An External Memory Approach to Computing the Maximal Repeats across Classes of DNA Sequences
†JING-DOOWANG*
Department of Computer Science and Information Engineering, Asia University, Taiwan
ABSTRACT
This work presents an external memory approach to extract the maximal repeats from whole genome sequences with the statistics of these repeats across classes, where the definition of a class is determined from the statistics to be computed. A heuristic method consisting of a bucket-sort-like approach and the Chinese term extraction approach is adopted. The bucket-sorting method is used to sort the suffixes of DNA sequences stored in files, and the term extraction is used to extract maximal repeats by scanning the sorted suffixes while computing the statistics of these repeats. The statistics of these repeats across classes might be useful for sequence classification and species identification.
Key words: maximal repeat, external memory, genomic comparison.
1. INTRODUCTION
The current growth of computation power is likely to be far beyond the need for biologists to handle a whole genome comparison efficiently among organisms simultaneously (Mount, 2004). A bottleneck in the computation for comparative genomics is the limit of the main memory available in a general computer, thus limiting the size of data sets to the capacity of the main memory. However, genome sequences can be compared across species, in the vector space model (Ricardo &
Berthier, 1999; Salton & McGill, 1983), by selecting representative repeats (patterns) whose frequency of distribution across classes (species) is biased to present genome sequences as vectors. Hence, many well known machine learning approaches (Alpaydin, 2004; Mitchell, 1997) in the vector space model could be applied to the bioinformatics problems (Miller, 2001). The statistics of repeats across classes might be useful for sequence classification (Atalay & Cetin-Atalay, 2005; Mukhopadhyay, Tang, Huang, & Palakal, 2003; Sandberg et al., 2001) and species identification from microarray-based detection (Chizhikov et al., 2002;
Gardner et al., 2004; Herbert, Stoeckle, Zemlak, & Francis, 2004).
The analysis of repeats (Edgar & Myers, 2005; Price, Jones, & Pevzner, 2005;
Subramanian et al., 2003) plays an important role in comparative genomics. Studies of repeat extraction include STRING (Parisi, Fonzo, & Aluffi-Pentini, 2003), REPuter (Kurtz, Choudhuri, Ohlebusch, Stoye, & Giegerich, 2001), MUMmer (Deogun, Ma, & Yang, 2003), FORRepeats (Lefebvre, Lecroq, Dauchel, &
Alexandre, 2003) and SRF (Sharma, Issac, Raghava, & Ramaswamy, 2004).
STRING is used to find tandem repeats in DNA sequences by alignment.
†A pre-version of this work had been published in the conference (Wang, 2005).
*E-mail: [email protected]
FORRepeats is a heuristic approach that uses a data structure called Factor Oracle to find repeats. Sharma et al. (2004) used Spectral Repeat Finder (SRF), a discrete Fourier transformation, to identify repetitive DNA sequences. REPuter and MUMmer detect repeats by the suffix tree (Gusfield, 1997). However, the suffix tree suffers from a large memory consumption (Kurtz, 1999; Monostori, Zaslavsky,
& Schmidt, 2002). Therefore, approaches that adopt suffix array instead of suffix tree have been presented (Abouelhoda, Kurtz, & Ohlebusch, 2004; Kärkkäinen &
Sanders, 2003; Kim, Sim, Park, & Park, 2003; Ko & Aluru, 2003).
To compute the statistics of repeats extracted from whole genomes across classes (species), the internal memory approaches, e.g., using a suffix tree or a suffix array, are not adopted in a general computer with limited main memory (Noble & Weir, 2001) if the amount of memory needed to hold the data structure and the statistics of these repeats is over the memory capacity because of disk I/O problems (Silberschatz, Galvin, & Gagne, 2000). Choi and Cho (2002) proposed a workbench called SequeX, an external memory approach, which is based on the Static SB-tree (SSB-tree) (Ferragina & Grossi, 1999), to analyze common n-mers for whole genome sequences from 72 microbial genomes. However, generating a program to construct the SSB-tree is not a trivial task, because of the need to handle disk accesses efficiently. Furthermore, they aimed to have the common n-mers but did not have the statistics of repeats with variable lengths across classes (species).
Conversely, the value of n usually ranges from 5 to 20 when computing statistics using the n-mers approach (Fofanov et al., 2004). The statistics of the large numbers of patterns generated by larger values of n, e.g., n = 100, are difficult to store.
