours, the samples are recorded the absorbance at 490nm by using a ELISA plate reader.
2.9 Trypan Blue exclusion assay
To analyze the number of viable cells present in a cell suspension, we seed
1x105 cells per well and transfection for 24 hours. By trypsinizing the cells and resuspensed in 1X PBS, we mixed 10μl cell suspension and 10μl trypan blue at room temperature. Then the cell suspension is filled into two side of a hemacytometer counter. All cells are counted (clear and blue).
𝑐𝑒𝑙𝑙 𝑣𝑖𝑠𝑖𝑏𝑖𝑙𝑡𝑦 =𝑛𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝑐𝑙𝑒𝑎𝑟 𝑐𝑒𝑙𝑙𝑠
8 × 2 × 104
(Formula.1)
𝑐𝑒𝑙𝑙 𝑑𝑒𝑎𝑡ℎ 𝑟𝑎𝑡𝑒 = (1 − 𝑛𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝑐𝑙𝑒𝑎𝑟 𝑐𝑒𝑙𝑙𝑠
𝑛𝑢𝑚𝑏𝑒𝑟 𝑜𝑓 𝑡𝑜𝑡𝑎𝑙 𝑐𝑒𝑙𝑙𝑠 ) × 100%
where
𝑡𝑜𝑡𝑎𝑙 𝑐𝑒𝑙𝑙𝑠 = c𝑙𝑒𝑎𝑟 𝑐𝑒𝑙𝑙𝑠 + 𝑏𝑙𝑢𝑒 𝑐𝑒𝑙𝑙𝑠
Total cells indicate the clear cells and blue cells are count simultaneously. After con- verting with the Formula.1, the number of total cells are number of clear cells and blue cells.
(Formula.2)
2.10 primer list
Name Sequences (5’ → 3’)
EGFP_F(EcoRI) CCCCGGAATTCATGGTGAGCAAGGGCGAG
EGFP_R(PstI) AAAACTGCAGTTACTTGTACAGCTCGTCCATGCC mCherry_F(Ba
mHI)
GCCCGGATCCATGGTGAGCAAGGGCGAGG
mCherry_R(ClaI )
GCCCATCGATTTACTTGTACAGCTCGTCCATGCC
miR-142-5p MBS_P1
GAT CTA GTA GTG CTT TCT ACT TTA TGA GTA GTGCTT T CT ACT TTA
miR-142-5p MBS_P2
TAC TCA TAA AGT AGA AAG CAC TAC TCA TAA AGT AGA AAG CAC TAC TAG
miR-142-5p MBS_P3
TGA GTA GTG CTT TCT ACT TTA TGA GTA GTG CTT TCT ACT TTA TGA GAT
miR-142-5p MBS_P4
ATC TCA TAA AGT AGA AAG CAC TAC TCA TAA AGT AGA AAG CAC
hBax-β_F(SalI) GCC CGT CGA CAT GGA CGG GTC CGG GGA G hBax-
β_R(BglII)
GCC CAG ATC TTC AGA CAC GTA AGG AAA ACG CAT TAT AGA CCA C
Procaspase-7_F GCCCGTCGACATCGCCATGGCAGATGAG
Procaspase-7_R CGGGATCCCGGGCTATTGACTGAAGTAGAGTTCC cleaved-
CASP7_F(SalI)
GCC CGT CGA CAC ATA TCA GTA CAA CAT GAA TTT TGA AAA GC
cleaved-
CASP7_R(BglII )
GCC CAG ATC TCT ATT GAC TGA AGT AGA GTT CCT TGG
Procaspase-3_F GCCCGAATTCATGGAGAACACTGAAAACTCAGTGGAT- TCAAAATC
Procaspase-3_R GCCCGGTACCTTAGTGATAAAAATAGAGTTCTTTTGTGAG- CATGG
cleaved-
CASP3_F(SalI)
GTC GTC GAC ATG TCT GGA ATA TCC CTG GAC AAC AG
cleaved-
CASP3_R(BglII )
GCC CAG ATC TTT AGT GAT AAA AAT AGA GTT CTT TTG TGA GCA TGG
EGFP_F(BglII) CCC CAG ATC TCA TGG TGA GCA AG
Chapter 3 Results
3.1 Comparison between full-length and truncated cyclin B1 promoter
Cyclin B1 promoter is known to regulate cells to entry the G2 phase and M phase.[10]
According to previous data [22], the experiment to block cells in G2/M phase is to cul- ture cells in medium containing 5 μM STLC (S-trityl-L-cysteine, Merck Millipore) for
24 hours. STLC is a potent tumor growth inhibitor that has been identified to target the Human mitotic kinesin Eg5, an essential motor for the formation of the bipolar mitotic
spindle.[26] Compared with blocking at G1/S phase with double thymidine block, this is not the ideal experiment, as cells are mainly at M phase, and some cells may begin to undergo apoptosis. Hence, we decided to synchronize the cells at S/G2 phase by harvest-
ing cells 2 hours after double thymidine block. The result shows that both 342 CBP (truncated cyclin B promoter) and 949 CBP (full-length cyclin B promoter) [11] have increased level of expression at G2 (Fig.4a, 4b). Furthermore, the percentage of the mCherry-positive cells increased at S/G2 phase than these at G0 phase. (Fig.4c)
Taken together, the results confirm that both 342 CBP and 949 CBP acti-
