國立台灣大學醫學院免疫學研究所 碩士論文
Graduate Institute of Immunology College of Medicine
National Taiwan University Master Thesis
研究 mTOR 分子於胃幽門螺旋桿菌調控胃上皮細胞對 TRAIL 細胞凋亡訊息傳導所扮演之角色
Study the role of mTOR in regulation of Helicobacter pylori- induced TRAIL apoptosis signaling in Human gastric epithelial
cells
林怡孜 Yi-Tzu Lin
指導教授: 許秉寧 博士
Adviser: Ping-Ning Hsu, M.D. /Ph.D.
中華民國 一百零七年 七月
July, 2018
誌謝
寫下誌謝的此時,都還記得當初抱著忐忑的心來詢問許老師實驗室的事 情,彷彿老師才剛親切地與什麼都不懂的我介紹小鼠跟大鼠的差異,然後我就 畢業了。非常感謝許老師這兩年間的教導與包容,除了實驗上紮實的邏輯訓 練、更是教導我嚴以待己的人生態度。而若說許老師是嚴父,朱清良老師就像 慈母般,常常替我打氣加油,提點我沒注意到的小細節。謝世良與吳明賢老師 也在我進度報告時提供建議,幫助我提升論述的精準度且更完整了我的論文。
我也要感謝陪伴我走完這兩年的實驗室夥伴:Chris,總是親切地處理很多 雜事,陪我討論實驗和抱怨大大小小的事;老蕭,讓我晚上做實驗總是不孤 單,還有安慰我哭的時候總是溫柔的剛剛好;全醫師,常常問我實驗的缺漏讓 我想了很久,還有分享人生經驗給我;思穎,陪著我一起討論實驗,還有只有 我們了解的菌很難養;致宏跟威延,一搭一唱的逗我開心還有介紹遊戲給我。
另外還有免疫所的好朋友們:明勳、貴云、子暄、陳名、凱富、羅筠…和可愛 的學弟妹們,你們總是用溫柔體貼,把我的負能量吸收又轉化成讓我繼續走下 去的動力。一個一個比我更在乎我的實驗,陪我討論、替我解決問題。免疫所 真的非常溫暖,在這裡的每一天對我來說都彌足珍貴。還有所外的朋友:可 風,柔程,天妤,彥伶…,告訴我人生不只有實驗,還有體驗生活中的美好與 確幸。
最後我要感謝我的母親,無怨無悔的支持我追尋我的夢想,在我低潮的時 候,安慰我的不勇敢;在我高興時更加倍替我歡喜,並且接受我一切的不完 美。還有承樺,總是鼓勵我說我並沒有想像中的那樣差勁、和默默地陪伴。在 免疫所的這兩年要感謝的真的太多了,我感謝這一切所有的安排,與你們的際 遇,都大大豐富了我的人生風景,謝謝。
中文摘要
胃幽門螺旋桿菌(Helicobacter pylori)是一種世界上常見的細菌感染源之一,
主要棲息在人體胃部,並且已被發現與許多胃部疾病的發生有關,例如胃炎、胃 潰瘍、黏膜層淋巴瘤、胃癌。其中,過去研究發現感染胃幽門螺旋桿菌的表皮細 胞其細胞凋亡的程度增加與胃幽門螺旋桿菌所導致的胃部病變有關。先前實驗室 的研究顯示,TRAIL 能誘導感染胃幽門螺旋桿菌的胃上皮細胞進行大量細胞凋 亡,進一步我們發現胃幽門螺旋桿菌透過負調控 Akt 的磷酸化程度使得 AIP4 蛋 白活性增加,導致重要的調控蛋白 FILPs 進行泛素化降解,進而細胞無法阻礙 TRAIL receptor 下游死亡複合物的形成,導致胃上皮細胞容易被 TRAIL 誘導細 胞凋亡。於此篇我們更利用 AIP4 基因剃除後發現 AIP4 分子的確參與在調控胃 幽門螺旋桿菌誘導之 TRAIL 細胞凋亡之訊息傳導途徑中。此外,在此篇研究中 我們也探討了胃幽門螺旋桿菌如何調控 Akt 的磷酸化,我們發現胃幽門螺旋桿菌 會降低 Akt 的上游分子 mTOR 的表現量,並且我們發現隨著感染時間增長,mTOR 的表現量降低與 Akt 去磷酸化程度、FLIPs 的表現量降低呈現正相關,因此我們 推測 mTOR 可能也參與在胃幽門螺旋桿菌所誘發的胃上皮細胞 TRAIL 敏感性提 高。我們同時也利用 mTOR 抑制劑與 mTOR siRNA 轉染證明若於胃上皮細胞抑 制 mTOR 活性則會導致細胞內 FLIPs 表現量下降且同時誘導細胞對 TRAIL 敏感 度提升,此現象如同模擬胃幽門螺旋桿菌感染的情況。再者,我們過度表現 mTORC2 於人類胃上皮細胞後發現藉由於細胞內增強 mTOR-Akt-FLIP 訊號傳遞 路徑可以使人類胃上皮細胞抵禦胃幽門螺旋桿菌所誘發之 TRAIL 引起的細胞凋 亡。在此篇研究中我們定義出 mTOR 分子於胃幽門螺旋桿菌的致病機制中扮演 重要角色,並且提供其成為將來治療感染胃幽門螺旋桿菌的分子標的之可能性。
Abstract
Helicobacter pylori (H. pylori) is one of the most common pathogens that inhabits
in human stomach, which has been known for being related to amounts of gastric
disease such as gastritis, peptic ulcers, gastric MALT lymphomas and gastric cancer. It
has been reported that the etiology of H. pylori is associated with the enhancement of
cell apoptosis in gastric epithelial cells. Previous study in our laboratory has shown that
H. pylori confers susceptibility to TRAIL-mediated apoptosis in human gastric
epithelial cells through decreased Akt phosphorylation, which results in activating the
AIP4 and leads to FLIPs degradation sequentially. In this study, we have further
demonstrated that knockout of AIP4 in AGS cells mitigated H. pylori-induced TRAIL-
mediated apoptosis, which has clarified that Akt-AIP4-FLIPs pathway participated in
H. pylori-induced TRAIL apoptosis signaling. Besides, in order to investigate how H.
pylori regulates Akt phosphorylation state, we have searched for the upstream
molecular of Akt and found mTOR expression decreased while H. pylori co-cultured
with gastric epithelial cells. Moreover, we found that the augmentation of mTOR-Akt
pathway by overexpressing mTORC2 in gastric epithelial cells reduced H.pylori-
induced TRAIL-apoptosis signaling. In this study, we address the role of mTOR in H.
pylori-induced TRAIL apoptosis signaling and provide a new insight for the
pathogenesis of H. pylori.
Contents
口試委員會審定書 ...i
誌謝 ... ii
中文摘要 ... iii
Abstract ... iv
Introduction ... 1
1. Helicobacter pylori (H. pylori) ... 1
2. Pathogenesis of H. pylori ... 2
3. TRAIL-mediated apoptosis ... 4
4. H. pylori enhances TRAIL-induced apoptosis signaling in gastric epithelial cells 6 5. The Akt pathway in regulation of FLIPs expression ... 6
6. mTOR signaling in regulation of FLIPs expression ... 8
Rationales ... 10
Materials and Methods ... 11
Results ... 25
I. H. pylori enhanced the apoptosis of AGS cells while treat with TRAIL, and phosphorylation of Akt as well as FLIPs expression in AGS cells decreased concurrently. ... 25
II. The expression of FLIPs was decreased and enhanced TRAIL-mediated apoptosis in AGS cells after knockdown of Akt... 26
III. Knockout of AIP4 reduced H. pylori-induced TRAIL-mediated caspase-8 activation. ... 27
IV. mTOR expression was decreased after H. pylori co-cultured. ... 28
V. Inhibition of mTOR reduced the expression of FLIPs and induced TRAIL- mediated apoptosis in AGS cells. ... 29
VI. Enhancement of mTOR-Akt signaling pathway reduced H. pylori-induced TRAIL-mediated apoptosis signaling. ... 30
VII. H. pylori-induced autophagy could be attenuated while enhanced mTOR-Akt
signaling pathway in AGS cells. ... 31
Discussion ... 33
I. H. pylori-induced TRAIL-mediated apoptosis through inhibiting the intracellular mTOR-Akt signaling pathway. ... 33
II. H. pylori downregulated FLIPs expression through reducing Akt phosphorylation. ... 36
III. H. pylori downregulated FLIPs expression through Akt-independent mechanisms. ... 40
IV. The upstream regulation of mTORC2 activity and the possible pathway for H. pylori to disrupt the mTORC2 activity in AGS cell lines. ... 41
Conclusion ... 44
References ... 45
Figures ... 55
Introduction
1. Helicobacter pylori (H. pylori)
Helicobacter pylori (H. pylori) is one of the most common pathogens that infects
approximately 50% of the population in worldwide (Tsai and Hsu, 2017). It was first
cultivated successfully in 1982 by Australian researchers: Barry J. Marshall and Robin
Warren. They have deciphered the crucial role of H. pylori in the aetiology of gastric
inflammation, duodenal ulcer, gastric ulcer, and strongly associated with
adenocarcinoma and lymphoma (Cave, 1997; Marshall and Warren, 1984). Hence, H.