This study uses the maximal repeat (Gusfield, 1997) to reduce the redundancy of repeats to decrease the output generated. A maximal pair (Kurtz et al., 2001) in a string S isapairofidenticalsubstringsα and β in S such thatthecharacterto the immediate left,resp.right,ofα isdifferentfrom thecharacter to the immediate left, resp.right,ofβ.Thatis,extending α and β in eitherdirection destroystheequality ofthetwo strings.A maximalrepeatα isa substring ofS thatoccursin amaximal pair in S. For instance, given a sequence “kabcyiazabczabcyrxak,”the patterns
“abc”and “abcy”are maximal repeats, while “ab”and “bc”are not. This work attempts to extract the maximal repeats from whole genome sequences while computing the statistics across classes using a general computer with limited memory, i.e., a 2-GB main memory. A class is defined in advance according to the statistics to be computed. The proposed approach (Wang, 2005) is simple and straightforward, and is easily scalable to parallel processing. A heuristic method consisting of a bucket-sort-like approach (Cormen, Leiserson, Rivest, & Stein, 2002; Futamura, Aluru, & Kurtz, 2001) and the Chinese term extraction approach (Wang & Tsay, 2002) is presented to extract the maximal repeats, and to compute the statistics of those patterns across classes. Bucket-sorting is used to sort the suffixes of the DNA sequences; and term extraction is used to extract maximal repeats while computing the statistics of these repeats across classes. To handle the large number of DNA sequences across classes (with a limited amount of memory), the suffixes of DNA sequences are first presorted by partitioning these suffixes into
a given number 4kof groups (files), where k is the length of the common prefix of the suffixes in one group. The suffixes within each group are then sorted individually. After sorting all the suffixes in each group, the scanning process of the Chinese term extraction approach is then adopted to extract the candidate maximal repeats with the right boundary verification. To obtain the candidate maximal repeats with the left boundary verification, the above processes are rerun with the suffixes of the reversed DNA sequences. The maximal repeats are those candidate maximal repeats that pass both the right and the left boundary verifications. Notably, the computation can easily be sped up by assigning the jobs of sorting suffixes and extracting candidate maximal repeats to different processors according to the value of k.
Experiments were performed on six species, namely the SARS coronavirus, Escherichia coli, Saccharomyces cerevisiae (Yeast), Plasmodium falciparum, Caenorhabditis elegans and Arabidopsis thaliana. The lengths of the DNA sequences ranged from several thousand (bp) to over 100 million (bp). In practice, we used the suffixes as the front l, say 50, characters of every suffixes instead of the whole suffixes. The value of k was determined manually such that the sorting of the suffixes in one group could be performed efficiently with limited memory. That is, the length of maximal repeats extracted by our approach is between k and l. To evaluate the variant values of k and l, experiments were performed with k set to 1, 2, 3 and 4, and l set to 25, 50, 100, 200, 500 and 1000. Notably, the length value l = 1000 of maximal repeats was much longer than either that of patterns extracted by the slide-window methods (Karlin, Campbell, & Mrazek, 1998) or that of primer designs (Rocha, Medeiros, Monteiro, Gon¸calves, & Marinho, 2004).
The remainder of this paper is organized as follows. Section 2 describes the processes of maximal repeat extraction. Section 3 provides the experimental results.
Sections 4 and 5 present the conclusions of this study and discuss the results.
2 METHODS
A bucket-sort-like approach (Cormen et al., 2002) is adopted to sort the suffixes lexicographically. A modified Chinese term extraction approach (Wang &
Tsay, 2002) is then used to extract the candidate maximal repeats while computing the statistics of these repeats across classes. Finally, the maximal repeats are selected as those candidate maximal repeats that passed both the right and the left boundary verifications. These repeats and their statistics are stored as plain text files. The definition of a maximal repeat is given in Section 2.1. The proposed approaches are described in detail in Sections 2.2. 2.3 and 2.4.
2.1 Notions
Let S be a string of length |S| = n over an alphabet ∑= {A, C, G, T, N}. S[i]
denotes the ith character of S, where I[1, n]. For i ≤ j, S[i, j] denotes the substring of S starting with the ith and ending with the jth character of S. Substring S[i, j] is
denoted by the pair of positions (i, j). The length of the substring S[i, j] is l = j − i + 1. A pair of substrings {(i1, j1), (i2, j2)} forms an exact repeat if and only if (i1, j1) ≠ (i2, j2) and S[i1, j1] = S[i2, j2]. An exact repeat {(i1, j1), (i2, j2)} is called maximal if and only if S[i1−1] ≠S[i2−1] and S[j1+ 1] ≠S[j2+ 1] (Gusfield, 1997; Kurtz et al., 2001).
2.2 Sorting the Suffixes
The suffixes are sorted in two steps, as described in Section 2.2.1 and Section 2.2.2. In the first step, the suffixes were generated and stored in temporary files according to their prefixes. The front l, say 100, characters of each suffix were applied instead of an entire suffix. In the second step, the suffixes were sorted in one file by the shell system call sort.1 After sorting, the suffixes were stored in temporary files according to their prefixes.