vate upon entering the cell cycle. According to the previous data [27], 342 CBP and 949 CBP show similar expression level in G0 phase. As the fluorescence also in- creased significantly at G1/S in 949CBP, 342 CBP is chosen as our sensor since the
342 CBP is more specific to M phase [22]. In this study, we also showed that there are more numbers of activated cells in 342 CBP than in 949 CBP at S/G2 phase.
(Fig.4c)
3.2 The degree of apoptosis triggered by hBax-β, cleaved-caspase-3 and cleaved-
capsepase-7
To further know whether toxic gene(s) can trigger the epithelial cells death or not, we chose to test three well-known genes in the apoptotic pathway, human Bcl-2–associated X protein hBax (hBax-β) and two caspase family (caspase-3 and caspase-7), as our candidate genes. Both caspase-3 and caspase-7 are truncated by
removing their pro-domains. According to previous study, truncation mutant of caspase-7 at amino acid 57 can kill cells more effectively then other mutants.[18]
(Fig.3) Therefore, in this study, we test whether the expression of hBax-β, truncated caspase-3 and truncated caspase-7 can be used for killing cancer cells.
First, three toxic genes were constructed into the expression plasmid with bi-
directional CMV (constitutive) promoter, pBI-CMV1, with EGFP simultaneously expressed as the marker for transfection. (Fig.5A) Result shows that constitutive ex- pression of hBax-β caused significant killing in both HeLa cells and 293T cells.
(Fig.5B) The apoptotic cells were blebbing with no EGFP detected. We can observe same degree of apoptosis with truncated caspase-7. We did not see a difference be- tween with or without miR-142 MBS for hBax-β and truncated-caspase-7. This is expected, as miR-142 are of low expression in HeLa cells and 293T cells.[27]
To further enhance the killing ability, we cotransfected the hBax-β and trun-
cated-caspase-7 (both in pBI-CMV1 plasmid). As there were already significant death for hBax-β and truncated-caspase-7 alone in HeLa and 293T, we did not see a visible increase in death in combination of these two. Combination experiments can be per- formed in death-resistant cells, e.g. A549. In conclusion, overexpression of hBax-β can effectively trigger apoptosis, and is therefore chosen as the toxic gene in later
experiments.
3.3 Activation of complete circuit in HeLa cells
Next, we tested the complete circuit (Figure 1B) in HeLa cells. Since in the Tet- On system, the inducer, Doxycycline, switches on the circuit, the first thing to confirm
is whether the inducer can regulate the system. We cotransfected the Tet-On 3G pro- tein driven by 342CBP and hBax-β driven by pTRE3G-BI into HeLa cells, and the result shows that this system can be controlled by Tet-On system (Fig 6). The hBax-
β gene was expressed when the inducer was present, as evident from the amount of cell death and EGFP, with or without miR-142 MBS. Moreover, we confirmed the result with time-lapse image (Figure 7). The first picture was taken immediately after the addition of doxycycline into the medium. It is evident that the circuit was activated in 30 min, as reported by both the mCherry and EGFP signal. Nevertheless, only a
proportion of the fluorescent cells rounded up (a feature of apoptosis). What’s more, the morphology of apoptosis was displayed 2 hour after the addition of doxycycline.