pylori was classified as class I carcinogen (IARC, 1994). H. pylori is a Gram-negative,
spiral-shaped bacterium which inhabits in human stomach. The transmission of H.
pylori is commonly through person-to-person by saliva, or be spread by fecal
contamination of food and water (Aziz et al., 2015; Goh et al., 2011; Khalifa et al.,
2010). Unlike other bacteria, H. pylori could survive in the harsh acidic environment in
the stomach due to the production of urease that neutralizes stomach acid. Besides,
H.pylori relies on its tail-like flagella to move around; and the helical shape facilitates
H. pylori to penetrate the mucoid covered on gastric epithelial cells in order to colonize
in stomach (Keilberg and Ottemann, 2016). Although the high prevalence of H. pylori,
most of patients remain asymptomatic in the lifetime. The clinical consequences of
H.pylori-infected patient are controlled by complicated interactions between the
polymorphisms in host, bacteria, and environmental parameters (Backert et al., 2016;
Kusters et al., 2006). The current treatment of H. pylori is taking advantage of
antibiotics such as amoxicillin and clarithromycin in order to eradicate H. pylori.
However, several cases have reported that H. pylori eradication therapy failed due to
the development of resistance in bacteria (Garza-Gonzalez et al., 2014; Megraud, 2004,
2007). Therefore, clarifying the molecular mechanism inside the pathogenesis of H.
pylori is imminent to meet the therapeutic needs.
2. Pathogenesis of H. pylori
The pathogenicity of H. pylori is mediated through the action of a variety of
bacterial virulence factors on host epithelial cells including but not limited to, urease,
vacuolating cytotoxin A (VacA), cag pathogenicity island, cytotoxin-associated gene A
(CagA), peptidoglycan outer membrane proteins (e.g., BabA, SabA, OipA) and γ-
glutamyltranspeptidase (GGT) (Peek and Crabtree, 2006; Polk and Peek, 2010;
Teymournejad et al., 2017; Valenzuela et al., 2013). H. pylori infection dysregulates the
intracellular signaling pathways. For instance, once the CagA protein translocated into
cells, the NF-κB pathway and MAP kinase would be activated via EGFR activation
(Diaz et al., 2018). Besides, VacA has been reported to disrupt the mitochondrial
functions, stimulate cell apoptosis and exert cell autophagy (Ashktorab et al., 2008;
Palframan et al., 2012; Raju et al., 2012; Terebiznik et al., 2009).
Moreover, it has been already observed that H. pylori enhances both proliferation
and apoptosis rate in the gastric biopsy specimen from H. pylori-induced gastritis
patients, which compared to the secondary gastritis patients or non-inflamed controls
(Jones et al., 1999; Jones et al., 1997; Moss et al., 1996). The following research has
also authenticated the main cause of H. pylori-induced gastric disease is through the
alteration of the balance between proliferation and apoptosis leading by H. pylori
infection (Peek and Blaser, 2002). Furthermore, the apoptosis augmented by H. pylori
stimulates the proliferation of gastric epithelial cells to maintain the integrity of
epidermal barrier. This may increase the gene mutation rate in hyper-proliferative
epithelial cells, and thus facilitate cells to become cancer-prone (Ashktorab et al., 2008;
Xia and Talley, 2001). As mention above, the outcome of H. pylori-infection hinges on
the bacteria virulence factors, host gastric mucosal factors and environment (Backert et
al., 2016; Kusters et al., 2006). Previous reports have shown that gastric-infiltrating T
cells are selectively increase during infection, especially resemble the phenotype of T
helper 1 (Th1). Those T cells secret Th1 cytokines such as interferon-γ (IFN-γ) and
tumor necrosis factor-α (TNF-α) in order to promote other pro-inflammatory cytokines
release, which further enhances H. pylori-induced apoptosis (Bamford et al., 1998;
Karttunen et al., 1995). On the other hand, H. pylori increases the expression of Fas in
gastric epithelial cells. This also gives rise to the activation of apoptosis in gastric
epithelial cells through additional Fas/FasL interaction with infiltrating T cells (Rudi et
al., 1998). These finding indicate that H. pylori is capable of modulating the immune
response, therefore inducing apoptosis signaling cascade in gastric epithelial cells
(Wang et al., 2000; Wu et al., 2004).
3. TRAIL-mediated apoptosis
TRAIL (tumor necrosis factor-related apoptosis inducing ligand; Apo2L) is
discovered in 1995, which has been identified as a member of TNF superfamily through
its sequence homology to TNF-α (Wiley et al., 1995). The canonical role of TRAIL is
well-known for inducing cell apoptosis through interaction with TRAIL receptor 1
(TRAIL-R1; DR4) and TRAIL receptor 2 (TRAIL-R2; DR5). Upon TRAIL binding to
TRAIL-R1 or TRAIL-R2, the intracellular death domain (DD) of TRAIL-R adopts a
conformation change, which endows the ability to recruit the intracellular adaptor
molecule FAS-associated death domain protein (FADD). FADD further recruits the
initiator caspases-8/10 via their death effector domain (DED) and forms the death-
inducing signaling complex (DISC), which results in active the caspase-8/10
afterward(von Karstedt et al., 2017). Although TRAIL is enable to induce apoptosis in
a variety of transformed cell lines in vitro, the normal primary cells exhibit resistance
against TRAIL (Ozoren and El-Deiry, 2002).
There are several mechanisms for cells to withstand the TRAIL-inducing apoptosis
(Lu et al., 2006). For instance, the FLICE-like inhibitory protein (FLIP, also known as
CFLAR) is one of the negative regulator of TRAIL-mediated apoptosis. FLIP is a death
effector domain-containing protein that could be recruited to DISC. Since it is a
caspase-8 homologue that could compete with caspase-8 for binding to FADD, it
impedes the activation process of caspase-8 and prevents TRAIL-mediated apoptosis
(Hersey and Zhang, 2001). FLIP has 13 distinct spliced variants, three of which are
expressed as proteins: 55 kda long form FLIP (FLIPL), 25kda short form FLIP (FLIPs),
24 kda form FLIPR (Bagnoli et al., 2010; Micheau, 2003; Safa et al., 2008; Safa and
Pollok, 2011). The difference between FLIP isoforms indicates distinct regulatory role
of FLIPL and FLIPs. FLIPs inhibits TRAIL-induced DISC formation and caspase-8
activation, hence inhibit apoptosis (Poukkula et al., 2005; Yu and Shi, 2008). While
FLIPL is controversial for preventing apoptosis due to the dual functions: when
expressed at high-levels, it inhibits Fas-induced caspase-8 activation; however, it
enhances caspase-8 activation when the expression level is low (Budd et al., 2006; Li
et al., 2006).
4. H. pylori enhances TRAIL-induced apoptosis signaling in gastric epithelial cells
Previously, our laboratory has shown that gastric infiltrating T cells express TRAIL
by the analysis of gastric biopsy of H. pylori-infected patients. Further study have
demonstrated that H. pylori enhances TRAIL-induced apoptosis (Wu et al., 2004); the
molecular mechanism has not being fully decoded, nevertheless. We already
ascertained the H. pylori-induced TRAIL-mediated apoptosis in gastric epithelial cells
is dependent on activation of downstream signaling of caspase-8, and further transmit
the death signals to mitochondria to break the resistance to apoptosis. Furthermore, we
found H. pylori raises the TRAIL-sensitivity in gastric epithelial cells through
downregulating FLIPs, which sparked off the initiation of caspase signaling cascade
via DISC, and eventually undergo apoptosis (Lin et al., 2014; Tsai and Hsu, 2017).
Recently the emerging clues in our laboratory point out that H. pylori might decrease
FLIPs expression through dephosphorylating Akt. However, how H. pylori interferes
intracellular Akt phosphorylation state still remains a puzzle.
5. The Akt pathway in regulation of FLIP
sexpression
The Ser and Thr kinase Akt, which is also known as (protein kinase B), was first
identified as an oncogene and served as the downstream of phosphoinositide 3 kinase
(PI3K) (Bellacosa et al., 1991; Staal, 1987). Akt is involved in several cellular processes
including cell survival, proliferation and growth (Brunet et al., 1999; Fruman et al.,
1999; Kodaki et al., 1994; Manning and Toker, 2017). Activation of Akt is a multistep
process that involves both membrane translocation and phosphorylation.