2.2.1 Generating suffixes and storing in files
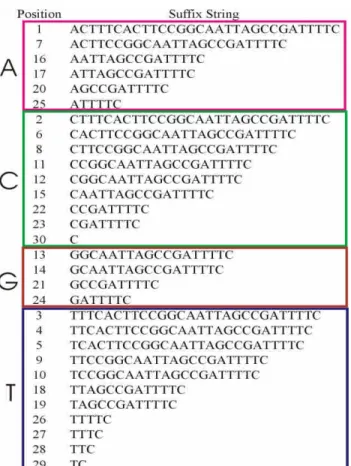
Since the alphabet of DNA sequences is limited to {A, C, G, T}, the suffixes were partitioned into 4kfiles according to the prefix of these suffixes, where k is the length of the common prefix. The value of k was chosen manually such that the process for sorting suffixes in Section 2.2.2 could be run efficiently while handling the files with the maximum suffix size. Intuitively, the value of k was increased if the available memory for computation of sorting was low. The front l characters of each suffix were stored, instead of the whole suffixes, such that the length of maximal repeats ranged from k to l. That is, the suffixes were partitioned into temporary files according to the prefix, which consisted of the front l characters of suffixes, instead of storing the starting positions of suffixes in the original sequences. For example, Figure 1 shows the suffixes of the sequence
“ACTTTCACTTCCGGCAATTAGCCGAT TTTC”which has a length of 30.
Notably, suffixes can be derived from only one sequence with 30 characters. In this case, as shown in Figure 2, the k = 1 and the number of files is 41= 4.
2.2.2 Sorting the suffixes in files
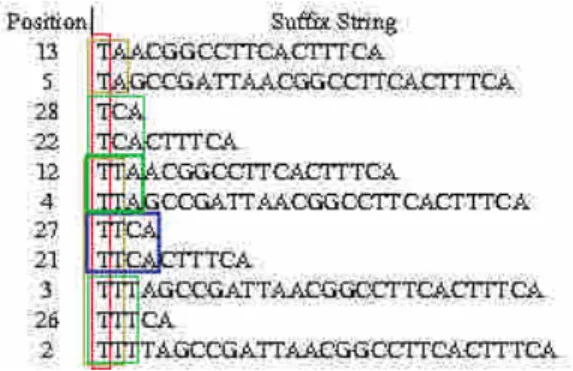
The suffixes were sorted individually in one file, e.g., A_Suffix. In this study, for simplicity, the suffixes in one file were sorted by using the shell system call sort, for example, as sort A_Suffix > A_SuffixSorted. The original file, A_Suffix in this case, was removed immediately after sorting. Similarly, the files with sorted suffixes, e.g., A_SuffixSorted, were also removed after extracting the maximal repeat, as described in Section 2.3. Figure 3 shows the sorted suffixes from the four files. Notably, the efficiency of sorting of those suffixes might be further improved as string sorting is a widely studied topic (Choi & Cho, 2002; Futamura et al., 2001;
Tsay, 2001). Furthermore, the speed of suffix sorting can be increased by suffix array (Kim et al., 2003; Ko & Aluru, 2003; Kärkäinen & Sanders, 2003;
Abouelhoda, Kurtz & Ohlebusch, 2002) without external memory. This study did
1 Written by Mike Haertel and Paul Eggert, Copyright 2004 Free Software Foundation, Inc.
not focus on this part, because the bottleneck of the computation occurred during the process of extracting the candidate maximal repeats, as described next in Section 2.3.
2.3 Extracting the Candidate Maximal Repeats with the Statistics
The scanning process of the Chinese term extraction approach (Wang & Tsay, 2002) was modified to extract the candidate maximal repeats while calculating the statistics across classes according to the labels of the suffixes specified by the user.
In this study, the candidate maximal repeats and their corresponding statistics were stored as plain texts in files named according to their prefixes. However, this step could be further improved by a database approach or specific data structures for the verification process described in Section 2.4. To make the paper self-contained, Appendix A describes the procedure for extracting the candidate maximal repeats.
Figure 1. Suffix examples.
Figure 2. Partitioning Suffixes into A, C, G and T groups (k = 1).
The suffixes of DNA sequences, resp. reversed DNA sequences, were scanned lexicographically. The longest common prefix of consecutive suffixes were selected as the candidate maximal repeats with the right, resp. the left, for boundary verification. For example, Figure 4 shows the partial of sorted suffixes whoseprefix beginswith “A,”and thepatterns“A,” “ACTT”and “ATT”were candidate maximal repeats with the right boundary verification. Notably, the patterns“AC,”“ACT”and “AT”did notpasstherightboundary verification.A stack with push and pop operations can be used to compute not only the term frequency of one repeat, but also the statistics of that repeat extracted if each suffix is concatenated with a specific label. For example, class identity Ciis derived from the ith class, as shown in Figure 5, when generating the suffixes files, as described in Section 2.2. Figure 6 shows the statistical results, that the value of “cnt”provides the term frequency and the frequency distribution of terms “A,” “ACTT” and
“ATT”acrossclassesC1, C2and C3.
Figure 3. Suffixes sorted in groups (k = 1).
Figure 4. The candidate maximal repeats with right boundary verification.
Figure 5: Suffixes concatenated with class identity.
Frequency Distribution across Classes
cnt C1 C2 C3
A 6 3 2 1
ACTT 2 0 2 0
ATT 2 1 0 1
Figure 6. The frequency distribution of terms across classes from Figure 5.