Most of cells successfully underwent cell death in 24 hours. However, there were still some EFGP+ cells alive, which means that cancer cells with higher death threshold (i.e. more resistant) still have the chance for escaping our circuit.
Subsequently, the other two toxic genes mentioned above were also constructed in the system to compare the degree of apoptosis. Quantification of apoptosis for all circuits is shown in Figure 8. Taken together, our results show that the genetic circuit has the ability to activate apoptosis in HeLa cells, and the expression of hBax-β trig- gered by TetOn system appears to be the best choice.
3.4 Insulator between two promoters
The ultimate question for this study is whether the miR-142 expression in hematopoietic cell lines, such as HL-60 and Jurkat, can repress the toxic signaling in our circuit. We attempt to address this, however, the problem we met is that most suspension cells were killed by electroporation in cotransfection of our circuit. Also,
the efficiency of cotransfection is very low. To solve this problem, we plan to first build stable cell line of TetOn-3G protein driven by 342CBP and constitutively ex- pressing the mCherry. This cell line (mCherry+ cells) can be selected with flow cy- tometry. Then, the line can be used to transfect the plasmid with the expression of hBax-β driven by TRE promoter, using EGFP as the marker for transfection, or an-
other selection for stable cell lines.
Nevertheless, another problem appeared in this design. The pTRE3G-Bi plas- mid we used cannot express EGFP without doxycycline. The addition of doxycycline also turns on the expression of hBax-β that leads to cell death. In other words, we cannot use EGFP+ to select for stable cells with this second layer of expression. There-
fore, we aim to create a new vector, containing a constitutive promoter and an induc- ible promoter, to express EGFP without toxic signaling. Using pBI-CMV1 as the
initial vector of our new vector (pTRE-CMV), we constructed a co-expression cas- sette with mCherry under the control of CMV promoter and the EGFP under the con- trol of TRE promoter (Fig. 9)[28]. However, the EGFP signal was present without doxycycline, suggesting that there might be interference between the constitutive CMV and the inducible TRE promoter (Fig. 11A)
To diminish the interference between the two promoters, we chose to test a previously characterized insulator, the DNA sequences at the 5’ end of the chicken β- globin HS4 (cHS4) locus in chicken’s chromosome, to prevent the unexpected signal emanating from their surrounding environment.[24, 25, 29] (Fig.10) We compared the effect of inserting zero, one and two copies of the cHS4 insulator and found that the
cHS4 core element can dose-dependently decrease the interference between the two promoters (i.e. influence from CMV promoter). (Fig.11). Quantifying the expression level of two reporters, the mCherry triggered by CMV promoter shows no different after adding the doxycycline. (Fig.12A) Insulator significantly weaken EGFP expres- sion, and it can be recover after inducer exist.(Fig.12B,12C) In this result, the trans-
fected HeLa cells are observed the lower fluorescent expression level with insulator
than without it. Moreover, we quantify the mCherry triggered by CMV promoter and
EGFP triggered by TRE promoter. mCherry shows same degree no matter the doxycy- cline exist or not, which means the constitutive promoter doesn’t effect by TRE pro-
moter. On the other hand, the expression level of EGFP less about 8-fold of construct without insulator. Then, cultured in the doxycycline-containing medium, the EGFP with insulator is recover the expression level like the no insulator one. Taken together, the insulator actually has the ability of stopping the leakage effect happened of two
promoters.
Taken together, the insulator actually has the ability to block the activation of
two promoters and CMV promoter still can constitutively without effected by insula- tor. TRE promoter can reach the same expression level of EGFP in the medium con- taining doxycycline.
Chapter 4 Discussion and Future Work
This study is inspired by a previous study that uses a customizable set of endogenous microRNAs as the multi-input sensor to achieve selective targeting of HeLa cell among the other cell lines [8]. The synthetic regulatory genetic circuit, called a cell-type “clas-
sifier”, is consisted of several miRNA binding site and LacI-controlled promoter CAGop as the upstream sensors. It is a promising platform that can be a novel anti-cancer treat- ment by using the artificial DNA constructs to specifically turn on the output when cer- tain conditions are matched.