Mechanistically, class I PI3K transduces upstream signaling from receptor tyrosine
kinases (RTKs) and G protein-coupled receptors (GPCRs) by phosphorylating the
phosphoatidylinositol-4,5-bisphophate (PI-4,5-P2) to generate phosphatidylinositol-
3,4,5-trisphosphate (PIP3) (Auger et al., 1989; Whitman et al., 1988). PIP3 serves as a
critical lipid second messenger that recruits Akt and Pyruvate Dehydrogenase Kinase
1(PDK1) to dock in the plasma membrane (Toker, 2012; Toker and Cantley, 1997;
Vanhaesebroeck and Waterfield, 1999). PDK1 phosphorylates the activation loop of
Akt at Thr308 and the full activation is accomplished by mTOR Complex 2 (mTORC2)
on phosphorylating the Ser473 site of Akt (Sarbassov et al., 2005; Saxton and Sabatini,
2017). In addition, there are several phosphatases which are known for controlling Akt
activation such as PIP3 phosphatase PTEN(Phosphatase and tensin homolog)
(Maehama and Dixon, 1998), Akt Ser473 phosphatase PHLPP(Leucine-rich repeat
protein phosphatase) and Thr308 phosphatase PP2A(Protein phosphatase 2)(Liao and
Hung, 2004).
Akt has been reported to have an impact on the regulation of FLIPs expression. For
instance, Akt is found to regulate FLIPs expression through ubiquitination by Itch (E3
Ubiquitin-Protein Ligase Itchy Homolog, also known as AIP4) in GBM cell lines and
xenograft (Panner et al., 2009); another report also shows that Akt controls the FLIPs
expression through administering the FLIPs mRNA accumulation in
mono/polyribosome (Panner et al., 2005) or directly affects the FLIP transcriptional
level (Nam et al., 2003). Akt upregulation was also observed with enhanced FLIPs
expression in tumor cells, thus exerting the resistance to TRAIL-mediated apoptosis to
cells including the gastric cancer cell line (Nam et al., 2003).
6. mTOR signaling in regulation of FLIPs expression
mTOR (The mammalian target of rapamycin) was first identified as the direct target
of rapamycin in 1994 (Brown et al., 1994; Sabatini et al., 1994; Sabers et al., 1995).
mTOR is a serine/threonine protein kinase in the PI3K-related kinase (PIKK) family,
which could form two distinct protein complex as mTOR complex 1 (mTORC1) and 2
(mTORC2) and participate in diverse signaling pathways. mTORC1 is composed of
three core components: mTOR, Raptor (regulatory protein associated with mTOR), and mLST8 (mammalian lethal with Sec13 protein 8; GβL). The downstream signaling
network of mTORC1 includes protein synthesis, cellular metabolism and protein
turnover. While mTORC1 regulates cell growth and metabolism, mTORC2 masters the
proliferation and cell survival through phosphorylating several protein kinase of the
AGC (PKA/PKG/PKC) family, especially known for activating Akt, a key effector of
insulin/PI3K signaling (Saxton and Sabatini, 2017). There are several evidences
demonstrated that mTOR regulates c-FLIP expression. In the study of bladder cancer
cells, utilizing metformin, which inhibits mTOR with activating autophagy,
downregulates c-FLIP levels and contributes toward the enhancement of TRAIL-
sensitivity (Zhang et al., 2014). Another report shows that mTOR controls FLIPs
translation and TRAIL sensitivity in glioblastoma multiforme cells (Panner et al., 2005).
In addition, mTORC2 was certified to be involved in c-FLIP degradation through E3
ubiquitin-protein ligase CBL(c-cbl) (Zhao et al., 2013). Taken together, these data
support the importance of mTOR in mediating TRAIL-resistance in cells.
In the preliminary data, we have found the expression of mTOR decreased while
co-cultured with H. pylori. Since there are several evidences show that mTOR is
capable to regulate the FLIPs expression. This finding inspired us to explore the role of
mTOR in H. pylori-mediated TRAIL sensitivity in human gastric epithelial cell line,
especially focus on mTOR-Akt signaling axis.
Rationales
It has been known that the pathogenesis of H. pylori is associated with induction
of cell apoptosis. We have already demonstrated that H. pylori confers the susceptibility
to TRAIL through decreasing of FLIPs expression, thus disabling gastric epithelial cells
to prevent excess caspase-8 activation. Moreover, the preliminary results in our
laboratory have revealed that H. pylori may negatively regulate Akt phosphorylation,
which resulted in FLIPs ubiquitination and decreased the expression of FLIPs.
Eventually, H. pylori enhanced TRAIL-mediated apoptosis in gastric epithelial cells.
However, the regulation of Akt phosphorylation while H. pylori infection still remains
unclear. We screened the upstream molecules that have been reported to affect the
activation of Akt. Among these upstream molecules, we found that mTOR expression
decreased during H. pylori infection. Base on this finding and the reports which have
indicated that mTOR enabled to regulate c-FLIP expression, we consider mTOR as a
possible molecule that is regulated by H. pylori and play a role in H. pylori-induced
TRAIL apoptosis signaling in gastric epithelial cells.
Materials and Methods
➤Materials 1. Cells
Human gastric epithelial cell line (AGS) were maintained in RPMI 1640 medium,
supplemented with 10% Fetal Bovine Serum (FBS) and L-glutamine at 37℃ in 5%
CO2 incubator.
2. Bacteria
H. pylori strains ATCC 43504 and HM-9 were kindly provided by Dr. Ming-Shiang
Wu. H. pylori was passaged on 5% sheep blood agar plates without antibiotics at 37℃
in micro-aerophilic incubator (10% CO2, 5% O2 and 85% N2) for 1-2 days culture. For
each experiment, H. pylori was scrapped from agar plates and re-suspended in 15 ml
Brucella broth with antibiotics and cultured for 12-18hrs in micro-aerophilic incubator.
The H. pylori was pellet at 3000rpm for 10 minutes and discarded the supernatants. The
pellet was then re-suspended with 1ml 1X DPBS and measured the O.D. 600 value by
spectrophotometer (1 O.D.= 109 CFU/ml). The re-suspended pellet was co-cultured in
complete RPMI-1640 medium without antibiotics with cells.
3. Antibodies
Primary Antibodies
#9272 Anti-Akt Cell signaling Technology, Danvers, USA
#9271 Anti-phospho-Akt (Ser473) Cell signaling Technology, Danvers, USA
#9275 Anti-phospho-Akt (Thr308) Cell signaling Technology, Danvers, USA
#12117 Anti-ITCH (D8Q6D) Cell signaling Technology, Danvers, USA
#9746 Anti-Caspase-8 (1C12) Cell signaling Technology, Danvers, USA
#56343 Anti-FLIP (D5J1E) Cell signaling Technology, Danvers, USA
#2983 Anti-mTOR (7C10) Cell signaling Technology, Danvers, USA
#2974 Anti-mTOR (Ser 2481) Cell signaling Technology, Danvers, USA
#2114 Anti-Rictor (53A2) Cell signaling Technology, Danvers, USA
#2280 Anti-Raptor (24C12) Cell signaling Technology, Danvers, USA
#9234 Anti-phosphor-S6K
(Thr389) (108D2)
Cell signaling Technology, Danvers, USA
#2708 Anti-p70-S6K (49D7) Cell signaling Technology, Danvers, USA
#12860 Anti-mSin1 (D7G1A) Cell signaling Technology, Danvers, USA
NB100-2331 Anti-LC3 Novus Biologicals, Colorado, USA
MAB1501 Anti-β-actin, clone C4 Merck Millipore, Billerica, USA
640941 APC-Annexin A5 BioLegend, San Diego, USA
Secondary Antibodies
#7074 Anti-rabbit IgG, HRP Cell signaling Technology, Danvers, USA
#7076 Anti-mouse IgG, HRP Cell signaling Technology, Danvers, USA
4. Recombinant protein
Recombinant TNF-related apoptosis-inducing ligand (TRAIL)
PeproTech, Rocky Hill, USA
5. Vector
peGFP-N2
peGFP-N2 with inserted human Akt cDNA by Shih-Chia Huang
peYFP-C1-mTOR Addgene Cambridge, Massachusetts, USA
pRK5-HA-YFP-rictor Addgene Cambridge, Massachusetts, USA
myc-Rictor corrected Addgene Cambridge, Massachusetts, USA
pcDNA3-Flag mTOR wt Addgene Cambridge, Massachusetts, USA
6. siRNA
SignalSilence® Akt siRNA I #6211 Cell signaling Technology, Danvers, USA
SignalSilence® Control siRNA #6568 Cell signaling Technology, Danvers, USA
MISSION® mTOR siRNA (siRNA ID: SASI_Hs01_00203145)
Sigma-Aldrich, St Louis, Missouri, USA
MISSION® Rictor siRNA (siRNA ID: SASI_Hs02_00366683)
Sigma-Aldrich, St Louis, Missouri, USA
MISSION® Raptor siRNA (siRNA ID: SASI_Hs01_00048380)
Sigma-Aldrich, St Louis, Missouri, USA
MISSION® siRNA Universal Negative control
Sigma-Aldrich, St Louis, Missouri, USA
7. Transfection reagent
GenJetTM Plus DNA In Vitro Transfection reagent
SignaGen Laboratories, Maryland, USA
LipofectamineTM 3000 reagent Invitrogen, California, Carlsbad, USA
LipofectamineTM RNAiMAX reagent
Invitrogen, California, Carlsbad, USA
jetPRIME® in vitro DNA & siRNA transfection reagent
Polyplus transfection, Illkirch, France
8. Medium and buffers
RPMI-1640 medium
Corning®RPMI-1640 culture medium containing 10% fatal bovine serum and L-
glutamine with or without Antibiotic-Antimycotic (Gibco, USA)
Anaerobic Blood agar
CDC anaerobic blood agar which is supplemented with 5% sheep blood agar is
purchased from BD biosciences, USA.