2.4 Verifying the Candidate Maximal Repeats
The maximal repeats are those candidate maximal repeats that pass both the right and the left boundary verifications. The maximal repeats and their statistics were output as plain texts into files. The repeats with left boundary verification were loaded from files into a hash, and each repeat was verified with the right boundary verification as a maximal repeat if its reversed strings existed in that hash.
As shown in Figure 7,the patterns“ACTT”and “ATT”weremaximalrepeats, because the corresponding reversed patterns,“TTCA”and “TTA,”also passed the left boundary verification. In practice, the number of maximal repeats might rise exponentially with increasing genome size. Therefore, for simplicity, the files consisting of the repeats with left boundary verification were divided into p partitions, and the repeats were loaded into the files within one of p partitions in turn into a hash. That is, the processes above might need to run p times.
3. EXPERIMENTAL RESULTS
Experiments were performed on DNA sequences of whole genomes from six species, namely, the SARS coronavirus, E. coli, Saccharomyces cerevisiae (Yeast), P. falciparum, C. elegans and A. thaliana. The statistics were computed using one server with 2*Xeon 2.4 GHz CPU, 2 GB RAM, an SCSI hard Disk and Red Hat Linux 9.0.
3.1 The Computation Time with Different Species
As shown in Table 1, the lengths of these DNA sequences ranged from several thousand (bp) to over 100 million (bp). For example, the time for computing the statistics of maximal repeats of A. thaliana was about 72 hours and 38 minutes, and the number of maximal repeats extracted was 124, 462, 282. The verification step used p = 16 when processing A. thaliana. The negative strand was also considered by concatenating the sequence with its reversed-complement sequence.
Figure 7. The candidate maximal repeats with left boundary verification.
Table 1. The statistics of DNA sequences for maximal repeats extraction
Species Length(bp) Total Time
(HH: MM:SS) # of Maximun Repeats
SARS 29,736 0:00:37 32,358
Ecoli 4,639,675 1:51:02 5,021,132
Yeast 12,070,527 11:20:47 12,835,695
P. falciparum 22,820,308 20:16:16 22,821,291
C. elegan 100,096,025 60:01:07 103,730,344
A. thaliana 119,186,497 72:37:48 124,462,282
3.2 The Computation Time with Different Values of k and l
To evaluate the computation time with different values of l, the value of k was first set to 1, and an experiment was performed on the genome sequences of Yeast at different values of l, namely 25, 50, 100, 200, 500 and 1000. As shown in Table 2, the total computation time was about 39 hours and 31 minutes when l = 1000, and the extraction process took the largest amount of computation time; the computation time of each process increased with rising l.
This part of computation could be further improved by sorting the suffixes according to the indexes of suffixes in the files, instead of sorting the suffixes
directly. Second, as shown in Table 3, l was set to 50 and k was set at 1, 2, 3 and 4.
The total computation time was similar for all values of k, although reducing the number of suffixes per file might speed up the sorting process.
Table 2. The computation time for the different values of l when k = 1 (Yeast)
The fornt l characters of suffixes
Processes 25 50 100 200 500 1000
Partition Suffixes (right boundary) 00:03:58 00:04:28 00:05:22 00:07:50 00:13:57 00:24:30 Partition Suffixes (left boundary) 00:04:21 00:04:25 00:05:27 00:07:40 00:14:00 00:24:48 Sort Suffixes (right boundary) 00:11:44 00:13:59 00:18:18 00:29:04 01:10:26 02:06:16 Sort Suffixes (left boundary) 00:11:32 00:14:07 00:18:24 00:29:28 01:10:21 02:06:05 Extract Candidate Maximal Repeats
(right boundary)
03:44:51 04:07:22 04:44:08 06:00:17 09:34:27 15:37:41
Extract Candidate Maximal Repeats (left boundary)
03:47:19 04:07:39 04:45:48 05:57:23 09:37:52 15:23:43
Verify the Candidate Maximal Repeats
02:04:58 02:07:06 02:09:01 02:15:43 02:34:23 03:27:36
total 10:08:43 10:59:06 12:26:28 15:27:25 24:35:26 39:30:39
Table 3. The computation time for the different values of k when l = 50 (Yeast)
The length k of common prefix
Processes k=1 k=2 k=3 k=4
Partition Suffixes (right boundary) 00:04:28 00:04:37 00:04:32 00:04:39
Partition Suffixes (left boundary) 00:04:25 00:04:33 00:04:32 00:04:42
Sort Suffixes (right boundary) 00:13:59 00:13:46 00:12:08 00:11:54
Sort Suffixes (left boundary) 00:14:07 00:13:06 00:12:21 00:11:54
Extract Candidate Maximal Repeats (right boundary)
04:07:22 04:03:36 04:05:58 04:06:09
Extract Candidate Maximal Repeats (left boundary)
04:07:39 04:05:55 04:04:13 04:03:06
Verify the Candidate Maximal Repeats 02:07:06 02:07:43 02:07:08 02:06:29
total 10:59:06 10:53:16 10:50:52 10:48:53
3.3 Intra-Chromosomal Comparison
To observe the frequency distribution of maximal repeats across the 16 chromosomes of Yeast, each chromosome was considered as one class. Let class frequency (cf) be the number of classes, in this case species in which the pattern appears, and let term frequency (tf) be the number of the pattern appears, and let the length (len) be the length of maximal repeat.
