國立臺灣大學生命科學院生命科學研究所 碩士論文
Graduate Institute of Life Science College of Life Science
National Taiwan University Master Thesis
依受質結合區探討 MTH1 與抑制劑之交互作用研究
Investigation of interaction between MTH1 and synthesized inhibitors based on substrate binding site
李昱璇 Yu-Hsuan Lee
指導教授: 鄭貽生 博士 Advisor: Yi-Sheng Cheng, Ph. D.
中華民國 108 年 6 月
致謝
能夠順利完成論文,首先要感謝指導老師鄭貽生老師的協助和建議,也很謝 謝口試委員何孟樵老師和徐駿森老師撥空前來並很仔細的提出研究上的問題和指 導。
念研究所的3 年過程中雖然經過了一些事情而有了波折,碩一決定換實驗室
後誤打誤撞進入完全不熟悉的領域。雖然那時候一直覺得自己因為一時間的情緒 做出不理性的抉擇,並不斷地懷疑自己能不能順利的從研究所畢業。不過很幸運 有遇到有很多幫忙和支援我的人,不管是實驗室的學長和同學們還是新竹同步輻
射中心的beamline staff 們,都作為我在實驗生活中的一大助力。
此外,在從研究所畢業的這個時刻,我想特別感謝我的家人讓我在上來台北 念研究所的這段期間給予我經濟和心靈上最大的支持。
李昱璇 謹識於 台灣大學生命科學院 2019 年 6 月
Contents
口試委員會審定書 ... i
致謝 ... ii
Contents ... iii
List of Figures ... vi
List of Tables ... viii
中文摘要 ... ix
Abstract ... x
List of abbreviations ... xii
Chapter 1 Introduction ... 1
1-1 Carcinogenesis: Distinguishing cancer cells from normal cells ... 1
1-1-1 ROS homeostasis in cancer development and apoptosis ... 1
1-1-2 Anticancer drug discovery exploiting ROS-mediated signaling pathways in cancer cell ... 2
1-2 Practical aspects in cancer therapeutic development ... 3
1-2-1 Identification and validation of molecular targets in cancer cell ... 3
1-2-2 High-throughput screening in searching for potential drug candidates and hit- to-lead optimization campaign ... 4
1-2-3 Biochemical and biophysical approaches in hit-to-lead optimization campaign ... 5
1-2-4 Thermodynamic profiles of protein-ligand complex formation ... 5
1-3 MutT homolog 1 (MTH1)- potential target for anticancer drug ... 7
1-3-1 Physiological function of MTH1 and ROS-modulated apoptosis ... 7
1-3-2 Protein structure of MTH1 ... 8
1-3-3 MTH1 inhibitor development- perspectives and directions ... 9
1-4 Specific Aims and objectives ... 10
Chapter 2 Experimental section ... 12
2-1 Materials ... 12
2.2 Methods ... 12
2-2-1 Transformation ... 12
2-2-2 Protein Expression ... 13
2-2-3 Affinity Chromatography ... 13
2-2-4 IC50 Determination ... 14
2-2-5 Isothermal titration calorimetry ... 15
2-2-6 Size-exclusion chromatography ... 15
2-2-7 Crystallization and soaking ... 16
2-2-8 X-ray data collection ... 17
2-2-9 Model building and Refinement ... 17
Chapter 3 Results ... 19
3-1 MTH1 protein purification ... 19
3-1-1 Affinity Chromatography ... 19
3-1-2 Size-exclusion chromatography (SEC) ... 19
3-2 Determination of binding affinity profiles and structure-activity relationship (SAR) ... 20
3-2-1 Fragment-based drug screening and optimization performed with 2- aminopyrimidine-based scaffold ... 20
3-2-2 Binding affinity profile and SAR study of scaffold 1-derived compounds .. 21
3-2-3 Binding affinity profile of scaffold 2-derived compounds ... 22
3-3 Thermodynamic study on 2-aminopyrimidine-based compounds derived from scaffold 2 ... 23
3-3-1 Enthalpy-driven binding of 2-aminopyrimidine-based compounds to MTH1 ... 23
3-3-2 Phenomenon of Enthalpy-Entropy Compensation (EEC) ... 24 3-3-3 Rationalization of observed EEC phenomenon through insights provided by atomic model of MTH1 in complex with 2-aminopyrimidine-based compounds . 25 3-4 Structural analysis of MTH1 complex structure bound with 2-aminopyrimidine -
based compounds ... 28
3-4-1 Conserve binding mode of pyrimidine-based compounds in the binding site of MTH1 ... 28
3-4-2 Pyrimidine moiety ... 29
3-4-3 R1 and R2-substituent ... 30
3-5 Superposition of MTH1 complexed with product 8-oxo dGMP and compound 29, 30 ... 30
3-5-1 Absence of affinity gain with extended substituent toward the entry of binding pocket ... 31
3-6 Notable difference in interaction pattern of heterocyclic substituent unraveled by deviation in position occupied by ligand ... 32
Chapter 4 Discussion ... 34
4-1 Functional mechanism of MTH1 protein and structural relevance to selectivity toward its substrate ... 34
4-2 The binding energetics underlying complex formation ... 35
4-2-1 Intrinsic phenomenon of EEC hinders the elevation of binding affinity ... 36
4-2-2 Proposed theories for explanation of contradictory effect of enthalpic and entropic component of Gibbs binding free energy ... 37
4-3 Structure-aided ligand design: perspectives and opportunities ... 38
4-3-1 Binding pattern observed in published MTH1-ligand complexes ... 39
4-3-2 Overview of available space left unexplored in MTH1 binding pocket in regard of information derived from explored regions ... 41
Chapter 5 Conclusion ... 43
References ... 45
Tables and Figures ... 51
Appendices ... 91
List of Figures
Figure 1. Schematic depiction of simplified work flow and objectives of this study .... 60 Figure 2. Purification of MTH1 protein from E.coli cell lysate ... 62 Figure 3. Binding affinity profile of scaffold 1 derivative compounds ... 64 Figure 4. Binding affinity profile of scaffold 2 derivative compounds ... 66 Figure 5. Thermodynamic study of scaffold 2 derivatives binding to MTH1 protein ... 68 Figure 6. Phenomenon of EEC observed in scaffold 2 derivatives binding to MTH1 protein ... 70 Figure 7. Comparison of thermodynamic profiles from structurally related compounds with different substituents incorporated in the R2-position ... 72 Figure 8. Comparison of thermodynamic profiles from structurally related compounds with different substituents incorporated in the R1-position ... 74 Figure 9. Phenomenon of enthalpy-entropy compensation gleaned from protein-ligand complex structures ... 76 Figure 10 Comparison of binding mode of 11 structurally related 2-aminopyrimidine- based compounds uncovered in x-ray crystallographic study. ... 78 Figure 11. Binding pose of MTH1 product 8-oxo dGMP, compound 29 and 30 in bound state gleaned from crystal structure complexed with MTH1 ... 80 Figure 12. Mutual alignment of MTH1 complex structures bound with substrate,
compound 29 and 30 ... 82 Figure 13. Binding mode of compound 22, 24, 29 in MTH1 binding pocket and
structural rearrangement of side chains from Phe27 and Met81 ... 84
Figure 14. Binding mode of compound 12, 26, 30 in MTH1 binding pocket and
structural rearrangement of side chains from Phe27 and Met81 ... 86 Figure 15. Binding mode of compound 20, 24, 25, 26 in MTH1 binding pocket and structural rearrangement of side chains from Phe27 and Met81 ... 88 Figure 16. Binding mode of compound 12, 22, 29, 30 in MTH1 binding pocket and structural rearrangement of side chains from Phe27 and Met81 ... 90
List of Tables
Table 1. Structure and related biochemical data of scaffold 1-derived compoundsa ... 51 Table 2. Structure and related biochemical data of scaffold 2-derived compoundsa ... 52 Table 2. Structure and related biochemical data of scaffold 2-derived compoundsa (continued) ... 53 Table 3. Thermodynamic profiles of scaffold 2-derived compounds determined with iTC ... 54 Table 4. Data collection and refinement statistics for MTH1 crystal structure complexed with 11 structurally-related 2-aminopyrimidine-based compounds ... 55 Table 4. Data collection and refinement statistics for MTH1 crystal structure complexed with 11 structurally-related 2-aminopyrimidine-based compounds (continued) ... 56 Table 4. Data collection and refinement statistics for MTH1 crystal structure complexed with 11 structurally-related 2-aminopyrimidine-based compounds (continued) ... 57 Table 4. Data collection and refinement statistics for MTH1 crystal structure complexed with 11 structurally-related 2-aminopyrimidine-based compounds (continued) ... 58