BBL broth
2.8% Brucella broth supplemented with 10% fatal bovine serum, 6μg/ml
Vancomycin and 2μg/ml Amphotericin B
Triton X-100 lysis buffer
1% Triton-X, 50mM Tris-HCl, 5mM EDTA in ddH2O
5X SDS sample buffer
250mM Tris-HCl (pH6.8), 5% β-mercapitalethanol, 0.02% Bromophenol blue,
30% glycerol, 10% SDS
10X running buffer
192mM Glycine, 25mM Tris-HCl, 1% SDS, pH=8.3
10X transfer buffer
195mM Glycine, 240mM Tris-HCl, 1.185% SDS, pH=8.4. Dilute to 1X transfer
buffer with 20% Methanol before usage.
10X washing buffer
100mM Tris-HCl (pH=7.4), 9% NaCl, 2% Tween-20
1X sorting buffer
1X HBSS(Ca2+/Mg2+ free) with 1mM EDTA, 25mM HEPES, 2% FBS, pH= 7.0
1X AnnexinV binding buffer
Diluted from 10X AnnexinV binding buffer from BD biosciences, USA.
9. Chemicals
Ammonium persulfate Fluka, Switzerland
Amphotericin B Sigma-Aldrich, St Louis, Missouri, USA
Acrylamide Bio-Rad, Hercules, California, USA β-mercapitalethanol Sigma-Aldrich, St Louis, Missouri, USA
Bovine serum albumin H.M. biologicals, 桃園市, 台灣
Brucella broth Becton Dickinson, Franklin Lakes, New Jersey, USA
DMEM with high glucose Gibco, USA
Dimethyl sulfoxide Sigma-Aldrich, St Louis, Missouri, USA Dulbecco’s phosphate-buffered saline Corning, Newark, USA
EDTA Watson Biotech, 新北市, 台灣
Fetal bovine serum Corning, Newark, USA
Glycerol Sigma-Aldrich, St Louis, Missouri, USA
Glycine Affymetrix, Santa Clara, California, USA
HEPES Corning, Newark, USA
Kanamycin MDBio, Inc., 台北市, 台灣
Metformin Sigma-Aldrich, St Louis, Missouri, USA
MG132 Cell signaling Technology, Danvers, USA
Methanol Merck, Darmstadt, Germany
Miller’s LB broth power Becton Dickinson, Franklin Lakes, New Jersey, USA
Opti-MEM® medium Gibco, USA
Phosphatase inhibitor cocktail2 Sigma-Aldrich, St Louis, Missouri, USA
Propidium iodide Sigma-Aldrich, St Louis, Missouri, USA
Phosphatase Inhibitor cocktail3 Sigma-Aldrich, St Louis, Missouri, USA
PP242 Selleckchem.com, Houston, USA
Prestained protein marker BIOTOOLS, 新北市, 台灣
Protease inhibitor cocktail Sigma-Aldrich, St Louis, Missouri, USA
Rapamycin Selleckchem.com, Houston, USA
RIPA buffer Omics Bio, 新北市, 台灣
Sheep blood 銳誠企業股份有限公司, 新北市, 台灣
Sodium bicarbonate Affymetrix, Santa Clara, California, USA
Sodium chloride Affymetrix, Santa Clara, California, USA
Sodium dodecyl sulfate SERVA, Heidelberg, Germany
TEMED Affymetrix, Santa Clara, California, USA
Torin1 Cayman Chemical, Michigan, USA
Tris-base Affymetrix, Santa Clara, California, USA
Triton-X Affymetrix, Santa Clara, California, USA
Trypan blue H.M. biologicals, 桃園市, 台灣
Tween-20 Affymetrix, Santa Clara, California, USA
Vancomycin Sigma-Aldrich, St Louis, Missouri, USA
4-15% pre-cast gel Bio-Rad, Hercules, California, USA
➤Methods
1. Cell death detection
(I) AnnexinV/ Propidium iodide apoptosis assay
Cells were cultured in 24-well (3×104 cells/well) overnight. For measuring H.pylori
induced TRAIL-mediated apoptosis, cells were then treated with H. pylori (MOI=100)
or not for 12 hours, following the recombinant TRAIL treatment (40ng/ml) for 6 hours.
Cells were harvested by collecting the cell culture medium and the cells retained on the
dish are washed by 1X DPBS, following accutase treatment for 15-20 minutes to obtain
the residue cells on the dish. Centrifuge at 300×g, 5 minutes and discard the supernatant.
The cell pellet was washed by 1X DPBS three times and re-suspended by AnnexinV
binding buffer which contained AnnexinV-APC antibody and PI for staining 15 minutes
in the dark. Before analyze by flow cytometry, dilute cells into appropriate volume by
AnnexinV binding buffer.
(II) Cell death detection ELISA
Cells were cultured in 96-plate (1×104 cells/well) overnight. Then treated cells with H.
pylori (MOI=100) or not for 12 hours, following the recombinant TRAIL treatment
(40ng/ml) for 3 hours. Cells were harvested by centrifugation at 200×g for 10 minutes.
Discard the supernatant carefully and lyse cells in 200μl 1x working solution of lysis
buffer. After incubate with the lysis buffer for 30 minutes, centrifuge the cell lysate at
200×g for 10 minutes. The 20μl supernatant was transferred to streptavidin-coated
microplate and incubate with 80μl prepared immunoreagents (1/20 anti-DNA-POD
antibody, 1/20 anti-histone-biotin antibody and 18/20 incubation buffer) for 2 hours
with shaking. After washing three times with the incubation buffer, add 100μl ABTS
substrate solution into each well and keep the plate in the dark until development of the
color was sufficient for photometric analysis. The reaction was determined by ELISA
reader at 405nm.
2. Cell lysis
After cell were harvested and centrifuge into pellet, the appropriate volume of lysis
buffer was added to re-suspend the cell pellet and incubate on ice for 20 minutes with
vortexing occasionally. The cell lysate were centrifuge for 15 minutes at 15,000×g, 4℃.
The proteins are in the supernatant and store the supernatant at -70℃.
3. Western blot
Proteins were extracted and quantified by protein assay dye. Equal amount of proteins
of each sample (20-35μg/ml) in SDS sample buffer were boiled for 5 minutes at 100℃.
Load each sample into the gel and run the SDS-PAGE (6%-15%) at 100V, protein on
the gel is transferred to PVDF membrane which was pre-treated in 100% methanol at
400mA by wet transfer system for 60-90 minutes. After transferring, put the PVDF
membrane into the blocking buffer (5% non-fat milk or bovine serum albumin (BSA)
in TBST buffer) and block for 1 hour. The membrane was incubated with indicated
primary antibody diluted in blocking buffer at 4℃ overnight. After incubation, the
membrane was washed with TBST buffer three times for 10 minutes and incubated with
the certain horseradish peroxidase (HRP)-conjugated secondary antibody at room
temperature for 1 hour. Wash the membrane with TBST buffer three times for 10
minutes and develop the membrane by ECL system.
4. Real-time polymerase chain reaction (RT-PCR)
Total RNA form AGS cells was isolated using TRIzol (Invitrogen). cDNA was prepared
following the instructions provided by manufacture (iScriptTM cDNA synthesis kit).
The cDNA products were used to amplify target gene (mTOR, c-FLIPs and GAPDH)
using SensiMix SYBR Bo-ROX kit (Bioline). Data were acquired with PikoReal
(Thermo Fisher).
The PCR primers were:
Gene Forward Reverse
mTOR 5’-GCTTGATTTGGTTCCCAGGACAGT-3’ 5’-GTGCTGAGTTTGCTGTACCCATGT-3’
FLIPs 5’- GCAGGGACAAGTTACAGGAATGT-3’ 5’-GGACAATGGGCATAGGGTGT -3’
GAPDH 5’-GTGAACCATGAGAAGTATGACAA -3’ 5’-CATGAGTCCTTCCACGATAC -3’
5. siRNA transfection
Different siRNA transfection reagent was used to conduct experiments under the
instructions provided by manufacturer. For Akt knockdown experiment, 1.5×105 AGS
cells were seeded in 6-well (2c.c. medium per well). After 24-hours culture, dilute 110-
220 picomoles Scramble/Akt siRNA into 200μl jetPRIME® buffer (referred as a final
concentration of 50nM-100nM per well). Then add 4μl of jetPRIME® reagent and
incubate for 10 minutes at room temperature. Add the transfection mix to the cells in
serum containing medium drop wise. Cells were harvest after 24-36 hours post
transfection. For scramble/mTOR/Rictor/Raptor knockdown experiment, 2×105 AGS
cells were seeded in 6-well (2c.c. medium per well). After 24-hours culture, dilute
lipofectamine® RNAiMAX reagent (7.5μl, 9μl, 12μl) and scramble/ mTOR/ Rictor /
Raptor siRNA (15, 30, 40 picomoles) into Opti-MEM® medium, respectively. Add
diluted siRNA into diluted lipofectamine® RNAiMAX reagent and follow the 5 minutes
incubation at room temperature. Then add siRNA-lipid complex to cells dropwisely.