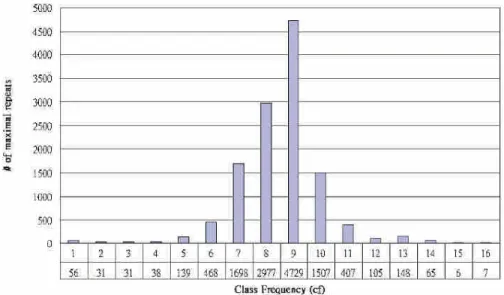
Figure 8 shows the statistics of class frequency of maximal repeats with tf ≥ 10 and len ≥ 30. Among these maximal patterns, for example, there were 56 patterns which appeared only in one chromosome, and 7 patterns which appeared in all 16 chromosomes. These observations might be useful for evaluating intra-chromosomal comparisons.
Figure 8.The class frequency of maximal repeats of yeast with tf ≥ 10 and len ≥ 30 (k = 3, l = 50).
3.4 The Statistics of Maximal Repeats across Species
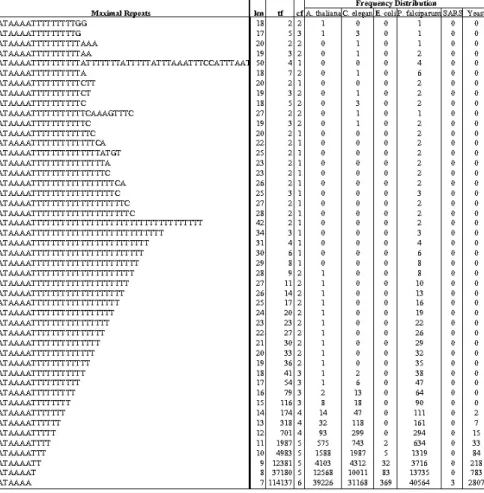
Distinctive patterns form the class markers if some patterns just appear in one class but not others. Table 4 shows the statistics of the maximal repeats across six species based on one species representing one class. Distinctive maximal patterns were extracted from the DNA sequences of these species. For example, the pattern
"TATAAAATTTTTTTTTTATTTTTTTATTTTTATTTAAATTTCCATTTAAT"
with len = 50 and tf = 4 appeared only in the P. falciparum while pattern
"TATAAAATTTTTTTTTTTCAAAGTTTC", with tf = 2 and cf = 2, appeared in both C. elegans and P. falciparum. Notably, the patterns with tf = cf = 2 could not be detected from one species alone without making the comparison across species.
4. CONCLUSIONS
An external memory approach is presented to extract maximal repeats from DNA sequences with the statistics of frequency distribution across classes, thus making genome sequence comparisons across species possible in the vector space model (Ricardo & Berthier, 1999). The proposed approach is not limited to the size of main memory available in a general computer when considering a large amount of sequences such as genomes from several species. Indeed, this approach has been used to extract the representative patterns for virus classification and achieved about 75% accuracy (Wang, 2004a; Wang, 2004b) where representative patterns were those maximal repeats whose frequency distribution across genus (classes)
were biased. The proposed approach could be applied to parallel computing with PC-clusters by partitioning the computation into different PCs according to the prefix of the suffixes. We will discuss that work in the future.
Table 4. The statistics of the partial maximal repeats across six species (k = 4, l = 50)
5. DISCUSSION
Section 5.1 analyzes the time complexity of the proposed approach and Section 5.2 discusses the practical limits to the values of k and l.
5.1 Time Complexity Analysis
The proposed approach consists of four main steps, (1) partitioning the suffixes into groups (files), (2) sorting the suffixes in one group (file), (3)
extracting, and (4) verifying the candidate maximal repeats. The time complexity of these steps is discussed as follows.
Suppose that the total length of DNA sequences is N, and that the first l characters instead of the entire suffix are extracted. Each suffix is divided into k partitions according to its prefix. In Step (1), assuming that the operating system can open 4k files concurrently, the partitioning of suffixes into 4k files can be achieved with time complexity O(Scan(N, l)), where the Scan(N, l) scans the DNA sequences and outputs the suffixes with the first l characters into 4kfiles according to the first k characters of the prefix of these suffixes. In Step (2), the time complexity to sort suffixes in 4kfiles is approximately O(4kSortString(N/ 4k, l)), assuming that the suffixes are uniformly distributed in each file, where SortString(|S|, l) is the time complexity of sorting |S| of length l.