中文摘要
Mut T homolog 1 (MTH1) 為 Nudix 水解酶超家族的成員,在癌細胞的增生中
扮演關鍵的角色。先前研究指出 MTH1 適合作為抗癌藥物的標靶,並已有 MTH1
抑制劑的體外以及體內成效被發表。本論文為深入探討 MTH1-抑制劑複合體的結
合機制,以高通量藥物片段篩選具有抑制MTH1 的小分子,以其中一組作為骨架,
進一步設計修飾2-氨基嘧啶衍生物共 14 個,依嘧啶環上 5 位碳乙基的有無分類為
兩個群組,並分別解析MTH1 與 11 個 2-氨基嘧啶衍生物複合體結構,輔以生化及
熱力學分析,以探討MTH1 與抑制物複合體結合作用之多方位資訊。
根據已解出之MTH1 與受質複合體結構,可將 MTH1 之活性口袋區初步劃分
為 三 個 特 定 區 域 。MTH1 之 受 質 8-oxo dGTP (8-Oxo-2’-deoxyguanosine-5’- Triphosphate)具有三部分官能基分別為鹼基,去氧核糖以及三磷酸,在鹼基部分主
要由 Asp119,Asp120 及 Trp117 的側鏈鍵結,此區域也是這些抑制劑在結合口袋
中的主要穩定作用力,旁側存在由多個疏水性胺基酸 Phe72,Phe74 及 Phe139 側
鏈形成之空間,本實驗中將抑制劑的官能基延伸至由Phe72,Phe74,Phe139 形成
的空腔時,並無法提升抑制劑結合能力。從受質的 8 號位置氧原子位於 Phe27 與
Met81 的側鏈附近,且設計抑制劑亦與 Phe27 及 Met81 可形成凡得瓦力交互作用,
本區將可提供與MTH1 專一性結合。根據本研究系列合成抑制物之抑制效率分析,
當抑制物延伸至受質之三磷酸的區域時,其結合能力降低三個級數,由 Lys23,
Glu52,Glu55,Glu56 及 Glu100 組成的三磷酸結合區在催化水解反應時,會與金 屬離子結合並形成水分子網絡,本區可作為未來延伸區域,以提高專一性結合。最 後,本研究以受質結合區結構、熱力學分析及酵素動力學探討設計抑制劑的方式,
將可作為未來在設計新穎抑制劑時參考依據。
Abstract
Mut T homolog 1 (MTH1) is a member of Nudix hydrolase superfamily and engages in proliferation of cancer cells as critical determinant. In previous study, it was proposed that MTH1 serves as ideal anticancer target for anticancer therapeutic development , and in vitro and in vivo efficacy were reported for several MTH1 inhibitors.
In this study, to elucidate the mechanism of MTH1-inhibitor complex formation extensively, high-throughput screening (HTS) was performed on fragment library intended to identify small molecule compound with moderate inhibitory activity against MTH1. From identified hits, one hit compound was utilized as scaffold and a series of 2- aminopyrimidine derivatives comprising 14 structurally-related compounds were subsequently synthesized based on HTS results. These 2-aminopyrimidine-based compounds are categorized into two groups based on the presence of ethyl substituent on pyrimidine ring. Explicitly, MTH1 complex structures bound with eleven 2- aminopyrimidine derivatives were obtained and supplemented with statistics from biochemical and thermodynamic analyses to complement the intricate binding interaction of MTH1-inhibitor complex from multiple aspects.
Based on MTH1-substrate complex structure, the substrate binding pocket can be divided into three subregions which are occupied by base, deoxyribose and triphosphate group of substrate 8-oxo dGTP, respectively. In the base binding region, the bonds are formed by side chains of Asp119, Asp120 and Trp117 which are essential in binding of synthesized inhibitors in the binding pocket. Adjacent to the base binding motif, a minor cavity formed by side chains of hydrophobic residues Phe72, Phe74, Phe139 allows further exploration with substituents. However, no enhanced binding affinity was observed in exploring this cavity. Additionally, in the analysis of MTH1-inhibitor complex structures, it was observed that residues Phe27 and Met81 form van der Waals
interactions selectively with synthesized compounds upon formation of complex.
These residues sit in the proximity of 8-oxo group from substrate which was inferred to contribute to specificity of MTH1 through formation of favorable interactions.
Intrinsically, as implied in structure-activity relationship study, incorporation of ligand moiety which occupies triphosphate binding region of MTH1 reduced binding affinity by three orders of magnitude. Structurally, triphosphate binding region constituted of Lys23, Glu52, Glu55, Glu56 and Glu100 facilitates catalysis of hydrolytic reaction through formation of water molecule network coordinated by metal ions. Therefore, it is indicated that this subregion can be further exploited for enhancement of specificity of inhibitors.
In conclusion, through investigation of MTH1 substrate binding pocket with structural analysis, thermodynamic studies and enzyme kinetic analysis in this study, directions are provided for design of novel inhibitors in the future.
List of abbreviations
Asn Asparagine
Asp Aspartic acid
cDNA complementary DNA
Cryo-EM Cryogenic electron microscopy
DMSO Dimethyl sulfoxide
DNA Deoxyribonucleic acid
dNTP Deoxynucleoside triphosphates
DTT Dithiothreitol
E.coli Escherichia coli
EEC Enthalpy-entropy compensation FPLC Fast protein liquid chromatography Glu Glutamic acid
Gly Glycine
H-bonds hydrogen bonds
HEPES 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid IC50 The half maximal inhibitory concentration
IPTG Isopropyl β-D-1-thiogalactopyranoside ITC isothermal titration calorimetry
kDa kilodalton
Kd dissociation constant
Ki inhibitory constant
LB Lysogeny broth
Leu Leucine
LiSO4 Lithium sulfate
Lys Lysine
Mg acetate Magnesium acetate MgCl2 Magnesium chloride MTH1 MutT homolog-1
NaCl Sodium chloride
NCS Non-crystallographic symmetry NMR Nuclear Magnetic Resonance
NSRRC National Synchrotron Radiation Research Center PDB Protein Data Bank
PEG Polyethylene glycol Phe Phenylalanine
PMSF Phenylmethane sulfonyl fluoride PPi Pyrophosphate
RNA ribonucleic acid
ROS Reactive oxygen species
SAR Structure–activity relationship
TEMED N,N,N',N'-tetramethylethane-1,2-diamine Thr Threonine
TLS Translation–Libration–Screw-rotation Tris 2-amino-2-(hydroxymethyl)propane-1,3-diol
Tris-HCl 2-amino-2-(hydroxymethyl)propane-1,3-diol hydrochloride Trp Tryptophan
Tween-20 Polysorbat 20 Tyr Tyrosine Val Valine
2-oxo dATP 2'-deoxy-2-hydroxyadenosine
8-oxo dGTP 8-oxo-2'-deoxyguanosine-5'-triphosphate
Chapter 1 Introduction
1-1 Carcinogenesis: Distinguishing cancer cells from normal cells
Cell, the most basic functional unit of living organism, propagates by numerous biochemical reactions which are modulated by intricate network of intersecting signal transduction pathways (Evan and Vousden 2001). The signaling pathways which involve in cell growth, proliferation and apoptosis are integral modulators in maintaining physiological activities and fates of individual cell. As the fine-tuned modulation network is interrupted through dysregulation of participating biological molecules, carcinogenesis of normal cell is elicited as consequence.
In general, cancer is regarded as a group of immortal cells attributed to their dysregulated growth resulting mainly from defects in signal transduction pathways. In comparison to normal cells which undergo cell cycle and enter apoptosis under control of regulated molecular network. It is noteworthy that reprogramming of signaling network is essential in cancer as these immortal cells thrive in environment lethal to their normal counterparts (Harris et al., 2011).
1-1-1 ROS homeostasis in cancer development and apoptosis
The immortality of cancer cells is proposed to arise from altered molecular network modulating physiological events. To trigger and facilitate proliferation of cancer cells in microenvironment hostile to normal cells (Amoedo et al., 2013), adaptation of gene regulation in favor of the rigorous growth is pivotal for the survival of cancer cells.