Cells were harvest after 24-36 hours post transfection.
6. Plasmid DNA transfection
The plasmid DNA was purified by NucleoBond® Xtra Maxi EF (MACHEREY-NAGEL,
Germany). 1.2×106 AGS cells were seeded in 10 cm-plate overnight. Before
transfection, replace the medium freshly (5c.c. per plate). For each 10 cm-plate, dilute
5μg plasmid DNA and 15μl GenJetTM Plus reagent into 250μl serum-free DMEM with
high glucose, respectively. Add the diluted GenJetTM Plus reagent immediately to the
diluted DNA solution all at once and gently pipette up and down 3-4 times. Incubate
the transfection complex for 10 minutes at room temperature and add 500μl
GenJetTM/DNA mixture drop-wise onto the medium. After 15-18 hours replace the
medium by fresh RPMI-1640 supplemented with 10% FBS.
7. Cell sorting
After 36 hours transfection, the transfected cells on 10-cm plate were washed by 1X
DPBS and detached by accumax (Innovative Cell Technologies, USA). Cells were
collected by 1X sorting buffer with PI and centrifuge at 400×g, 3mins. Discard the
supernatant and re-suspend cell pellet in appropriate volume of sorting buffer (usually
3-5×107 cells were re-suspend in 1 ml sorting buffer). Cells were sorted on FACSAria
(BD Bioscience, San Jose, CA) through the service provided by the Flow Cytometric
Analyzing and Sorting Core (the First Core Laboratory, National Taiwan University
College of Medicine). The sorted cells in RPMI-1640 supplemented with 10% FBS and
antibiotics were centrifuge at 400×g, 5mins and wash by RPMI-1640 supplemented
with 10% FBS without antibiotics. The cells were seeded in 12-well plate (2×105/ per
well) overnight. The cell number was counted before H. pylori co-culture.
8. Statistics
Statistical analysis of experimental groups was performed by unpaired Student’s t-tests.
P<0.05 was considered significant. ( P value ≦0.05 = *, ≦0.01 = **, ≦0.001 = ***,
≦ 0.0001 = ****) The quantification of Western blots was performed by ImageJ.
Results
I. H. pylori enhanced the apoptosis of AGS cells while treat with TRAIL, and
phosphorylation of Akt as well as FLIPs expression in AGS cells decreased
concurrently.
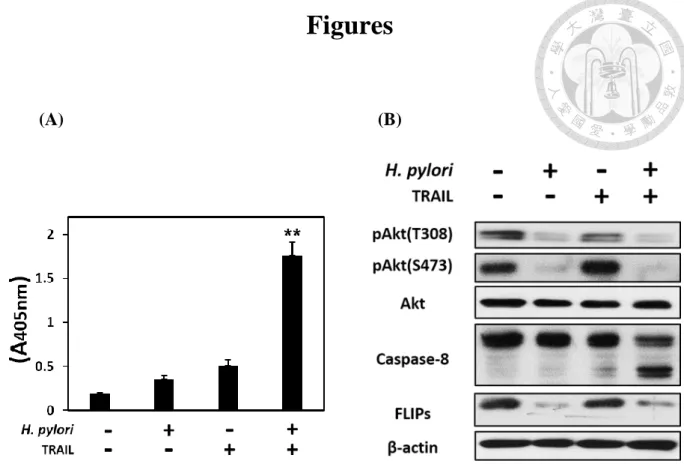
Previous study has reported that, the apoptosis of AGS cells increased after co-
culture with H. pylori for 12 hours and treat with TRAIL (40ng/ml) sequentially (Lin
et al., 2014). To start with, we confirmed the TRAIL-mediated apoptosis was severely
increased during co-culturing the human gastric epithelial cell line (AGS) with H. pylori, but couldn’t be induced when only incubated with either H. pylori or TRAIL (Fig. 1A).
Also, the pro-caspase-8 was cleaved into active form only when co-cultured with H.
pylori and TRAIL engagement simultaneously. This result was consistent with the
previous result in our laboratory. Furthermore, the phosphorylation of Akt in both of
S473 and Thr308 site, were decreased while H. pylori co-cultured, and this phenomenon
was correlated with FLIPs downregulation (Fig. 1B).
Moreover, previous study has shown that H. pylori-induced gastric epithelial cell
apoptosis is associated with increased of Fas receptor expression (Jones et al., 1999).
Besides, TRAIL shows sequence homology to Fas ligand (Wiley et al., 1995). We
wondered that whether Fas ligand exerts the same effect as TRAIL while co-cultured
with H. pylori. However, the enhancement of apoptosis only occurred while treated
with TRAIL but not Fas ligand. This result highlighted the importance of TRAIL-
mediated apoptosis while H. pylori infection in AGS cells (Fig. 2).
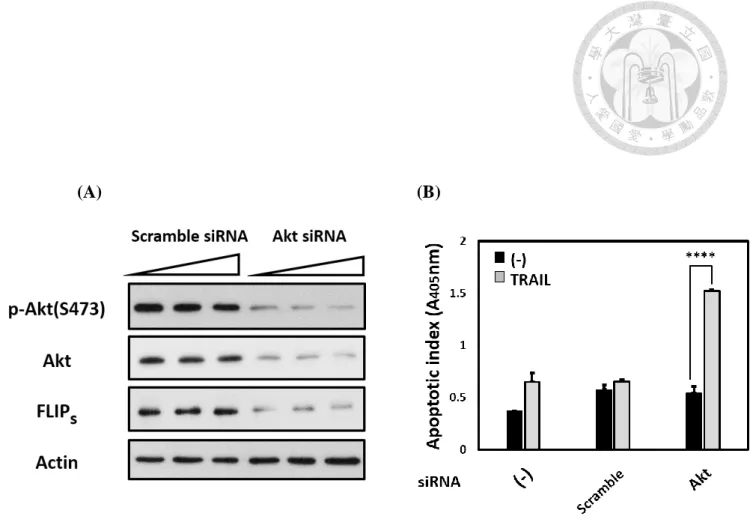
II. The expression of FLIPs was decreased and enhanced TRAIL-mediated
apoptosis in AGS cells after knockdown of Akt.
FLIPs is known to play a crucial role in preventing TRAIL-mediated apoptosis (von
Karstedt et al., 2017). Furthermore, our laboratory has reported that H. pylori
downregulated FLIPs expression, hence increasing TRAIL-sensitivity in AGS cells
(Lin et al., 2014). Associated with our observation in the previous section, we wondered
that whether Akt is able to regulate FLIPs expression in AGS cells. In order to clarify
the correlation between Akt and FLIPs, we transfected Akt siRNA in order to
knockdown Akt expression in AGS cells. The result has shown that decreased of Akt
expression resulted in FLIPs expression downregulated (Fig. 3A). Besides, loss of Akt
also enhanced the TRAIL-mediated apoptosis in AGS cells (Fig. 3B). Our results have
demonstrated that Akt enabled to regulate FLIPs expression and prevented TRAIL-
mediated apoptosis in AGS cells. Moreover, these data suggested that loss of Akt
activity is a possible pathway for H. pylori to enhance TRAIL-mediated apoptosis in
AGS cells.
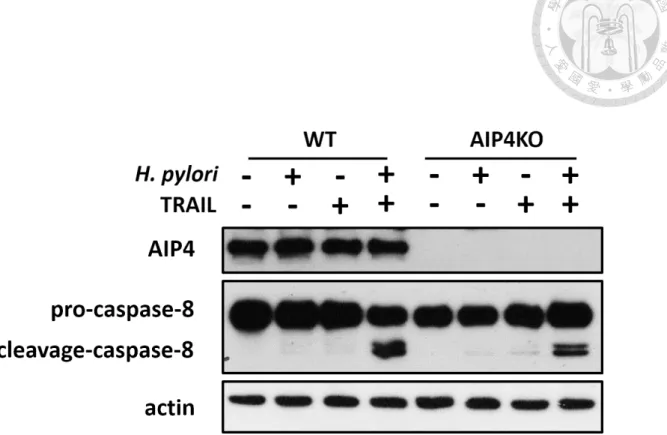
III. Knockout of AIP4 reduced H. pylori-induced TRAIL-mediated caspase-8
activation.