In Step (3), the analysis of the time complexity for the extraction of candidate maximal repeats consists two parts. The first part involves reading the next consecutive string and determining the longest common prefix. The second part adopts the stack operations, push and pop, to handle statistics concerning significant terms. The complexity of the first part is proportional to the number of the sorted strings and the suffix length. The second part might need amortized analysis (Lee, Tseng, Chang, & Tsai, 2005) for further investigation. Conversely, the memory space needed for this procedure is independent of the number of the sorted strings. This is because these strings are read one by one from files, and the related statistics are accumulated by a stack requiring only a small amount of memory. The process of determining the longest common prefix can be further improved as described in (Kasai, Lee, Arimura, Arikawa, & Park, 2001).
Since the number of maximal repeats might increase exponentially, the verification in Step (4) might need be repeated p times to load the candidate maximal with left boundary verification from one of p partitions into a hash, and to verify the maximal repeats with right boundary verification as maximal repeats.
The value of p is determined manually by monitoring the percentage of main memory used while this step is processed. However, this process can be avoided by storing the maximal repeats with left boundary verification into a database.
5.2 Limitation of the values of k and l
In this study, the length of maximal repeats ranged from k to l. The value of k is increased if the computation for sorting suffixes encounters main memory limitations. However, the value of k is limited to 5 in this study because the default maximum number of opening files is 45= 1024 in Linux 9.0 if the kernel is not recompiled. To remove this limitation on the value of k, parallel computing with PC-clusters can be applied by partitioning the computation into different PCs according to the prefix of the suffixes of DNA sequences. An experiment was performed in this study with the different values of l ranging from 25 to 1000. In (Wang et al., 2002), the length of an oligonucleotide was 70 for microarray-based detection of viruses. That is, the length of maximal repeats extracted in this study might be long enough to distinguish one species from the others.
ACKNOWLEDGEMENT
The author would like to thank the National Science Council of the Republic of China (Taiwan) for partially supporting this research under Contract No. NSC 93-2213-E-468-003. The center of Bioinformatics of Asia University is appreciated for setting up the computing environment. I would like to thank Professor. Ka-Lok Ng for gathering the genomic sequences as well as Prof. Pei-Jun Chang for providing valuable suggestions. The anonymous referees are commended for offering their valuable suggestions. Prof. Wen-Hsiang Tsai, Miss Wan-Ching Hung and Prof. Ted Knoy are appreciated for his editorial assistance.
APPENDIX A: THE PROCEDURE OF CANDIDATE SIGNIFICANT TERM EXTRACTION
The goal of this procedure (Wang & Tsay, 2002) was to extract the longest common prefix of consecutive strings by scanning the strings which were sorted lexicographically in one file while computing the statistics of those terms extracted.
Figure 10 shows the procedure of candidate significant term extraction. The initial input, SortedSuffixStringList, of the procedure is the sorted suffixes with the least string, when arranged in a lexical order. The function NextString moves to the next sorted suffix and returns the corresponding string; the function SamePrefix returns the length of the longest common prefix of string, “Str1” and string, “Str2.” TopRecord is the points to the top record of the stack. The operations, (S14) and (S32),outputthestatistics,e.g.,term frequency “cnt”ofsignificantterms.
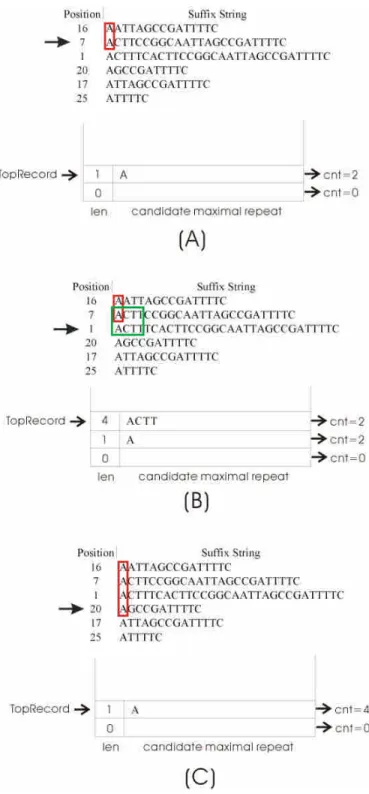
Some steps in the candidate significant term extraction are shown below. For example, as shown in Figure 9(A), The “A” whose length is 1 is a candidate significantterm and hasthevalueof“cnt”as“2”when scanning to thesuffix with the value of position as 7. When the next suffix, with the value of position as 1, is scanned, as shown in Figure 9(B), the longest common prefix of two adjacent suffixesisobtained as“ACTT,”whoselength is4 and exceedsthelength 1 in the field, "len", at the top record of the stack. Then, the new record with “ACTT”is pushed into the stack. Note that the terms, “AC” and “ACT,” are not pushed according to the definition of maximal repeat. When the next suffix is scanned, as shown in Figure 9(C), the longest common prefix of two adjacent suffixes is shown as“A,”whoselength is1 and isshorterthan thatof4,atthetop record of the stack.
Therefore,therecord with theterm,“ACTT,”ispopped and thestatistics,“cnt= 2,”ofthatterm areoutput.Becausetheterm “A”isasubstring of“ACTT,”the statistics concerning “ACTT”must be included in those concerning the term,“A.”