Compared with our design, we only chose one endogenous miRNA (miR-142;
hematopoietic-high) marker and Tet-On system controlled by cell cycle regulatory pro- moter as our sensors to distinguish the dividing hematopoietic cell lines form non-hem- atopoietic cell lines. Whether miR-142 is selective enough is still an open question. Also, some cells transfected with the complete circuit (Fig 6; EGFP+ cells) can still escape
death upon induction. Hence, we also plan to simultaneously express two toxic genes linked by a “self-cleaving” P2A peptide [30, 31] to enhance the killing and to avoid the interference since the bi-directional of vector.
Another problem we encountered was the transfection efficiency of hematopoietic cells. These suspension cells are too difficult to transfect by either lipofectamine or elec-
mCherry+), however, the number of cells that survived the electroporation was too low to do further experiments, especially in this case that we want to compare the degree of cell death (data not shown). We also have attempted to generate stable 342CBP_Te- tOn+mCherry-expressing HL-60 and Jurkat cell lines with transient transfection. Unfor- tunately, in that one experiment, transfected cells were very sick and could not survive
for more than one week. We plan to use lentivirus to solve this problem in the near future.
Chapter 5 Figures
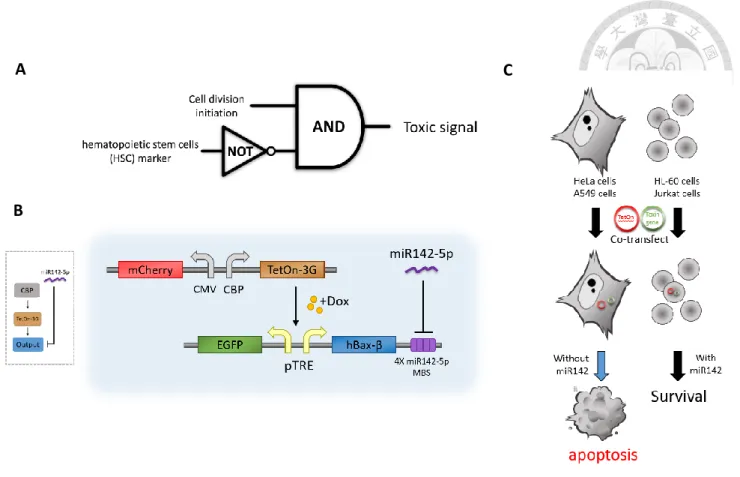
Fig1. The diagram of conception and basic design
As our mainly idea of the specificly selectivity gene circuit apporched by synthestic biology, whetehr the cell division initiated and selection marker exist are the most impotaint characteristics. (A) Schematics of a AND gate between two sensors. (B) An
illustration of actral setup in DNA seqence. hBax-β, an example of output. (C)Selectivity of circuit in different cell line.
A
B
C
Fig.2 The flow chat with fusing 4 repeats miR-142 microRNA binding site.
The sequnces of miR-142 are seperaed into two pairs of pre-annealed oligonucleotides
which contain one sticky end and one blunt end of restrcition enzyme at P1 and P4 sequences (BglII and EcoRV). By using the T4 PNK to transfer the γ-phosphate from ATP to the 5’-OH group, miR-142 MBS can ligate with vector to create a new recombi- nation vector.
Fig.3 The aligements of procaspase-3 and cleaved caspase-3 ; procasepase-7 and
truncated caspase-7.
To accelerate the protein maturation, the prodomain of both caspase-3 and caspase-7 is cleavaged initialy. Morever, we chose the truncated c-caspase-7 mutated at amino acid 57, is reported that triggers apoptosis frequenly than other mutant type. (A) The procaspase-3 gene is cloned form whole genome in the DH5α strain. Compared with the DNA sequence reported online, we construct the activated caspase -3 by removed the
prodomain. (B) To construct the truncated caspase-7, the prodomain is removed first.
Refer to the supporting information from previous study, amino acids 57-303 of caspase- 7 shows the morphologies of apoptosis after activated and causes the significant cell A
B
death than others mutant. Hence, we design the primers which can bind on caspase-7 sequence started form amino acid 57 to remove the amino acid 1-56.