Several mechanism has been reported to participate in Akt-FLIPs pathway in
TRAIL-mediated apoptosis. One of them has been demonstrated that was modulated
by AIP4, an ubiquitin E3 ligase, to stimulate FLIPs degradation through proteasome
pathway. Previous data in our laboratory has shown that H. pylori infection resulted in
activating AIP4 activity (Woan-Yu Lin, 2016). In order to prove the role of AIP4 in H.
pylori-induced TRAIL-mediated apoptosis, we adopted AIP4 KO AGS cells to
examine that whether knockout of AIP4 rescued H. pylori-induced TRAIL-mediated
apoptosis. Our data has shown that, compared to WT AGS cells, AIP4 KO AGS cells
have less active cleavage-caspase8 while H. pylori co-cultured and TRAIL treatment
(Fig. 4).
Although we have shown that knockout of AIP4 reduced the H. pylori-induced
TRAIL-mediated caspase-8 activation, we have found that there still remains a certain
amounts of active caspase-8 while co-cultured with H. pylori and treated with TRAIL.
We have sought for another E3 ubiquitin ligase cbl, which has also been reported to
stimulate FLIPs degradation in mTOR-dependent but Akt-independent manner (Zhao
et al., 2013). We have observed that co-cultured with H. pylori caused cbl expression
increased (Fig. 5A). In addition, the interaction between cbl and FLIPs increased while
H. pylori infection. However, knockdown of cbl failed to mitigate H. pylori-induced
TRAIL-mediated apoptosis (Fig. 5C).
IV. mTOR expression was decreased after H. pylori co-cultured.
In order to investigate that how H. pylori modulated intracellular Akt activity, we
have screened the upstream molecules which control the Akt activity. Among of them,
we have found the mTOR expression decreased was correlated with Akt
phosphorylation as well as FLIPs expression decreased while co-cultured with H. pylori
in time-dependent manner (Fig. 6A). Besides, we found that H. pylori infection not only
resulted in decreasing mTOR expression but also the expression of Rictor and mSin1,
which are the other obligate subunit for mTORC2 formation. Moreover, the
phosphorylation mTORS2481, an intact marker for mTORC2 activity (Copp et al., 2009),
reduced while H. pylori infection. These results have shown that H. pylori may
modulate Akt activity through hindering mTORC2 formation to suppress mTORC2
activity. We have also investigated that how H. pylori prompted mTOR expression
decreased. We found that H. pylori did not affect mTOR gene expression but promote
mTOR degradation through proteasome (Fig. 6C, 6D).
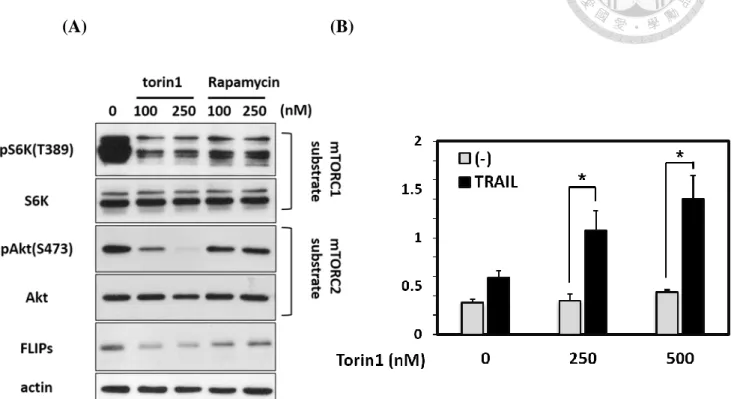
V. Inhibition of mTOR reduced the expression of FLIPs and induced TRAIL-
mediated apoptosis in AGS cells.
Since we have observed that mTOR expression decreased was correlated with Akt
de-phosphorylation as well as decreased of FLIPs expression. We wondered that
whether mTOR enable to regulate FLIPs expression in AGS cells. First, we utilized
mTOR inhibitor (Torin1 & Rapamycin) to investigate the relevance between mTOR
and FLIPs. We found that while using rapamycin, the mTORC1 inhibitor, caused the
slightly decreased of FLIPs expression but the expression of FLIPs was remarkably
reduced while treat with Torin1, the pan-mTOR inhibitor (Fig. 7A). Additionally, we
pretreated Torin1 to AGS cells and followed the TRAIL treatment afterward also
elevated the TRAIL-mediated apoptosis in gastric epithelial cells (Fig. 7B). This result
has revealed that pan-mTOR inhibition was able to heighten the TRAIL-mediated
apoptosis signaling in gastric epithelial cell via controlling FLIPs expression.
Moreover, in order to exclude the off-target effect while using the mTOR inhibitors,
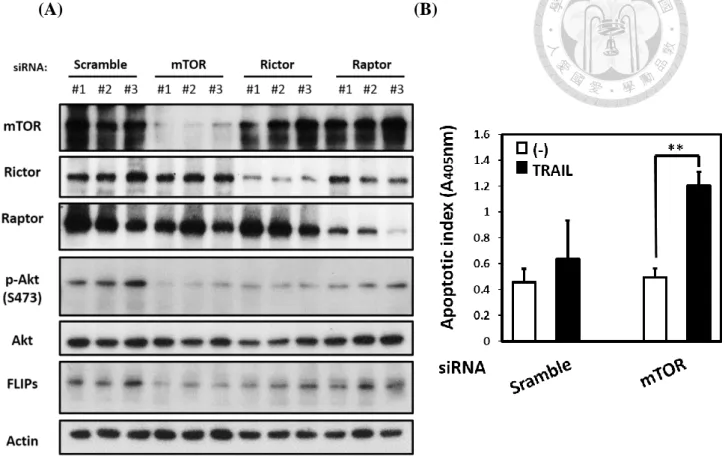
we also transfected mTOR siRNA into AGS cells. Knockdown of mTOR in AGS cells
strongly decreased the FLIP expression and enhanced the TRAIL-mediated apoptosis
in AGS cells (Fig. 8A, 8B). Since mTOR synergistic with other protein to form distinct
protein complexes, known as mTOR Complex 1 (mTORC1) and 2 (mTORC2) (Saxton
and Sabatini, 2017), we further discriminated the role of mTORC1/mTORC2 in
TRAIL-mediated apoptosis signaling. Knockdown of Raptor and Rictor, which is the
core protein of mTORC1 and mTORC2, to interfere the mTORC1/2 activity. While loss
of mTORC1 activity, the expression of FLIPs slightly reduced. However, while loss of
mTORC2 activity, the expression of FLIPs dramatically decreased (Fig. 8A). These
data demonstrated that mTOR, especially mTORC2, was able to regulate FLIPs
expression. In addition, mTOR provided resistance to TRAIL-mediated apoptosis in
AGS cells. Above all, these result implied that H. pylori may break the TRAIL-
resistance in AGS cells through inhibiting mTOR activity.
VI. Enhancement of mTOR-Akt signaling pathway reduced H. pylori-induced
TRAIL-mediated apoptosis signaling.
The data we performed have already shown that mTORC2 lost its function when
co-cultured with H. pylori. Since mTORC2 is the primary kinase for activating Akt, we
suspected the functional loss of mTORC2 resulted in the Akt de-phosphorylation while
H. pylori infection. First of all, in order to elevate the mTORC2 activity, we co-
overexpressed the peYFP-C1-mTOR/pRK5-HA-YFP-Rictor plasmid to enforce more
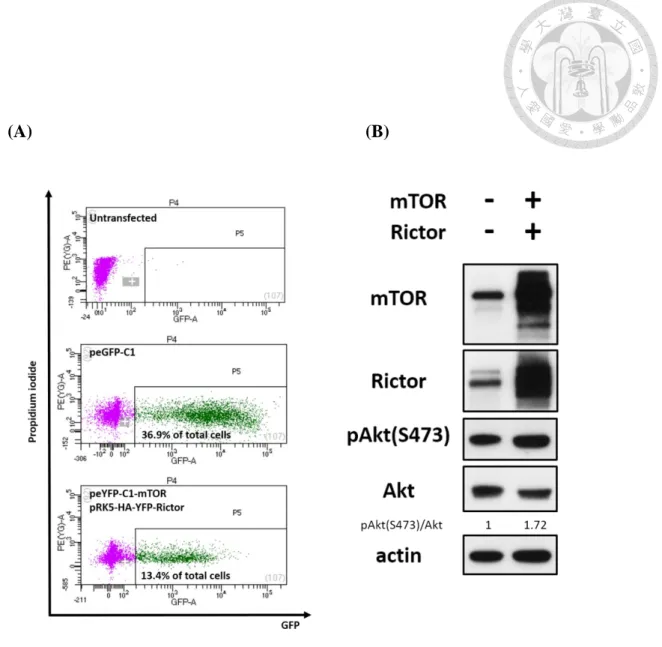
mTORC2 complex formation in gastric epithelial cells. We sorted out the GFP+ cells
and found the phosphorylation of AktS473, the downstream substrate of mTORC2
(Sarbassov et al., 2005), was increased compared to the vector group (peGFP-C1, GFP+
cells) (Fig. 9A, 9B). By taking advantage of this strategy, we found that while enhancing
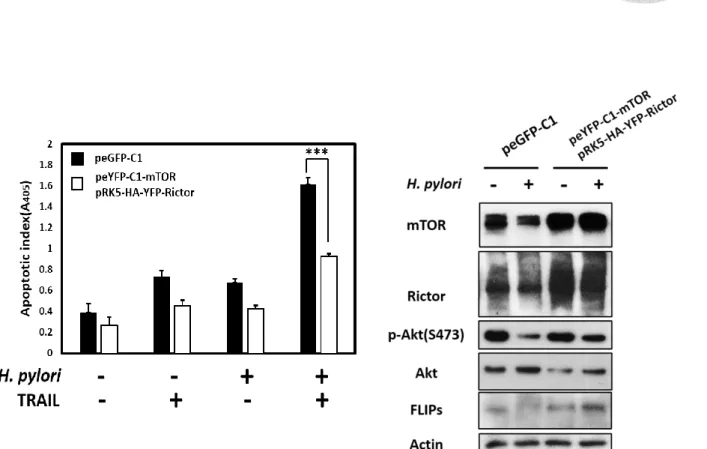
the mTORC2 activity, the H. pylori-induced TRAIL-mediated apoptosis was attenuated
in gastric epithelial cells (Fig. 10A). Furthermore, we found that augmentation the
mTORC2 activity sustained the Akt phosphorylation level while H. pylori infection
(Fig. 10B). These result demonstrated that mTORC2 was crucial for AGS cells to resist
with TRAIL-induced apoptosis while H. pylori infection through sustaining the
phosphorylation of Akt.