Under elevated rate of metabolic activity common in fast-growing cancer cells, reactive oxygen species (ROS) is excessively generated as byproduct of metabolism.
In comparison to normal cells which grow and proliferate under regulation of intrinsic molecular network, homeostasis of intracellular ROS is critical in maintaining cellular function of cancer cells with elevated ROS level (Liou and Storz, 2010).
To sustain the physiological functions of cancer cells, redox balance is maintained through production of antioxidant proteins in response to increased levels of ROS (Kumari et al., 2018). Furthermore, to complement the understanding of impact of ROS on cancer initiation, progression and apoptosis, the role of ROS in cancer development has been extensively reviewed earlier from the perspective of cellular metabolism (Cairns et al., 2011).
1-1-2 Anticancer drug discovery exploiting ROS-mediated signaling pathways in cancer cell
Explicitly, to facilitate cancer progression, redox balance related to modulation of intracellular ROS level is regarded as key player in cancer physiology (Waris and Ahsan, 2006). Therefore, to exploit this well-characterized feature of cancer cells for anticancer agent development, it has been proposed in numerous studies that establishment of knowledge derived from ROS-mediated cellular events facilitates identification of molecular targets for drug discovery campaigns (Moloney and Cotter, 2018).
ROS-mediated signaling pathway for elimination of cancer cells, strategies were developed based on established knowledge of ROS-mediated cancer progression.
Therefore, two opposite approaches featuring pro-oxidants and antioxidants which promote cell apoptosis through distinguished working mechanisms provide opportunities for anticancer agent development. Intended to elevate intracellular oxidative stress to induce cell apoptosis, pro-oxidants exert its effects through promoting accumulation of ROS in cancer cells. Oppositely, antioxidants inhibit signaling molecules from ROS-
mediated signal transduction pathway and suppress tumor growth via abrogation of ROS signaling (Trachootham et al., 2009).
1-2 Practical aspects in cancer therapeutic development
Attributed to intensive research on molecular mechanism for modulation of cellular events in cancer cells over decades, exploitation of established knowledges of cancer biology for anticancer agent development is fueled and several therapeutics including imatinib (Gleevec), gefitinib (Iressa), ceritinib (Zykadia), crizotinib (Xalkori) have been successfully developed in this period. In practical aspect, anticancer drug discovery is generally initiated from theoretical strategies with aims to restore programmed cell death in cancer cells (Stephen, 2005). As immortality of cancer cells stem from deregulation of apoptotic pathway, elimination of these immortal cells depend on intervention via therapeutic agents designed for targeting specific signaling molecules to elicit cell apoptosis.
Followed by concretization of abstract ideas derived from acquired knowledge of cancer biology, extensive search on druggable targets in molecular signaling pathway is performed as routine work in cancer drug discovery.
1-2-1 Identification and validation of molecular targets in cancer cell
In the initial stage of anticancer drug discovery campaign which spans various disciplines of scientific research, the most fundamental while influential issue is always concerned with identification of optimal molecular target with significant relevance to the disease. Recalls that the hypothesis which is supportive of the notion that modulating key molecular targets in cancer cells serves as dominant strategy in anticancer therapeutic development, through selection of specific targets for design of therapeutic agent several
problems emerge at this time point which may hinder further progress and success in anticancer drug discovery campaign.
To address the problems emerged from selection of exploitable targets in cancer cells, validation of identified molecular targets is the primary mission to be fulfilled to ensure that designed therapeutics is clinically effective through acting on selected target.
As paradigm of target validation, three lineages of approaches have been proposed and reviewed by John D. Benson et al. To investigate the formulated mechanism of therapeutic agent acting on target molecules which is related to establishment of its clinical efficacy, validation through setting up experimental trials and collection of data in genetic studies, cell-based and animal model systems plays a key role in the success of anticancer drug discovery campaign. As biological system is far more complex than simplified overview of signaling transduction pathways which modulate cellular events, rationalization of action of drug molecules designed for its intracellular targets in in vitro and in vivo systems is therefore fundamental in optimization of specificity, selectivity, physicochemical properties for drug-likeness and ADME profiles of designed drugs.
1-2-2 High-throughput screening in searching for potential drug candidates and hit-to- lead optimization campaign
To identify adequate starting point for development of anticancer drug, high- throughput screening (HTS) is routinely performed on established library comprised of synthesized chemicals in large amount. In regard of utility of identified hits from HTS in subsequent hit-to-lead optimization campaign, criteria for filtration of trivial fragment compounds in HTS library is set out to ensure the integrity of drug candidates.
Pragmatically, to complement this context assessment of structure-activity relationship (SAR) and absorption, distribution, metabolism and excretion properties is conducted
(Konrad et al., 2003).
Based on results from HTS performed in conditioned biochemical assays, identified hits with moderate affinity toward a given target are further optimized. Therefore, investigation of hit compounds in preset experimental trials is performed to evaluate possibilities of creating lead series with favorable drug properties and optimal selectivity profile toward its targets. As outcome of hit-to-lead optimization campaign, modifications on initial scaffold compound may lead to compensation effects in biochemical affinity for target and drug-likeness properties. Despite of this dilemma, it is to recognize that establishment of complementary profile for ideal drug is comprised of balanced biological and physicochemical properties.
1-2-3 Biochemical and biophysical approaches in hit-to-lead optimization campaign Throughout optimization program of hits identified from high-throughput screening on compound library, information related to interaction profiles of hit compounds with their target is on demand for development of more suitable drug candidate at this stage.
As hit-to-lead optimization campaign usually initiates with structure-activity relationship (SAR) studies conducted in biochemical assays. Validation of binding event and elucidation of exact interaction profile are important in decision making during generation of optimized lead series.
In practical terms, determination of such interaction profiles is dependent on several well-established biophysical techniques including x-ray crystallography, nuclear magnetic resonance spectrometry, surface plasmon resonance spectrometry and isothermal titration calorimetry (Renaud et al., 2016).
1-2-4 Thermodynamic profiles of protein-ligand complex formation
From the perspective of thermodynamics, association and dissociation of protein- ligand complex is always accompanied by fluctuation in energetic status compared to unbound state which is attributed to rearrangement of overall configuration involving formation and breakage of chemical bonds (Geschwindner et al., 2015). In practical, to gain more insight into details of protein-ligand association which is essentially highly dynamic in nature, through recording thermodynamic signal from binding event related to the formation of protein-ligand complex with isothermal titration calorimeter (iTC) which is praised for its convenience and simplicity in practical measurement (Schnapp et al., 2016). Furthermore, elucidation of driving forces facilitating the binding event is enabled through calculating the enthalpic and entropic component of Gibbs free energy which is given as ∆G=∆H-T∆S. As illustrated in published research articles (Klebe, 2015), issues associated with thermodynamic analysis of protein-ligand association in various independent case studies came into spotlight which in certain degree further complicate the interpretation of measured iTC data (Sharp, 2001). Consequently, making judgement solely on thermodynamic profiles of each drug candidate might be biased in some occasion. To overcome this dilemma, interpretation of thermodynamic signatures of compound series binding to its target molecule derived from iTC data set is complemented by x-ray structure of protein-ligand complex which yields more reliable information for subsequent optimization program.
1-2-5 X-ray crystallography-static picture of protein-ligand complex
In searching for bioactive compounds which can be exploited as therapeutic agent for diseases, molecular recognition of drug target driven by physical forces which contribute to specificity and selectivity of the binding interaction serves as key element in structure-based drug design (Kuhn et al., 2011) . In support of developing drug
molecule which is characterized by high affinity and specificity toward its target, x-ray structure of target macromolecule complexed with an array of small molecule inhibitors sheds light on functional mechanism of how drug molecules act on its interacting partner (Deller et al., 2015).
1-3 MutT homolog 1 (MTH1)- potential target for anticancer drug
It is well-recognized that identification of disease-relevant targets plays a key role in drug discovery campaign. In the case of anticancer drug development, identified targets stem mainly from apoptosis-relevant biomolecules which include the BCL2 family of anti-apoptotic proteins, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptors, inhibitor of apoptosis (IAP) proteins and MDM2 (Stephen, 2005). It is revealed that promoting apoptosis serves as main strategy in killing these immortal cells. Whereas, regarding the issue of intra-tumor heterogeneity proposed in previous studies, it is reasoned by Gad et al. that targeting genetic defects is limited and suggested that exploiting cancer phenotype may serve as more optimal strategy.