A
B
Fig.4 The expression level of reporter triggered by full length (949) or truncated
(342) cyclin B1 promoters at G0 and G2 phase
The first sensor is chosen because the abnormal division and unlimited proliferation are the most common feature in cancer cells. We sythcrozined the transfected HeLa cells by double thymidine assay and relased for 2 hours that allows cells to go into G2 phase. As the control group, we also block cells into G0 phase, let cells have no ability to activate the cell cycle by cultured in certain medium which is lack of nutrients for 72 hours.
Obseved the fluorescence by microscope, the reporter (mCherry) triggered by (A) truncated cyclin B1 promoter (342 CBP) and (B) full length cyclin B1 promoter (949CBP) shows significantly high expression in G2 phase than G0 phase. (C) The percentage of fluorescent-positive cells indicate that the 342 CBP can successfully
regulate the gene accroding the cell cycle.
C
B A
HeLa cells 293T cells
Fig.5 The transfected cancer cells which constitutively express the toxic gene results
in cell apoptosis in HeLa cells and 293T cells.
(A) The illustration of constructs containing different toxic gene and with or without miR-142-5p MBS located at the downstream triggered by CMV promoter. The EGFP reporter is used as transfected maker. (B) The image of cell morphology result that the overexpression of hBax-β gene and truncated caspase-7 gene can efficiently killing the cancer cells. The transfected cells expressing cleavage caspase-3 gene
show the EGFP and caused less cell death after transfected for 24 hours, which means that overexpression the cleavage caspase-3 alone doesn’t have the enough ability of killing the cancer cells. We also co-transfect the two toxic genes to know whether the killing ability is more powerful than expressed alone or not. In this re- sults, the cleavage caspase-3 and truncated caspase-7 shows the significant killing
co-work with hBax-β gene.
Fig.6 The inducible promoter triggers the toxic gene results in cell apoptosis.
The circuit is containing the tetOn-3G system regulated by 342 CBP and toxic gene activated by TRE promoter. If the Dox exist, the toxic proteins are produced and cause the cell death. This result explains the same conclusion of the data mention above (Fig.5B). We consider the hBax-β gene shows the most powerful killing than others and successfully induced the apoptosis happened. Control: transfected 342CBP_TetOn +
mCherry and pTRE_EGFP. Positive control: transfected 342CBP_TetOn + mCherry and pTRE_EGFP (treated with STLC for 24 hours)
Fig.7 Time lapse image of HeLa cells transfected pTRE3G-BI_ hBax-β+EGFP after
inducing for 24 hours by doxycycline.
As this result, the HeLa cells start producing the hBax-β and EGFP in just 30 minutes after inducing by Dox. After 24 hours, most positive-transfected cells round up and
blebbing that means that cells are going to apoptosis.
Fig.8 Measurement the cell viability and cell death rate by trypan blue exclusion
assay in HeLa cells.
To measure the cell survival rate or death rate , we stain the transfected cells with try- pan blue and calculate the living cells and death cell by Hemocytometer. All the cells are co-transfected with 342CBP-TetOn + mCherry and toxic gene derived by
pTRE3G-BI.( A) Cell viability. (B) cell death rate. This data shows that most of the living cells number are decreased except the pTRE_EFGFP + c-caspase3::miR-142-5p MBS. pTRE_EGFP indicate the survival cells and pTRE_EGFP(STLC) indicate the cell death control. ns, non-siginificant(P > 0.05);**,P ≤ 0.01;***, P ≤ 0.001;**** P ≤
0.0001 (P values)
A B
(%)
Fig.9 Flow chat of cloning the construct containing constitutive and inducible pro-
moter.
In order to make a vector that can continuously produce the reporter gene and activate the toxic gen only when we add the inducer, we combinate the pBI-CMV1 and
pTRE3G-BI by digested with XhoI and XbaI. There are one side consist of CMV en-
hancer and CMV promoter and another side consist of 7X tet operator and TRE prom- ter.
Fig.10 Designs of vector and two promoters and insulator.
(A) Illustration of 2x core element of insulator located between two different promoters.