VII. H. pylori-induced autophagy could be attenuated while enhanced mTOR-
Akt signaling pathway in AGS cells.
Reduced Akt phosphorylation also inhibits downstream mTORC1 signaling
pathway (Saxton and Sabatini, 2017). In the previous result, we have displayed that H.
pylori decreased mTOR expression as well as reduced Akt phosphorylation (Fig 1B,
6A, 6B). These data implied that H. pylori may also suppress mTORC1 downstream
signaling pathway. We have displayed that loss of mTORC1 activity also resulted in
FLIPs decreased, although it was not severed as strong as the effect of losing mTORC2.
mTORC1 is also known for controlling protein turnover through autophagy. Besides,
there has been reported that inhibition of mTOR with activating autophagy flux
downregulates c-FLIP levels and contribute toward the enhancement of TRAIL-
sensitivity (Xu et al., 2016; Zhang et al., 2014). Combined with the data of decreased
mTOR expression while infection with H. pylori (Fig. 6A), these data prompted us to
investigate about whether H. pylori-induced autophagy contributes to enhancement of
TRAIL-mediated apoptosis in AGS cells. Indeed, we found H. pylori infection induced
cellular autophagy (Fig. 11) by elevating the expression of LC3-II, an autophagy marker.
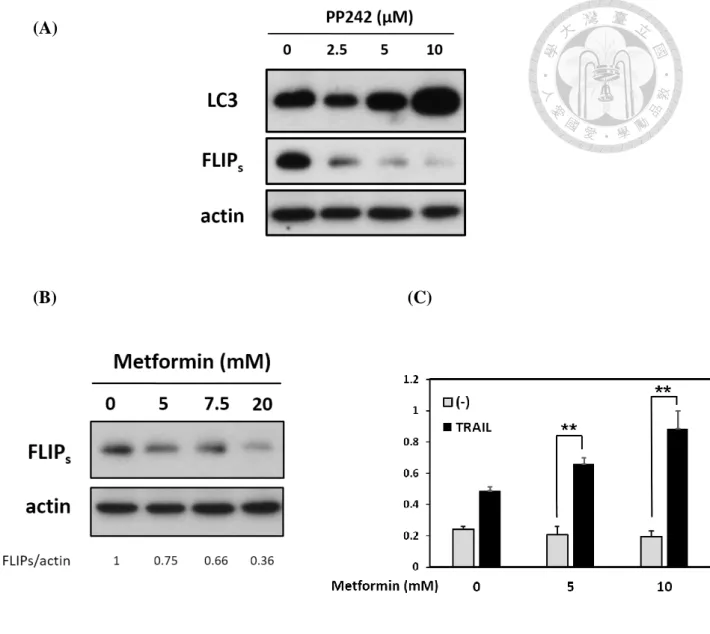
In order to examine whether mTOR inhibition was sufficient to induce autophagy, we
utilized PP242, the pan-mTOR inhibitor, to observe the effect of mTOR inhibition-
mediated autophagy in AGS cell lines. We found the expression of LC3-II has increased
as a dose-dependent manner. Besides, LC3-II expression increased was correlated with
FLIPs downregulation (Fig. 12A). We excluded the effect of mTORC2 by treating with
Metformin, an autophagy inducer, to address the relevance between autophagy and
FLIPs expression. We have also found that FLIPs expression reduced while activating
the autophagy flux (Fig. 12B). Besides, treating with Metformin also enhanced TRAIL-
mediated apoptosis (Fig. 12C). We further examined that whether the autophagy flux
could be turn off by enhancing the mTORC1 activity through overexpressing
mTOR/Rictor or Akt plasmid in gastric epithelial cells. We have found that enhancing
the mTORC2-Akt axis or Akt only successfully attenuated the autophagy process (Fig.
13A, 13B). Taken together, we have found that attenuation of autophagy process was
correlated with reducing H. pylori-induced TRAIL-mediated apoptosis (Fig. 13, Fig.
10).These data suggested that autophagy may also contribute in H. pylori-induced
TRAIL-mediated apoptosis.
Discussion
I. H. pylori-induced TRAIL-mediated apoptosis through inhibiting the
intracellular mTOR-Akt signaling pathway.
Based on the previous founding in our laboratory, Akt plays a crucial role in H.
pylori-induced TRAIL-mediated apoptosis. We wondered that how does H. pylori
reduce Akt phosphorylation state. In this study, we have found the expression of mTOR,
the core protein of mTORC1/2, decreased as H. pylori co-cultured. Besides, we found
the S2481 phosphorylation site of mTOR, which is an intact mTORC2 activity marker
also decreased (Copp et al., 2009). This data indicated that mTORC2 complex activity
was disrupted while H. pylori co-cultured. However, we haven’t investigated the details
about other obligate subunit such as the phosphorylation of mSin1, which has been
reported to associate with the positive feedback loop between Akt and mTORC2 (Yang
et al., 2015). It is crucial for us to further understand the H. pylori-modulated mTORC2-
Akt axis in order to realize the whole picture of H. pylori-induced TRAIL-mediated
apoptosis signaling.
Besides, we have developed a system to elevate the activity of mTORC2 by co-
overexpressing plasmids with mTOR/Rictor. At first, we tried to enhance the pan-
mTOR signaling pathway through overexpressing mTOR plasmid only. However, we
found the phosphorylation of AktS473 site decreased after overexpressed mTOR plasmid.
That may be the negative feedback exerted by mTORC1, which has been reported to
negatively regulate receptor tyrosine kinases (RTKs) signaling (Hsu et al., 2011; Yu et
al., 2011). Since mTORC2 is the upstream molecule of Akt, we turned to consider
enhancing mTORC2 activity to upregulate mTOR-Akt signaling pathway. We have
consulted several papers which also attempt to enhance mTORC2 activity by
overexpressing the plasmid of Rictor (Laugier et al., 2015; Masri et al., 2007). In line
with previous reports, we found that overexpression of rictor in AGS cell lines indeed
enhanced the Akt phosphorylation on S473 site. Furthermore, we got the largest
activation of Akt while co-overexpressing mTOR/Rictor plasmid simultaneously.
Therefore, we adopted this method to conduct the afterward experiment. But still, this
model existed some unresolved question: Does the mTOR/Rictor protein we have
overexpressed form the functional mTORC2 indeed? Although we have monitored the
mTORC2 activity by direct downstream substrate Akt, it may still remain other
unidentified pathway to enhance the Akt activity since the network signaling of mTOR
is complicated. To further clarify this question, we should observer whether there is
more interaction between mTOR and Rictor by co-immunoprecipitation while co-
overexpressing mTOR/Rictor plasmid.
As described previously, we found the negative impact on AktS473 phosphorylation
while overexpressing mTOR only. We got the largest Akt activation in our hands when
co-overexpressed mTOR and Rictor plasmid, despite the AktS473 phosphorylation does
not reach as same intensity as directly overexpressed Akt compared to previous result.
There may be two reasons: one is that the physiological regulation of mTORC2 toward
Akt is limited; another explanation is that our sorting strategy is imprecise (Fig. 9A).
We simply sorted out the GFP+ cells while co-overexpressing peYFP-C1-mTOR and
pRK5-HA-YFP-Rictor, however, we couldn’t identify every single cell expressing
YFP+ was homogeneously expressing mTOR and Rictor at same ratio since the
mTOR/Rictor protein expressed the same fluorescent protein. The possible solution is
to construct different fluorescent protein in one of these plasmids, and clarify the
appropriate ratio for mTOR: Rictor while enhancing the activity of mTORC2.