However, followed by launch of MTH1 inhibitor development undertaken by Gad et al., intensive debate on MTH1 as a validated anticancer target emerged as an issue for clinical evaluation of anticancer efficacy of drug candidates targeting MTH1 derived from lead optimization archives (Kawamura et al., 2016).
1-3-1 Physiological function of MTH1 and ROS-modulated apoptosis
As one of the members of Nudix hydrolase superfamily, MutT homolog 1 (MTH1) has become popular in cancer research recent years which is attributed to the fact that MTH1 has been validated as promising anticancer target in either in vitro or in vivo experimental assays from the results published in 2014 by Gad et al. However, to support
the statement that MTH1 is suitable to serve as an anticancer target, the function of MTH1 and its relevance in the pathogenesis of cancer should be further elucidated (Kettle et al., 2016).
At the beginning of this chapter, ROS is referred to as important modulator in the physiological function of cell. In relation to the uncontrolled proliferation of cancer cells, elevated level of ROS leads to cell apoptosis in the absence of antioxidants such as housecleaning enzymes. This information reveals that inhibition of the activity of these cellular components maintaining the proliferation of cancer cells can be utilized in developing strategy for cancer therapies (Luo et al., 2010). In the case of MTH1 which is characterized as housecleaning enzyme in removing nucleobases oxidized by ROS to prevent cancer cell from entering programmed cell death, is regarded as promising target for development of small molecule inhibitors used in treatment of cancer (Gad et al., 2014).
1-3-2 Protein structure of MTH1
As a member of Nudix hydrolase superfamily, the overall structure of MTH1 is made up of typical Nudix fold α- β- α sandwich structure composed of two α-helices and eight β-strands, in which a mixed β-sheet is sandwiched between two α-helices (Bessman et al., 1996). Beside secondary structure building up the whole structural architecture, atomic detail of the hydrolase is also illustrated as followed.
Firstly, with insight into the substrate binding pocket of MTH1, key residues Asn- 33, Asp-119 and Asp-120 participate in substrate recognition through hydrogen bonding interaction with base moiety of the substrate (Sakai et al., 2002) and therefore regarded as the most well-characterized feature for selectivity and specificity of MTH1 protein (Waz et al., 2017).
Besides, in the substrate binding pocket, a minor cavity made up of phenyl rings of residues Phe72, Phe74 and Phe139 is well-defined and located in the neighborhood from recognition element Asp119-Asp120 motif (Nissink et al., 2016). While in the near of the entry of MTH1 binding pocket, a Glu cluster composed of residues Glu52, Glu55 and Glu56, Glu100 which lies in the Nudix motif is featured as region for catalyzing the hydrolysis of the substrate 8-oxo dGTP and 2-oxo dATP via binding network made up of metal ion and water molecules (Mishima et al., 2004). In conclusion, structural features mentioned above is generally thought to be associated with substrate selectivity and specificity of MTH1.
1-3-3 MTH1 inhibitor development- perspectives and directions
From the time when the statement that hydrolase MTH1 serves as promising target for anticancer drug was proposed by Gad, H. et al., inhibitor development with aims to deplete MTH1 catalytic activity has been undertaken by several research groups during this period (Gad et al., 2014) (Huber et al., 2014) (Kettle et al., 2016) (Rahm et al., 2018).
With the launch of anticancer drug discovery campaign targeting protein MTH1 in cancer cells which is non-essential in its normal counterparts, numerous structural motifs with moderate affinity toward MTH1 have been identified. Therefore, structure-based drug design is propagated with an abundance of crystal structures of MTH1 protein in either bound or unbound state accessible on the website of Protein data bank (PDB).
Over years, MTH1 crystal structure complexed with lead series optimized from structurally-irrelevant hit compounds provide insights into varied interaction pattern of MTH1-inhibitor complexes (Gad et al., 2014) (Huber et al., 2014) (Kettle et al., 2016) (Rahm et al., 2018). From these independently conducted optimization campaigns, design of potent inhibitor with desired selectivity profile is guided by key binding interactions
in MTH1-substrate complex structure (Nissink et al., 2016) (Waz et al., 2017). Through characterization of structural components in MTH1 binding pocket, promiscuous binding behavior is unraveled and implies that selectivity profile of drug candidate might be affected without establishing specific binding interactions. In consistence with results from off-target effect reported for MTH1 inhibitor TH588 in spite of reasonable selectivity profile from test on a panel of 87 enzymes, GPCRs, kinases, ion channels and transporters (Gad et al., 2014). Therefore, selectivity profile of MTH1 inhibitor has been augmented by mimicking interaction pattern adapted by substrate molecule in MTH1 binding pocket (Kettle et al., 2016) (Rahm et al., 2018).
1-4 Specific Aims and objectives
To address the issue of promiscuous binding behavior observed for designed ligands targeting MTH1, key structural features in MTH1 binding pocket are explored with substituents to elucidate mechanism of protein-ligand complex formation.
In this study, a series of analogue compounds was synthesized based on 2- aminopyrimdine scaffold. Subsequently, structure-activity relationship (SAR) was established which provides information on affinity profiles of structurally-related compounds. In addition, visualization of atomic structure of MTH1-compound complexes and structural alignment with MTH1-substrate structure enables dissection of binding patterns observed for different ligands. Explicitly, underlying mechanism of formation of MTH1 complex structure is investigated by isothermal titration calorimetry (iTC) which reveals thermodynamic profiles of analyzed molecules (Figure 1).
Remarkedly, research on binding interaction of MTH1 with a series of structurally related compounds unravels conformational changes related to accommodation of different ligands. To apply these acquired information in further optimization program, it
is important to formulate binding motifs and interaction patterns contributing to specificity and selectivity of protein toward its binding partners in complex biochemical environment.
Chapter 2 Experimental section
2-1 Materials
For expression of MTH1 protein, plasmid with cDNA encoding human MTH1 used in this experiment is provided by Ph.D. candidate Cheng Peng in our lab. Test compounds in this study were designed and synthesized by professor Ji-Wang Chern and graduate student Zhe-Hwa Cheng from graduate institute of pharmacy, National Taiwan University.
Related experimental details regarding chemical design and synthesis is described in master thesis written by Zhe-Hwa Chen.
2.2 Methods
2-2-1 Transformation
Rosetta competent cells used in this experiment were prepared previously according to protocol of our lab and stored at -80℃ which serve as material for transformation experiment. Prior to each transformation, competent cells were thawed on ice for 10 minutes followed by reaction with 2 μl of plasmid DNA (approximately 75 ng/nl) which should be mixed by pipetting several times then leave the mixture on ice for 2 minutes.
The mixture was heat shocked in water bath at 42℃ for 90 seconds followed by incubation for 2 minutes on ice. The mixture made up of competent cells and plasmid DNA was recovered in 1 ml lysogeny broth (without antibiotics) and further incubated at 37℃ with shaking (180 rpm) for 1 hr which provide sufficient time for expression of antibiotic resistant genes. The competent cells were collected followed by incubation with centrifugation at 9,000 rpm for 3 minutes and resuspended in fresh LB medium. For plating the cells, LB-agar plate containing kanamycin and chloramphenicol were warmed up to room temperature previously and the plated cells are incubated at 37℃ overnight.
2-2-2 Protein Expression
The cDNA encoding human MTH1 was expressed from the pET2.8a vector using E. coli strain Rosetta. For large-scale expression of MTH1 recombinant protein, colonies of transformants on LB agar plate were selected and left to grow in 50 ml LB medium containing kanamycin and chloramphenicol at 37℃ for 8-12 hours. Prior to IPTG induction, 2 liters of LB medium should be freshly prepared and autoclaved. Subsequently, 5 ml of LB medium containing E. coli expressing the target protein is added to 500 ml autoclaved LB medium with appropriate antibiotics added and incubated at 37℃ for 2 hours with shaking at (180-200 rpm) until the O.D. value has reached 0.4-0.5. To induce the expression of MTH1 recombinant protein 1 M IPTG was added to a final concentration of 1 mM followed by incubation for 2.5 hours at 37℃ with vigorous shaking (200-220 rpm). The cells were harvested with centrifugation at 8,000 rpm for 20 minutes and during the process the temperature was kept at 4 ℃. Collected cell pellet was stored at 20℃.