Moreover, we anticipate the cell would change the color form red to yellow after doxycycline exist, which express the red and green fluorescent at the same time. (B) The design of constructing the vector containing the insulator. We use the SalI to
ligate with XhoI since they have the same cutting site. According to this design, there A
B
is a scar happened but it doesn’t effect next enzyme digest and we can inset another core element of insulator as much as we want.
A
B
Fig.11 The blocking ability of different copies of core elements of insulator in HeLa
cells.
There are three kind of copies between two of the promoters (A) no insulator (B) 1x core insulator (C) 2x core insulator are used as our test group. No matter the inducer
exist or not, the mCherry driven by CMV promoter is produced all the time in all the groups. The EGFP driven by TRE promoter is significantly decreased in insulator-con- tained group than no insulator group. As this result, we can simplify learn that this con- struct can actually block the leakage between two different type of promoters.
C
Fig.12 Quantify the expression level of reporter triggered by two promoters con-
taining different copies insulator.
(A) The construct (Fig.11A) and Tet-On system controlled by 342 CBP are co-trans- fected into HeLa cells. As the result, the expression level of mCherry triggered by
CMV promoter has no change after adding the doxycycline, which means the con- stitutive promoter can stably express the downstream gene. (B) Expression level of EGFP is significantly decreased that containing one or two copies core element of
insulator. (C) Adding the doxycycline is attended, EGFP can be up to the same ex- pression level as the one without insulator. As the conclusion, the problem of leak- age can be resolved by inserted the insulator between two promoters. (n=20) ns,
non-siginificant(P > 0.05); **** P ≤ 0.0001 (P values)
Chapter 6 Reference
[1] C. H. Topham and S. S. Taylor, "Mitosis and apoptosis: how is the balance set?," Current opinion in cell biology, vol. 25, no. 6, pp. 780-785, 2013.
[2] G. J. Kops, B. A. Weaver, and D. W. Cleveland, "On the road to cancer:
aneuploidy and the mitotic checkpoint," Nature Reviews Cancer, vol. 5, no. 10, p. 773, 2004.
[3] K. Chan, C. G. Koh, and H. Li, "Mitosis-targeted anti-cancer therapies: where they stand," Cell death & disease, vol. 3, no. 10, p. e411, 2012.
[4] J. R. Jackson, D. R. Patrick, M. M. Dar, and P. S. Huang, "Targeted anti-mitotic therapies: can we improve on tubulin agents?," Nature Reviews Cancer, vol. 7, no. 2, p. 107, 2007.
[5] K. E. Gascoigne and S. S. Taylor, "Cancer cells display profound intra-and interline variation following prolonged exposure to antimitotic drugs," Cancer cell, vol. 14, no. 2, pp. 111-122, 2008.
[6] S. A. Benner and A. M. Sismour, "Synthetic biology," Nature Reviews Genetics, vol. 6, no. 7, p. 533, 2005.
[7] P. E. Purnick and R. Weiss, "The second wave of synthetic biology: from modules to systems," Nature reviews Molecular cell biology, vol. 10, no. 6, p.
410, 2009.
[8] Z. Xie, L. Wroblewska, L. Prochazka, R. Weiss, and Y. Benenson, "Multi-input RNAi-based logic circuit for identification of specific cancer cells," Science, vol. 333, no. 6047, pp. 1307-1311, 2011.
[9] F. Lienert, J. J. Lohmueller, A. Garg, and P. A. Silver, "Synthetic biology in mammalian cells: next generation research tools and therapeutics," Nature reviews Molecular cell biology, vol. 15, no. 2, p. 95, 2014.
[10] K. S. Katula et al., "Cyclin-dependent kinase activation and S-phase induction of the cyclin B1 gene are linked through the CCAAT elements," Cell growth &
differentiation: the molecular biology journal of the American Association for Cancer Research, vol. 8, no. 7, pp. 811-820, 1997.
[11] A. Hwang, A. Maity, W. G. McKenna, and R. J. Muschel, "Cell cycle- dependent regulation of the cyclin B1 promoter," Journal of Biological Chemistry, vol. 270, no. 47, pp. 28419-28424, 1995.