Figure 9. Extract candidate maximal repeats.
Figure 10. Candidate significant term extraction.
REFERENCES
Abouelhoda, M. I., Kurtz, S., & Ohlebusch, E. (2002). The enhanced suffix array and its applications to genome analysis. In Proceedings of the Second International Workshop on Algorithms in Bioinformatics, 449–463.
Abouelhoda, M. I., Kurtz, S., & Ohlebusch, E. (2004). Replacing suffix trees with enhanced suffix arrays. Journal of Discrete Algorithms, 2(1), 53–86.
Alpaydin, E. (2004). Introduction to Machine Learning. Massachusetts, USA: The MIT Press.
Atalay, V., & Cetin-Atalay, R. (2005). Implicit motif distribution based hybrid computational kernel for sequence classification. Bioinformatics, 21(8), 1429–1436.
Chizhikov, V., Wagner, M., Ivshina, A., Hoshino, Y., Kapikian, A. Z., &
Chumakov, K. (2002). Detection and Genotyping of Human Group A Rotaviruses by Oligonucleotide Microarray Hybridization. Journal of Clinical Microbiology, 40(7), 2398–2407.
Choi, J.-H., & Cho, H.-G. (2002). Analysis of common k-mers for whole genome sequences using SSB-tree. Genome Informatics, 13, 30–41.
Cormen, T. H., Leiserson, C. E., Rivest, R. L., & Stein, C. (2002). Introduction to Algorithms (2nd ed.). Massachusetts, USA: MIT Press and McGraw-Hill.
Deogun, J. S., Ma, F., & Yang, J. (2003). A prototype for multiple whole genome alignment. In Proceedings of the 36th Hawaii International Conference on System Sciences (HICSS’03).
Edgar, R. C. & Myers, E. W. (2005). PILER: identification and classification of genomic repeats. Bioinformatics, 21(suppl 1), i152–158.
Ferragina, P., & Grossi, R. (1999). The string B-tree: a new data structure for string search in external memory and its applications. Journal of the ACM, 46(2), 236–280.
Fofanov, Y., Luo, Y., Katili, C., Wang, J., Belosludtsev, Y., Powdrill, T., Belapurkar, C., Fofanov, V., Li, T.-B., Chumakov, S., & Pettitt, B. M. (2004).
How independent are the appearances of n-mers in different genomes?
Bioinformatics, 20(15), 2421–2428.
Futamura, N., Aluru, S., & Kurtz, S. (2001). Parallel suffix sorting. In Proceedings 9th International Conference on Advanced Computing and Communications, 76–81.
Gardner, S. N., Lam, M. W., Mulakken, N. J., Torres, C. L., Smith, J. R., & Slezak, T. R. (2004). Sequencing Needs for Viral Diagnostics. Journal of Clinical Microbiology, 42(12), 5472–5476.
Gusfield, D. (1997). Algorithms on Strings, Trees, and Sequences: computer science and computational biology. New York, USA: Cambridge University Press.
Kärkkäinen, J., & Sanders, P. (2003). Simple linear work suffix array construction.
In Proceedings 13th International Conference on Automata, Languages and Programming, 943–955.
Karlin, S., Campbell, A. M., & Mrazek, J. (1998). Comparative DNA analysis across diverse genomes. Annual Review of Genetics, 32(1), 185–225.
Kasai, T., Lee, G., Arimura, H., Arikawa, S., & Park, K. (2001). Linear-time longest common- prefix computation in suffix arrays and its applications. In Proceedings of the 12th Symposium on Combinatorial Pattern Matching, (CPM'01), Lecture Notes in Computer Science, 2089, 181-192.
Kim, D. K., Sim, J. S., Park, H., & Park, K. (2003). Linear-time construction of suffix arrays. In Proceedings of the 14th Symposium on Combinatorial
Pattern Matching, 186–199.
Ko, P., & Aluru, S. (2003). Space efficient linear time construction of suffix arrays.
In Proceedings of the 14th Annual Symposium, Combinatorial Pattern Matching, LNCS, 200–210.
Kurtz, S. (1999). Reducing the space requirement of suffix trees. Software-Practice and Experience, 29(13), 1149–1171.
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Stoye, C. S. J., & Giegerich, R. (2001).
REPuter: the manifold applications of repeat analysis on a genomic scale.
Nucleic Acids Research, 29(22), 4633–4642.
Lee, R., Tseng, S., Chang, R., & Tsai, Y. T. (2005). Introduction to the Design and Analysis of Algorithms: a strategic approach. New York, USA: McGraw-Hill.
Lefebvre, A., Lecroq, T., Dauchel, H., & Alexandre, J. (2003). FORRepeats:
detects repeats on entire chromosomes and between genomes. Bioinformatics, 19(3), 319–326.
Miller, W. (2001). Comparison of genomic DNA sequences: solved and unsolved problems. Bioinformatics, 17(5), 391–397.