2-2-3 Affinity Chromatography
All buffer used in purification were prepared and filtered using Stericup (Millipore) with pore size of 0.22 μM in advance. Prior to cell lysis, the cell pellet stored at 20 ℃ was thawed on ice for 10-20 minutes followed by dissolving in lysis buffer (20 mM Tris- HCl pH 7.4, 500 mM NaCl, 10% Glycerol, 10 mM Imidazole, 2 mM TCEP) which is supplied with freshly prepared stock solution of 0.5 M PMSF and protease inhibitor cocktail (Roche). The process of cell lysis was conducted on sonicator (Misonix Sonicator 3000) under the condition of preset program (pulse-on time= 10 s, pulse-off time= 10 s, total=5 min). Lysis solution was kept on ice during the procedure to maintain the stability of target protein. To remove cell debris, cell lysis was centrifugated at 12,500 rpm for 25
minutes with temperature set at 4 ℃. As outcome of centrifugation, proteins were dissolved in supernatant with pellet formed by cell debris at the bottom. Prior to purification, Supernatant was filtered using Syringe Filter 0.22 μM (Millipore). To acquire purified protein solution containing only the target protein, purification of protein solution was performed using HisTrap FF column (GE healthcare) to capture his-tagged recombinant protein which was followed by elution with buffer containing 500 mM imidazole (Appendix 7). Purification of target protein was operated on ÄKTA Protein Purification Systems (GE healthcare), detailed settings in purification program are listed as below. flow rate=5 ml/min, pressure limit= 0.3 mpa, Equilibrium volume= 5 ml, Sample injection volume= 120 ml, wash volume= 15 ml, elution volume = 200 ml.
2-2-4 IC50 Determination
The inhibition activity of synthesized compounds against MTH1 protein is measured in the presence of substrate 8-oxo dGTP (Trilink Biotechnologies) and IC50
value of test compound is derived from dose-response curve fitted by data from inhibition assay. Firstly, compounds to be analyzed were dissolved in dimethyl sulfoxide (DMSO) followed by serial dilution in 1:4 starting from an initial concentration of 20 mM. For a single measurement, 12-15 dilutions of test compound were prepared prior to mixing with MTH1 protein and substrate 8-oxo dGTP which were diluted to final concentration of 4.8 nM and 100 μM in reaction buffer (10 mM Tris-acetate pH7.5, 40 mM NaCl,10mM Mg acetate,1 mM DTT and 0.005% Tween20), respectively. Followed by sample preparation, reaction mixture was incubated at 37 ℃ for 75 minutes. Released PPi was detected with PPiLight inorganic pyrophosphate assay kit from Lonza according to manufacturer’s instruction. Through several cycles of trial and error, adjustments were made to compounds with suboptimal fitting curve which should be diluted in a 1:3 or 1:2 dilution
series to give more optimal dose-response curve. As MTH1 enzyme activity is correlated to the amount of pyrophosphate released during the reaction catalyzed by MTH1 (Appendix 8), monitoring luminescence produced from PPi in a wide concentration range of inhibitor yields primary data which can be fitted into a dose-response curve using the analytical method non-linear regression supplied in GraphPad Prism software.
2-2-5 Isothermal titration calorimetry
Thermodynamic analysis of test compounds was performed on MicroCal 200 ITC (GE healthcare) under constant pressure at 25℃. For each measurement, MTH1 protein solvated in iTC buffer which is prepared according to the method of Gad et al. (100 mM Tris pH 8.0, 40 mM NaCl, 10 mM MgCl2, 0.005% Tween-20 and 1 mM Tris(2- carboxyethyl)phosphine hydrochloride) with DMSO proportional to the concentration of ligand solution added was loaded into 280 μl sample cell and no bubbles should be present to avoid signal produced by noise. To prepare ligand solution, ligand dissolved in DMSO (20-100 mM) was further diluted in iTC buffer to final concentration of 600-860 μM.
According manufacturer’s protocol, the concentration of DMSO should not surpass 5%
as thermodynamic signal would be disturbed. For generation of a binding isotherm with thermodynamic parameters including the dissociation constant (Kd), the enthalpy of binding (∆H), the entropy of binding (T∆S) and stoichiometry (n), 20-30 injections for a single measurement is required with the solution in the sample cell stirred during the entire measurement process at 1000 rpm. Analysis of data set was carried out using Origin 7 software. For curve fitting, one-site model was applied and calculated related statistics from the fitted binding isotherm (Appendix 10).
2-2-6 Size-exclusion chromatography
Prior to crystallization, His-tagged MTH1 proteins were digested with thrombin to remove His-tag at 4℃. To eliminate cleaved His-tag present in the protein solution, size-exclusion chromatography was performed to separate His-tag and MTH1 protein according to their molecular size. Before separation of target protein from other undesired substances in the solution with size-exclusion chromatography, buffer (20 mM Tris pH 7.4, 150 mM NaCl, 2 mM TCEP, 5% Glycerol) used in the experiment was prepared and filtered using Stericup (Millipore) with pore size of 0.22 mm. Experimental details regarding settings in FPLC machine is described as below. For size exclusion chromatography, pressure limit is set to 0.3 mpa followed by flow rate= 1ml/min;
equilibrium volume= 15 ml, elution volume= 120 ml, fraction size= 2ml. Protein sample was preloaded into syringe with a volume of 5 ml followed by injection into superdex75 gel filtration column (GE healthcare) during equilibrium.
2-2-7 Crystallization and soaking
Prior to crystallization, purity of MTH1 protein with his-tag removed was examined using SDS-PAGE on which no extra visible band should be present. For sample preparation, 50 mM MgCl2 was added to concentrated MTH1 protein (8-10 mg ml-1) to a final concentration of 5 mM. To acquire MTH1 crystals, hanging drop experiment was set up which is composed of drops (2μl) containing 1:1 mixture of protein solution and mother liquid (0.2 M LiSO4, 0.1 M sodium acetate pH 3.75, 30% PEG6000)to be diffused against reservoir solution (~1 ml) in the well at 22℃. Needle-like crystals appeared after 1-2 days. For soaking test compounds into MTH1 crystal, diffraction quality crystals were selected and transferred into reservoir supplemented with 5 mM ligand solution. In this case, crystals should be soaked for at least 16-24 hours to ensure that ligands are bound.
2-2-8 X-ray data collection
Soaked crystals were first sorted according to cracks appeared on the surface to avoid diffraction image with defects which directs to data set of bad quality. As qualifying criterium for ideal diffraction quality crystals, they should appear flawless without significant cracks and thick instead needle-like in their conformation. All crystals were flash-frozen in liquid nitrogen and mounted using cryo-loops with size of 0.1-0.2 mm.
Data collection was performed at BL13B1 and 13C1, National Synchrotron Radiation Research Center, Taiwan. Experimental settings for collection of a single data set is described as below: detector distance = 200-250 mm, exposure time= 2-60 s, frame width= 0.3-0.5∘, total oscillation angle= 80-120∘, wavelength=1.000, 0.976 Å for 13B1 and 13C1, respectively. Data sets were integrated and scaled with HKL2000 (Otwinowski et al., 1997).
2-2-9 Model building and Refinement
Processed x-ray data was primarily solved via molecular replacement using apo MTH1 structure available in Protein Databank (PDB ID: 3ZR1). To place ligand into reasonable position in the solved structure, coordinate and restraint files for ligand were first generated using eLBOW (Moriarty et al., 2009) which is a convenient tool built in Phenix (Adams et al., 2010) for preparing ligand files applied in subsequent structure determination procedure. Followed by ligand placement, several cycles of refinement were performed on current model to optimize the position of placed atom in corresponding calculated electron density map. In further stage of model building, placement of water molecules in solved structure was performed in Autobuild (Terwilliger et al., 2008). As water molecules were placed, a few rounds of refinement with TLS parameter and NCS restraint should be sufficient to complete the final model
with refined R values. Structure determination and refinement of all protein-ligand complex structures solved in the study were conducted with Phenix. Images of structural models were generated using software Pymol (Schrödiger).
Chapter 3 Results
3-1 MTH1 protein purification
The purification of target protein from cell lysate is intended to obtain research material for subsequent experimental trials with high purity. The complete procedure is composed of three individual parts which involves affinity column purification of his- tagged target protein followed by removing purification tag with thrombin digestion at interval stage before the final purification procedure of size-exclusion chromatography is performed.