[12] S. Ding et al., "Decreased microRNA‐142‐3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus,"
Arthritis & Rheumatism, vol. 64, no. 9, pp. 2953-2963, 2012.
microRNAs during TPA-induced differentiation of HL-60 cells," Biochemical and biophysical research communications, vol. 322, no. 2, pp. 403-410, 2004.
[14] S. Sharma, "Immunomodulation: A definitive role of microRNA-142,"
Developmental & Comparative Immunology, vol. 77, pp. 150-156, 2017.
[15] P. Landgraf et al., "A mammalian microRNA expression atlas based on small RNA library sequencing," Cell, vol. 129, no. 7, pp. 1401-1414, 2007.
[16] P.-Y. Chung, "A MicroRNA-based Apoptotic Circuit for Differential Killing of Non-Hematopoietic Cancer Cells," 2018.
[17] D. R. McIlwain, T. Berger, and T. W. Mak, "Caspase functions in cell death and disease," Cold Spring Harbor perspectives in biology, vol. 5, no. 4, p. a008656, 2013.
[18] E. Mills, X. Chen, E. Pham, S. Wong, and K. Truong, "Engineering a photoactivated caspase-7 for rapid induction of apoptosis," ACS synthetic biology, vol. 1, no. 3, pp. 75-82, 2011.
[19] Y. Liu et al., "Synthesizing AND gate genetic circuits based on CRISPR-Cas9 for identification of bladder cancer cells," Nature communications, vol. 5, p.
5393, 2014.
[20] T. Tenev, M. Marani, I. McNeish, and N. Lemoine, "Pro-caspase-3
overexpression sensitises ovarian cancer cells to proteasome inhibitors," Cell death and differentiation, vol. 8, no. 3, p. 256, 2001.
[21] S. Lowe, S. Rubinchik, T. Honda, T. McDonnell, J. Dong, and J. Norris,
"Prostate-specific expression of Bax delivered by an adenoviral vector induces apoptosis in LNCaP prostate cancer cells," Gene therapy, vol. 8, no. 18, p.
1363, 2001.
[22] M. Reitman, G. J. M. Felsenfeld, and c. biology, "Developmental regulation of topoisomerase II sites and DNase I-hypersensitive sites in the chicken beta- globin locus," vol. 10, no. 6, pp. 2774-2786, 1990.
[23] J. H. Chung, A. C. Bell, and G. Felsenfeld, "Characterization of the chicken β- globin insulator," Proceedings of the National Academy of Sciences, vol. 94, no. 2, pp. 575-580, 1997.
[24] N. Uchida, K. N. Washington, C. J. Lap, M. M. Hsieh, and J. F. Tisdale,
"Chicken HS4 insulators have minimal barrier function among progeny of human hematopoietic cells transduced with an HIV1-based lentiviral vector,"
Molecular Therapy, vol. 19, no. 1, pp. 133-139, 2011.
[25] K. Yahata et al., "cHS4 insulator-mediated alleviation of promoter interference
[26] S. Tcherniuk, R. Van Lis, F. Kozielski, and D. A. Skoufias, "Mutations in the human kinesin Eg5 that confer resistance to monastrol and S-trityl-L-cysteine in tumor derived cell lines," Biochemical pharmacology, vol. 79, no. 6, pp.
864-872, 2010.
[27] B.-H. Lian, "A Cascade of Synthetic Logic Gates for Differentiated Anti- mitotic Cancer Therapy," Master's Thesis, 2016. Institute of Molecular and Cellular Biology College of Life Science, National Taiwan University
[28] A. Chtarto et al., "Tetracycline-inducible transgene expression mediated by a single AAV vector," Gene therapy, vol. 10, no. 1, p. 84, 2003.
[29] A. G. West, M. Gaszner, and G. Felsenfeld, "Insulators: many functions, many mechanisms," Genes & development, vol. 16, no. 3, pp. 271-288, 2002.
[30] A. L. Szymczak et al., "Correction of multi-gene deficiency in vivo using a single'self-cleaving'2A peptide–based retroviral vector," Nature biotechnology, vol. 22, no. 5, p. 589, 2004.
[31] J. H. Kim et al., "High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice," PloS one, vol.
6, no. 4, p. e18556, 2011.