Mitchell, T. M. (1997). Machine Learning. New York, USA: The McGraw-Hill Companies, Inc.
Monostori, K., Zaslavsky, A., & Schmidt, H. (2002). Suffix vector: Space- and time-efficient alternative to suffix trees. In M. J. Oudshoorn (Ed.), Twenty-Fifth Australasian Computer Science Conference (ACSC2002).
Melbourne, Australia, ACS.
Mount, D. W. (2004). Bioinformatics: Sequence and Genome Analysis (2nd ed.).
Cold Spring Harbor, New York, USA: Cold Spring Harbor Laboratory Press.
Mukhopadhyay, S., Tang, C., Huang, J., & Palakal, M. (2003). Genetic sequence classification and its application to cross-species homology detection. The Journal of VLSI Signal Processing : Systems for Signal, Image, and Video Technology, 35(3), 273–285.
Noble, J., & Weir, C. (2001). Small Memory Software: Patterns for systems with limited memory. Reading, Massachusetts, USA: Addison Wesley Professional.
Parisi, V., Fonzo, V. D., & Aluffi-Pentini, F. (2003). STRING: finding tandem repeats in DNA sequences. Bioinformatics, 19(4), 1733–1738.
Herbert, P. D. N., Stoeckle, M. Y., Zemlak, T. S., & Francis, C. M. (2004).
Identification of birds through DNA barcodes. PLoS Biology, 2(10), e312.
Price, A. L., Jones, N. C., & Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics, 21(suppl 1), i351–358.
Ricardo, B. -Y., & Berthier, R. -N. (1999). Modern Information Retrieval. New York, USA: Addison Wesley.
Rocha, K., Medeiros, C., Monteiro, M., Gon¸calves, L., & Marinho, P. (2004).
Design of species-specific primers for virus diagnosis in plants with PCR. In Fourth IEEE Symposium on Bioinformatics and Bioengineering, 149–158.
Salton, G., & McGill, M. J. (1983). Introduction to Modern Information Retrieval.
New York, USA: McGraw-Hill Book Company.
Sandberg, R., Winberg, G., Branden, C.-I., Kaske, A., Ernberg, I., & Coster, J.
(2001). Capturing Whole-Genome Characteristics in Short Sequences Using a
Naive Bayesian Classifier. Genome Research, 11(8), 1404–1409.
Sharma, D., Issac, B., Raghava, G. P. S., & Ramaswamy, R. (2004). Spectral Repeat Finder (SRF): identification of repetitive sequences using Fourier transformation. Bioinformatics, 20(9), 1405–1412.
Silberschatz, A., Galvin, P. B., & Gagne, G. (2000). Applied Operating System Concepts (1st ed.). New York, USA: John Wiley & Sons.
Subramanian, S., Madgula, V. M., George, R., Mishra, R. K., Pandit, M. W., Kumar, C. S., & Singh, L. (2003). Triplet repeats in human genome:
distribution and their association with genes and other genomic regions.
Bioinformatics, 19(5), 549–552.
Tsay, L.-T. (2001). A study on sorting string suffixes of large-scaled text collection in external memory.Master’sthesis,DepartmentofComputerScienceand Information Engineering, National Chung Cheng University, Chiayi, Taiwan.
Wang, J.-D. (2004a). A case study of predicting the class of the unknown viruses of ssRNA positive-strand in NCBI. In The 21st Workshop on Combinatorial Mathematics and Computational Theory, 249–256.
Wang, J.-D. (2004b). DNA sequences classification via representative patterns. In The 7th Conference on Chinese Medicine and Pharmacy, Engineering Technology and Applications to Chinese and Western Medicine, 116–116.
Wang, J.-D. (2005). An external memory approach to compute the statistics of maximal repeats across classes from whole genome sequences. In 2005 National Computer Symposium, BIC1–2.
Wang, D., Coscoy, L., Zylberberg, M., Avila, P. C., Boushey, H. A., Ganem, D., &
DeRisi, J. L. (2002). Microarray-based detection and genotyping of viral pathogens. Microbiology, 99(24), 15687–15692.
Wang, J.-D., & Tsay, J.-J. (2002). Mining periodic events from retrospective Chinese news. International Journal of Computer Processing of Oriental Languages, A Special Issue on “Web and WAP Oriental Languages Multimedia Computing,”15 (4), 361–377.
Jing-Doo Wang received the B.S. degree in Computer Science and Information Engineering from the Tatung Institute of Technology in 1989, and the M.S. and Ph.D. degrees Computer Science and Information Engineering from the University of Chung Cheng in 1993 and 2002 respectively. He has been with Asia University (formerly Taichung Healthcare and Management University) since spring 2003, where he is currently an assistant professor in the Department of Computer Science and Information Engineering, and also holds a joint
appointment with the Department of Bioinformatics. His research interests were in the areas of Chinese text categorization, information extraction before 2003.
Recently, he focused on the bioinformatics, such as DNA sequence classification, significant pattern extraction and genomic sequences comparison.