3-1-1 Affinity Chromatography
The purified target protein MTH1 with his-tag was eluted from his-tag affinity column during wash volume 550-700 ml (Figure 2A). The purity of his-tagged MTH1 protein was examined in the following SDS-PAGE analysis (Figure 2B). In regard of band shown on SDS-PAGE gel which is indicative of protein degradation, experimental condition including incubation time and temperature during large-scale protein expression was revised and modified.
As a result, three independent experimental trials for large-scale protein expression were performed under condition of incubation temperature 22℃ for 14﹑16 hours and 37℃ for 2 hour. From Figure 2B it is demonstrated that protein purified from lysate of E.
coli cells incubated at 37℃ retains the integrality of his-tagged MTH1 protein compared to 22℃ which indicates incubation time and temperature might be the influential factors in protein degradation for this case.
3-1-2 Size-exclusion chromatography (SEC)
After purified his-tagged MTH1 protein has been obtained, his-tag for affinity chromatography was removed with thrombin digestion. To separate cleaved his-tag and MTH1 protein, size-exclusion-chromatography was performed on Superdex75 Gel Filtration column (GE healthcare) equilibrated with gel filtration buffer (20 mM Tris pH 7.4, 150 mM NaCl, 2 mM TCEP, 5% Glycerol). As the separation of MTH1 protein from other undesired substances present in sample solution depends on size of molecules, the standardized elution volume corresponding to representative molecular weight is given in the form of referential parameter as shown in Figure 2C. After the final stage of purification, the separated MTH1 protein was examined on SDS-PAGE gel which shifts from the position of molecular weight 20 kDa to approximately 18 kDa compared to his- tagged MTH1 protein (Figure 2D).
Consequently, purified MTH1 protein with his-tag removed (Figure 2E) serves as experimental material in later crystallization trial for x-ray diffraction experiment (Figure 2F).
3-2 Determination of binding affinity profiles and structure-activity relationship (SAR) 3-2-1 Fragment-based drug screening and optimization performed with 2- aminopyrimidine-based scaffold
In searching for scaffold compound with moderate binding affinity toward MTH1, high-throughput screening (HTS) on 2313 structurally irrelevant fragments was performed by Ph.D. candidate Cheng Peng from our lab. Consequently, four compounds were identified to be micromolar binders in the enzyme inhibition assay. From these identified hits, compound 6077639 was selected as structural skeleton for further optimization campaign in regard of its inhibitory activity on prostate cancer cell lines.
Provided that x-ray structure of MTH1 bound with compound 6077639 has been
solved by Ph.D. candidate Cheng Peng, notable interaction pattern which may contribute to favorable binding interaction are characterized. Attributed to observation from x-ray complex structure, systematical exploration of binding pocket using 2-aminopyrimidine scaffold was further performed to elucidate the correlation between interaction pattern of individual functionality and its contribution to binding affinity profile of inhibitor candidates.
For simplicity of description, the synthesized compound series in this study is categorized into two separate groups according to the presence or absence of ethyl group at C5 of the pyrimidine moiety. The parent compounds of these 2-aminopyrimidine derivative compound series are referred to as scaffold 1 and 2 in the following text.
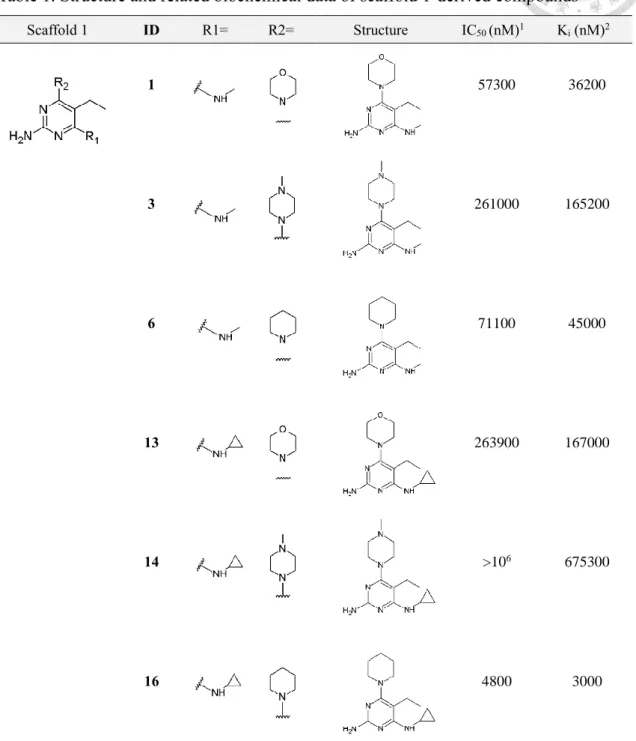
3-2-2 Binding affinity profile and SAR study of scaffold 1-derived compounds
For compounds optimized using scaffold 1 which has ethyl group incorporated at C5 of the pyrimidine ring (Figure 3A), dramatic loss in binding affinity was observed compared to hit compound 6077639 (Figure 3A).
In the six compounds derived from scaffold 1 with biochemical potency distributes unevenly in sub-millimolar IC50 range (Fig 3B-E), variation in binding affinity profile is noteworthy which approximately differentiates by a factor of 200 for the weakest and the most potent binder in this compound series.
In the 2-aminopyrimidine-based compound series optimized using scaffold 1, exploration of structure-activity relationships (SARs) was performed on the substituents in the R1- and R2-position. To complement this objective, compound 1, 3, 6, 13, 14 and 16 was designed and synthesized by our colleague Zhe-Hwa Cheng from graduate institute of pharmacy. In biochemical assay, compound 14 with 1-methylpiperazinyl incorporated in the R2-position exhibits millimolar activity toward MTH1 and is the
weakest binder in this structurally-related compound series. Whilst micromolar potency is determined for compound 16 substituted with piperidinyl group in R2-position. In summary, the observation described above suggests that influence of substituent in the R2-position is profound (Fig 3B-E). Nevertheless, in the case of substituent replaced in the R1-position which are N-methyl and N-cyclopropyl in this compound series, the impact on binding affinity profile is rather insignificant as observed in compounds pairs 1 and 13; 3 and 14; 6 and 16 which differentiate by a factor of 4 to 15 depending on substituent incorporated in the R2-position.
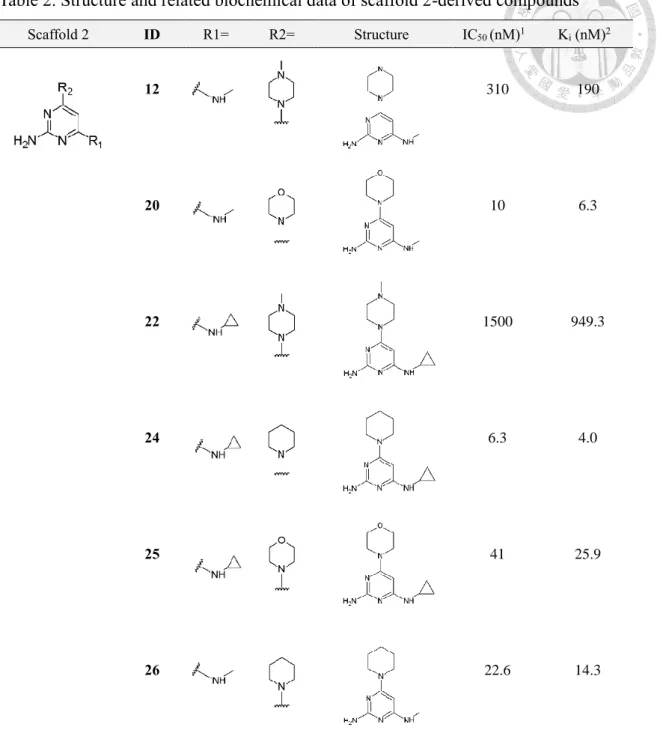
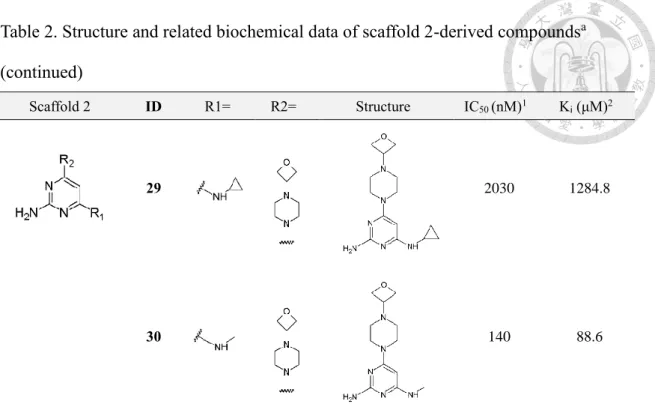
3-2-3 Binding affinity profile of scaffold 2-derived compounds
Followed by biochemical assay for characterization of binding affinity profiles of the 2-aminopyrimidine-based compound series derived from scaffold 1. It was notified that using scaffold 1 for optimization appears to hinder the enhancement of binding affinity toward MTH1 (Figure 3). In regard of this obstacle in the optimization campaign, ethyl group at C5 of the pyrimidine moiety was speculated to be the underlying cause for the dramatic loss in biochemical potency. To confirm the assumption that ethyl group at C5 of the pyrimidine moiety should be the underlying cause for weak activity of scaffold 1-derived compounds toward MTH1, A series of structurally-related compounds based on scaffold 2 (Figure 4A-B) with ethyl group at C5 of the pyrimidine moiety removed was designed and synthesized.