Besides, the previous study in our laboratory has shown that H. pylori also
dephosphorylated PTEN. The phosphorylated PTEN is proved to be less active (Song
et al., 2012). Thus, it implied that downregulation of Akt phosphorylation may also be
regulated by more active PTEN while H. pylori infection. We have conducted transient
knockdown of PTEN in AGS cell lines and followed the H. pylori co-cultured and
TRAIL treatment. The data showed that while PTEN knockdown, the H. pylori-induced
TRAIL-mediated apoptosis was mitigated (Szu-Ying, Chen. 2017). Yet it raised a
question: Whether PTEN or mTORC2-mediated pathway is dominant in H. pylori-
induced TRAIL-mediated apoptosis? Previous perspective may take PTEN and
mTORC2 as two independent events in regulation of Akt. However, there are increasing
evidence shows that PTEN also regulates mTORC2 directly or indirectly. Bhattacharya
et al., have observed that PTEN negatively regulates mTORC2 formation by promoting
Rictor phosphorylation (Thr1135), which is known to inhibit mTORC2 activity
(Bhattacharya et al., 2016; Julien et al., 2010). Thus, PTEN de-phosphorylation while
H. pylori infection may also contributes to the inhibition of mTORC2 activity (Fig. 6B).
Besides, knockdown of PTEN also gives rise to hyperactive mTORC2 activity
(Bhattacharya et al., 2016), hence rescuing H. pylori-induced TRAIL-mediated apoptosis couldn’t take it as a PTEN-dependent effect only. Also, PTEN counteracts
PI3K to dephosphorylate PIP3 (Song et al., 2012), Liu et al., have also observed that the
PH domain of mSin1 binds to the mTOR kinase domain in the absence of PIP3, thus
suppressing the mTOR kinase activity. Overall, loss of mTORC2 activity may also by
reason of H. pylori-induced PTEN activation.
II. H. pylori downregulated FLIPs expression through reducing Akt
phosphorylation.
Previous study in our laboratory has shown that H. pylori increased the activity of
AIP4 to ubiquitinate FLIPs through downregulating Akt phosphorylation. In this study,
we have further examined the role of AIP4 in H. pylori-induced TRAIL-mediated
apoptosis signaling. By taking advantage of AIP4 KO AGS cells, we have found that
H. pylori-induced TRAIL-mediated caspase-8 activation was mitigated while
compared to WT AGS cells. However, the AIP4 KO AGS cells did not fully block the
H. pylori-induced TRAIL-mediated apoptosis signaling (Fig. 4). This data suggested
that there may have other mechanisms which participated in FLIPs downregulation by
H. pylori. We have knockdown the Akt expression in AGS cells and found the FLIPs
expression was strikingly decreased (Fig. 3A). This result implicated that Akt activity
is crucial for regulating FLIPs expression. There are abundant evidences for displaying
the ability of Akt to regulate FLIPs but several of them elucidate the mechanisms in
details (Chen et al., 2001; Nam et al., 2003; Panka et al., 2001). Akt participates in
controlling translation process, post-translational modification and autophagy, and
those aspect of Akt have been reported to affect FLIPs expression (Panner et al., 2009;
Panner et al., 2005; Zhang et al., 2014). In this section, the different aspect of
controlling FLIPs expression by Akt would be discussed.
(a.) Translation
Akt is known to master the translation process through modulating mTORC1 activity.
In brief, PI3k-mediated activation of Akt leads to phosphorylation of TSC2, which
results in inhibition of the TSC1/2 complex. In the absence of TSC1/2, the small
GTPase Rheb is able to enhance mTOR activity and stimulate activation of downstream
targets of mTOR, the 70-kDa ribosomal S6 kinase 1 (S6K1) and the eukaryotic
translation initiation factor 4E (eIF4E) binding protein 1 (4EBP1). Both of S6K1 and
4EBP1 are master regulator of cellular translation process. Besides, Panner et al., have
shown that mTOR controls FLIPs translation and TRAIL sensitivity in glioblastoma
multiforme cells. Since H. pylori decreased mTOR expression and dephosphorylated
Akt sequentially (Fig. 1B, Fig. 6), we suspected H. pylori may also affect FLIPs
expression through mRNA level. However, we simply measured the FLIPs mRNA level
with/without H. pylori infection and found that there was no significant difference. This
data revealed that H. pylori did not affect the number of FLIPs mRNA, but it still can’t
exclude the possibility of H. pylori interfered the FLIPs translation process. As
described by Panner et al., they have found the ribosomal distribution of FLIPs mRNA
is critical for different GBM cell lines to exert TRAIL-resistance (Panner et al., 2005).
Whether H. pylori modulates the distribution of FLIPs mRNA in non-translating
monoribosomes or translating polyribosomes remains an interesting question to explore.
(b.) Post-translational modification
So far, the well-characterized pathway for the FLIPs regulation by Akt till now is
through modulating AIP4 activity (Panner et al., 2010). In this study, we have further
demonstrated that AIP4 participated in the H. pylori-induced TRAIL-mediated
apoptosis signaling. However, the H. pylori-induced TRAIL-mediated apoptosis did
not completely mitigate even if we knockouted AIP4 in AGS cells. Thus, we suspected
that there may have other undefined mechanisms which was regulated by Akt also
participate in H. pylori-induced TRAIL-mediated apoptosis signaling.
(c.) Autophagy
Akt de-phosphorylation inhibits the downstream mTORC1 signaling pathway. In this
study, we also found H. pylori infection induced cellular autophagy (Fig. 11). We have
found that enhancing the mTORC2-Akt axis or Akt only successfully attenuated the
autophagy process. We have shown that attenuation of autophagy process correlated
with reducing H. pylori induced TRAIL-mediated apoptosis, suggesting that autophagy
may contribute in H. pylori-induced TRAIL-mediated apoptosis. However, both methods we have utilized in this study (Overexpressed mTORC2/Akt) can’t solely
observe the effect of autophagy process on H. pylori induced TRAIL-mediated
apoptosis. To further resolve the impact of autophagy on H. pylori-induced TRAIL-
mediated apoptosis, knockdown of Atg5 or LC3 is a common technique to block the
autophagy process. Although we have demonstrated the importance of mTOR in H.
pylori-induced TRAIL-mediated apoptosis, the detail mechanisms inside are still worth
to investigate.
III. H. pylori downregulated FLIPs expression through Akt-independent
mechanisms.
There is a report that demonstrated mTORC2 is involved in regulation of FLIPs
degradation and sensitivity of TRAIL-induced apoptosis through c-cbl, an E3 ubiquitin
ligase (Zhao et al., 2013). Zhao et al., have demonstrated that PP242, an ATP-
competitive inhibitors of mTOR, cooperates with TRAIL to enhance apoptosis in
NSCLC cell lines. Besides, they identified the mTORC2 inhibition mediates cbl to
promote FLIPs degradation. Surprisingly, the author found that FLIPs downregulation
by PP242 could not be restored by constitutively active form of Akt. This paper
implicates that cbl-mediated FLIP degradation is a mTORC2-dependent, but Akt-
independent mechanism. We have also explored the possibility of cbl involved in H.
pylori-mediated TRAIL-mediated apoptosis, since AIP4 KO AGS cells couldn’t fully
block the activation of caspase-8 while H. pylori sensitizes TRAIL-mediated apoptosis
in AGS cells. However, we failed to rescue the H. pylori-mediated TRAIL-mediated
apoptosis by knockdown of cbl (Fig. 5). Although we have detected more interaction
between cbl and FLIP, cbl may have other physiological role on FLIPs but not
mediating FLIPs degradation in H. pylori-induced TRAIL-mediated apoptosis. These
data further support that mTOR-Akt-FLIP axis is dominant in H. pylori-induced TRAIL
apoptosis signaling.
There is another report shows that c-FLIP is a target of deltex1, an E3 ubiquitin ligase,
in gastric cancer, although they focus on studying the FLIPL expression. Their data also
show that overexpressed Deltex1 decreases both FLIPL/FLIPs expression in AGS cell
lines. Finally, they found Deltex1-mediated FLIPL degradation is through the
endosome-lysosomal pathway but not proteasome-independent degradation (Hsu et al.,
2018). Although our data have shown that H. pylori-mediated FLIP degradation is
mainly through proteasome (Fig. 6D), the possibility of deltex1 to participate in H.
pylori-mediated TRAIL-mediated apoptosis signaling is still another direction to
investigate.
IV. The upstream regulation of mTORC2 activity and the possible pathway for
H. pylori to disrupt the mTORC2 activity in AGS cell lines.
The upstream regulation of mTORC2 is not well-defined, although primarily
functions as an effector of insulin/PI3K signaling. Besides, the ribosome association is
also known for being mTORC2 activator (Kim et al., 2017; Sarbassov et al., 2005;
Saxton and Sabatini, 2017; Zinzalla et al., 2011). Despite a lots of unknown remains on
regulating mTORC2 activity, there are increasing evidence which have demonstrated
that the localization is important for mTORC2 activation. Liu et al., have also observed
the PH domain of mSin1 which binds to the mTOR kinase domain suppresses the