Expectedly, the removal of ethyl group indeed contributed to enhancement of the binding affinity profiles of 2-aminopyrimidine-based compound series derived from scaffold 2 (Figure 4C-F). The overall binding affinity profile of the compound series was determined in the nanomolar IC50 range. Nevertheless, despite of improved binding affinity toward MTH1, significant deviation in binding affinity profile within the
compound series is notified which differentiates by three orders of magnitude.
Among the substituents incorporated at R2, 1-(oxetan-3-yl) piperazinyl and 1- methylpiperazinyl group apparently prohibits the elevation of binding affinity of compound 12, 22, 29 and 30 which are replaced with substituents referred above. While compounds incorporated with either piperidinyl or morpholine substituent were found to be nanomolar binders in enzyme inhibition assay (Figure 4D,F).
Consequently, the comparison of binding affinity profiles in regard of the influence of substituents at either R1 or R2 reveals that the modulation of binding affinity is more correlated to substituents incorporated in the R2-position rather than R1-position. Of note, this finding is in coherence with the observation made in the 2-aminopyrimidine derivative compound series optimized based on scaffold 1 discussed previously (Fig 3).
3-3 Thermodynamic study on 2-aminopyrimidine-based compounds derived from scaffold 2
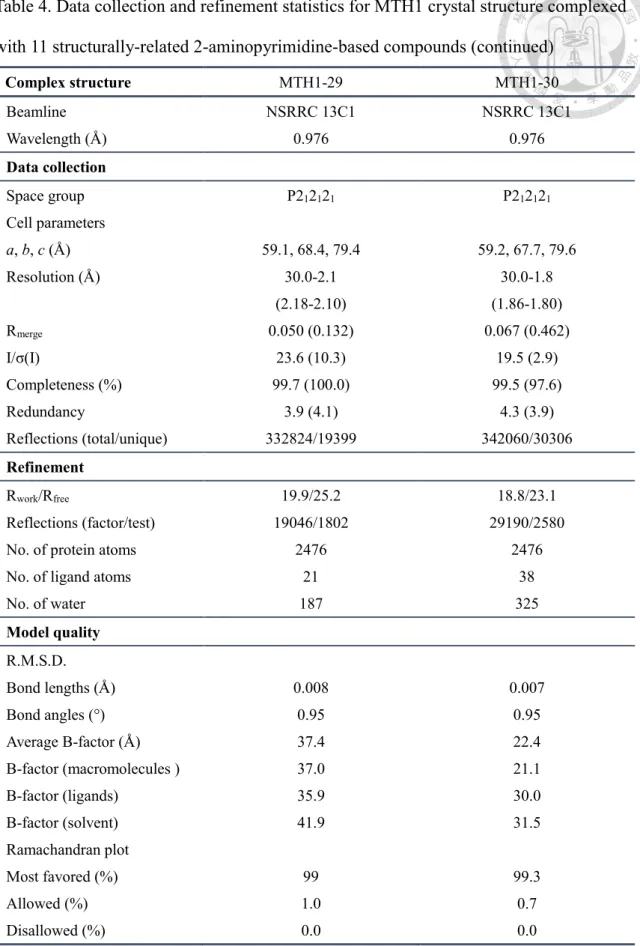
For establishment of complementary affinity profiles of optimized compound series, binding affinity of ligand toward MTH in the absence of substrate was measured using isothermal titration calorimetry (Figure 5A). In consideration of the detection limit of isothermal titration calorimetry (ITC) which lies within the range of 10-2-10-9 M according to instruction of manufacturer, the determination of dissociation constant (Kd) including related thermodynamic parameters was performed on 2-aminopyrimidine- based compound series optimized using scaffold 2 (Figure 4A,B) with weak binders derived from scaffold 1 excluded.
3-3-1 Enthalpy-driven binding of 2-aminopyrimidine-based compounds to MTH1 To determine the thermodynamic profile of the present compound series modified
using scaffold 2 with profound improvement in biochemical potency, isothermal titration calorimetry (iTC) was applied to record signals of heat released from chemical reaction involving breakage and formation of chemical bonds which allows calculation of thermodynamic parameters ∆H and -T∆S from titration curve fitted using 1:1 binding model (Appendix 10) . Consequently, an array of thermodynamic profiles for the 2- aminopyrimidine-based compound series binding to MTH1 was established (Figure 5B- I). Meanwhile, it is notable that binding affinity profile measured using iTC is in consistency with previously determined biochemical potency in enzyme inhibition assay (Figure 5J). In spite of similar structural skeleton, deviated binding mechanism is revealed in compound 12, 22, 29 and 30 (Figure 5K). While for compound 20, 24, 25 and 26 which are the most potent binding partners for MTH1 in this study, deviation in binding mechanism is absent (Figure 5L).
The plot of thermodynamic signatures demonstrates that the binding interaction between target molecule MTH1 and 2-aminopyrimidine based compound series is dominated by favorable enthalpic component of Gibbs free energy (Figure 6A). From another aspect, despite of significant enthalpic signal measured directly in biophysical analysis using iTC, the enthalpic contribution is always counteracted by a minor entropic component which eventually leads to similar binding affinity profile observed across the investigated compound series.
3-3-2 Phenomenon of Enthalpy-Entropy Compensation (EEC)
Within the 2-aminopyrimidine-based compound series which bind to MTH1 in an enthalpy-driven fashion, profound enthalpy-entropy compensation (EEC) was unraveled (Figure 6A,B) when enthalpic component (∆H) was plotted against entropic component (-T∆S) with correlation coefficient value R2= 0,94 (Figure 6B). Whereas, the correlation
which is indicative of enthalpy-entropy compensation is questionable in regard of numerous published research articles debating over the issue of empirical error present in the calculated thermodynamic parameters ∆H and -T∆S. To decipher the observed phenomenon which is often masked by errors rooted in the equation ∆G=∆H-T∆S applied in calculation of the entropic component ∆S which could not be measured experimentally, it was suggested that thermodynamic data should be handled with carefulness in respect to susceptible error in the experimental workflow described above.
3-3-3 Rationalization of observed EEC phenomenon through insights provided by atomic model of MTH1 in complex with 2-aminopyrimidine-based compounds
Assuming that behind the phenomenon of EEC there is certain physical mechanism modulating such intrinsic interplay between binding partners and solvent molecules in surrounding environment as proposed in numerous research works, the counteracting effect between thermodynamic parameters enthalpy and entropy (Figure 6C) may somehow be investigated with insights from atomic structure of protein-ligand complex which uncovers interaction pattern of binding partners.
Thus, to clarify the complex formation process described using thermodynamic parameters ∆H, -T∆S and Gibbs free energy ∆G which is the summation of enthalpy (∆H) and entropy (-T∆S), schematic illustration is presented in Figure 6D-F. Elaborately, enthalpic component of binding free energy involves formation of H-bonds and van der Waals contacts while entropic component is involved in conformational change and hydrophobic interaction which can hardly be measured using present experimental techniques.
3-3-3-1 Thermodynamic profiles of structurally-related compounds with substituent
explored on R2-position
As revealed in bar diagram from Figure 6A which demonstrates typical phenomenon of EEC observed frequently in compound series stem from optimization campaign. The relation between structural feature of structurally-related compounds and the effect of EEC is further investigated as the present compound series is systematically explored on R1 and R2-position with substituents.
Firstly, thermodynamic profiles of scaffold 2-derived compounds binding to MTH1 was categorized according to R1-substituent, which is underrepresented by compound 12, 20, 26, 30 and compound 22, 24, 25, 29 depicted as bar diagram displayed in Figure 7A- B. Subsequently, the thermodynamic parameters ∆H and -T∆S was plotted in diagram with correlation coefficient R2 indicated (Figure 7B-C). From the results of linear regression analysis on thermodynamic parameters of scaffold 2-derived compounds categorized based on substituent incorporated in R1-position, the R2 value for compounds with N-methyl group incorporated in R1-position is 0.99 while the presence of additional cyclopropane ring lead to decreased correlation coefficient value of R2= 0.86. For rationalization of this paradox, visualization of atomic details of protein-ligand complex is performed with x-ray crystallography. In Figure 7D, structural alignment between compound 20 (colored magenta) and 30 (colored yellow) unraveled deviated positioning of R2-substituent in MTH1 binding pocket. Meanwhile, this tendency was also observed in structural alignment of compound 25 (colored blue) and 29 (colored violet) in Figure 7E.
3-3-3-2 Thermodynamic profiles of structurally-related compounds with substituent explored on R1-position and structural relevance of EEC phenomenon
Opposed to arrangement of the scaffold-2 derived compounds in Figure 7 according
to substituent incorporated in R1-position as described above. The compound series is categorized into four groups according to substituent incorporated in R2-position as illustrated in Figure 8. Rearrangement of thermodynamic profiles reveals profound EEC upon replacement of R1-substituent in scaffold-2 derived compounds incorporated with four variants of substituents in R2-position. As plausible explanation for this intrinsic phenomenon, structural alignment of compounds from Figure8 A-D provides insights into binding mode adapted by these compounds in MTH1 binding pocket which suggests more extensive hydrophobic interaction between R1-substituent and phenyl cluster made up of Phe27, Phe72, Phe74, Phe139 upon replacement of N-methyl group with N-cyclopropyl group (Figure 8E-H).
3-3-3-3 The influence of size of substituent in the aspect of Enthalpy-Entropy Compensation
For scaffold-2 derived compounds investigated in thermodynamic study using iTC.
Comprehensive overview of thermodynamic profiles of representative compounds in combination with structural insights is presented in Figure 9 which is compiled of three individual parts. In this compact figure, thermodynamic profiles illustrated in bar diagram for compound 24, 29, 12, 30 (Fig 9E-H) reveal deviated pattern of thermodynamic signatures within the structurally-related compound series. In attempt to uncover the correlation between structural features of compound and deviated thermodynamic profiles, insight into the structural architecture of MTH1 binding pocket (shown in surface representation) bound with compound 24, 29, 12, 30 (shown in stick representation) is presented in Figure 9 A-D. To formulate conclusion for the compensation effect based on information from structural details of protein-ligand complex and related thermodynamic profiles, it should be first notified that as the size of
R2-substituent increases, less enthalpically favorable and more entropy-driven is the binding interaction which is observed in compound 29 and 30 in comparison to compound 24 and 12 (Figure 9I-L). Therefore, as already demonstrated in Figure 8, incorporation of N-cyclopropyl group in R1-position leads to more entropically favorable binding interaction.
3-4 Structural analysis of MTH1 complex structure bound with 2-aminopyrimidine - based compounds
Structural analysis of binding interaction was performed on eleven MTH1-inhibitor complex structures obtained via soaking preformed MTH1 crystals in ligand dissolved in DMSO and further diluted in reservoir solution (1:10). Similar binding pattern in above- mentioned protein-ligand structures is unraveled. For elucidation of underlying mechanism of the varying binding affinity profiles revealed in enzyme inhibition assay for determination of inhibitory activity of 2-aminopyrimidine-based compound series comprising fourteen structurally-related compounds, thorough inspection of interaction pattern in an array of structurally-related ligands binding to target molecule MTH1 protein provides plausible explanations for the structure-activity relationship (SAR) uncovered in biochemical assay (Figure 10).
3-4-1 Conserve binding mode of pyrimidine-based compounds in the binding site of MTH1
Upon inspection of protein-ligand complexes solved by x-ray crystallography, it was found that conserve binding mode is adopted by 2-aminopyrimidine-based compound series bound in the binding pocket of MTH1 (Figure 10B). To illustrate the details of general binding geometry and orientation of the 2-aminopyrimidine-based
compounds, common features of structural architecture of the 2-aminopyrimidine compound series are decomposed into (i) pyrimidine moiety and (ii) R1 and R2- substituent for ease of description.
3-4-2 Pyrimidine moiety
From previous results of high-throughput screening (HTS) performed on fragment library composed of 2313 compounds of divergent structural architecture purchased from CHEMBRIDGE, one bioactive hit compound was identified which is referred to as compound 6077639 in respect of ID code assigned by manufacturer. Followed by the discovery of this HTS hit compound, cocrystal structure of MTH1 bound with compound 6077639 determined to a resolution of 1.8 Å was resolved by Ph.D. candidate Cheng Peng which sheds light on the intrinsic pattern of protein-ligand interaction (Figure 10A).
At first glance, the binding mode is well-characterized which is mainly attributed to the pyrimidine moiety of ligand molecule which is held firmly in the bottom of binding pocket by the indole ring of Trp117 via π-π stacking interaction. H-bonds formed between recognition element Asp119-Asp120 binding motif and amino group of hit compound 6077639 also contribute to the stabilization forces in the protein-ligand complex (Figure 10A).
In the present optimized compound series, the pyrimidine moiety serves as anchor group which strengthens and rigidifies the orientation of ligand molecule which is related to restriction on bond angle of H-bonds. Simultaneously, systematical exploration with substituents on the pyrimidine moiety allows tracing of structural changes upon binding of the pyrimidine derivative compounds in the binding pocket of MTH1 in a rational fashion.
3-4-3 R1 and R2-substituent
Chosen for the subsequent optimization campaign from HTS, compound 6077639 serves as structural skeleton for the present 2-aminopyrimidine-based compound series (Figure 10A). For modification of hit compound 6077639, C4 and C6 of the pyrimidine moiety are assigned as R1 and R2-substituent in the following passages and explored with several functionalities. In the case of R1-substituent, which occupies the cavity formed by phenyl rings of Phe27, Phe72, Phe74 and Phe139, interacts with these side chains through hydrophobic contacts. On the other hand, from the pyrimidine moiety anchored by indole ring of Trp117, substituent at R2-position expends the skeleton of small molecule compound toward the entrance of binding pocket and forms either hydrophobic interaction or van der Waals contact with several rigid components of binding pocket made up of side chains of Tyr7, Thr8, Leu9, Lys23 and Asn33. (Figure 11C).
After thoroughly inspecting the structural details revealed in binding pocket of MTH1 bound with different ligand molecules obtained via x-ray crystallography. The high similarity of binding mode observed in protein-ligand complexes determined in this study further hampers the formulation of rationales for the largely deviated binding affinity profiles of the 2-aminopyrimidine-based compound series. Nevertheless, there remains some notable features to be explored and may provide plausible explanation for the varying inhibitory activity of the present 2-aminopyrimidine-based compounds against MTH1.
3-5 Superposition of MTH1 complexed with product 8-oxo dGMP and compound 29, 30 In the binding pocket of MTH1, compound 29 and 30 overlay well with MTH1 product 8-oxo dGMP which serves as natural ligand exploited for drug design (Figure 14A-B). To dissect the interaction pattern of above-mentioned ligands, contact between
![Table 3. Thermodynamic profiles of scaffold 2-derived compounds determined with iTC ID N (sites) K d [× 109 M -1 ] * ∆G [kcal/mol] ∆H [kcal/mol] -T∆S [kcal/mol] 6077639 1.69 199 ± 12 -9.16 ± 0.16 -7.34 ± 0.08 -1.8 ± 0.08 20 0.89](https://thumb-ap.123doks.com/thumbv2/9libinfo/9608726.634227/70.892.151.781.137.509/table-thermodynamic-profiles-scaffold-derived-compounds-determined-sites.webp)