國立臺灣大學理學院化學研究所 碩士論文
Department of Chemistry College of Science
National Taiwan University Master Thesis
設計與合成磷酸基甘油酸衍生物 來抑制細菌轉醣酶
Design and Synthesis of Phosphoglycerate Derivatives for Inhibition of Bacterial Transglycosylase
吳蕙如 Huei-Ru Wu
指導教授:方俊民 博士 Advisor: Jim-Min Fang, Ph.D.
中華民國 105 年 6 月
June, 2016
中文摘要
抗生素的濫用造成許多具有多重抗藥性的細菌大量繁衍,嚴重威脅到人類的存 亡,發展出新穎且有效的抗生素便有著刻不容緩的需求。細菌的細胞壁是由肽聚醣 所組成,其生合成涉及了一個重要的酵素”轉醣酶”的催化,轉醣酶裸露在細胞膜 外側,因此藥物不需經由通過細胞膜便可直接抵達酵素進行抑制,故以轉醣酶為目 標而開發的新型抗生素是極具吸引力的。
我們設計並合成出一系列具有潛力的轉醣酶抑制劑,利用帶有不同取代基的聯 苯結構模擬單體 lipid II 在進行轉醣化過程中所形成過渡態之電荷與結構,並搭配 轉醣酶的天然抑制劑 moenomycin 的特徵基團”磷酸甘油酸”為基礎架構,利用較 短碳鏈的磷酸甘油酸衍生物來改善抑制劑的生體可用率,再以胺基或醯胺基連接 鏈結合上述兩個片段。
成功合出一系列具有潛力的轉醣酶抑制劑後,我們透過高效液相層析‒轉醣酶 活性分析法及最低抑菌濃度檢測來確認所合成之抑制劑是否具有良好的抑菌效果。
在一系列磷酸甘油酸衍生物中,聯苯上帶有能產生氫鍵官能基並具有胺基連接鏈 的化合物,其轉醣酶活性檢測具有最佳抑制性在 100 M 有 76%抑制性,但因為活 體實驗較酵素實驗複雜,因此該化合物的最低抑菌濃度大於 100 M。綜合本文實 驗結果,我們認為帶有能產生氫鍵官能基的聯苯結構及能產生正價性質之胺基連 接鏈是相當重要的,因此在未來化合物的結構衍生修飾上,我們將保有這些特性,
並藉由更改磷酸甘油酸上的脂質鏈來增加化合物及酵素之間的作用力,期望能開 發出具有更佳抑制效果的轉醣酶抑制劑。
Abstract
The abuse of antibiotics results in dramatic increase of multi-drug resistant bacteria which become a serious threat to human health. Developing novel and effective antibiotics against resistant bacteria is undoubtedly required. Bacteria cell wall is constituted by peptidoglycan which is formed involving the catalysis by an important enzyme transglycosylase (TGase). TGase is located on the external of cell membrane, so inhibitors can reach TGase without entering cytoplasm. Therefore, TGase becomes an attractive target for development of new antibiotics.
We designed and synthesized a series of potential TGase inhibitors that contain a biphenyl moiety with different substituents to mimic the oxonium transition state during the lipid II transglycosylation. A phosphoglycerate moiety which is a characteristic group of moenomycin bearing a curtailed aliphatic chain is incorporated into the designed inhibitors to improve the bioavailability. The two above-mentioned moieties are connected by amide or amine linker.
Some potential transglycosylase (TGase) inhibitors were synthesized and subjected to the HPLC-based TGase fluorescence assay and MIC assay. Among all the synthesized phosphoglycerate derivatives, a compound bearing an amine linkage and a biphenyl moiety with four hydroxyl substituents showed the best TGase inhibitory activity of 76%
at 100 M. The cell experiment is usually more complicated than the enzymatic assay due to more controlling factors. Hence, this compound was inactive to bacteria, showing MIC values above 100 M. Among all of the transglycosylase inhibitors designed here, we considered that the phosphoglyceric acid, amine linkage and multiple substituted biphenyl are important moieties to provide the desired interactions with the active site of TGase. To further explore more effective TGase inhibitors, we will keep the important properties of current compounds but elongate the lipid substituent to improve the interaction with TGase.
Table of Contents
Abstract in Chinese ... I
Abstract in English ... III
Table of Contents ... V
Index of Figures ... VIII
Index of Schemes ... X
Index of Tables ... XI
Abbreviations ... XII
Chapter 1. Introduction ... 1
1.1 Background ... 1
1.2 Gram-positive bacteria and Gram-negative bacteria ... 1
1.3 Biosynthesis of peptidoglycan ... 3
1.4 History of antibiotics ... 6
1.5 Penicillin-binding proteins (PBPs) and structure of transglycosylase ... 10
1.6 Mechanism of transglycosylation ... 13
1.7 Development of transglycosylase inhibitors ... 16
1.8 Research of transglycosylase inhibitors in our laboratory ... 25
1.9 Analytic methods for assessment of transglycosylase activity ... 27
1.9.1 Radioactive labeling ... 27
1.9.2 Fluorescence labeling for HPLC ... 27
1.9.3 Surface plasmon resonance ... 30
1.9.4 Fluorescence anisotropy ... 31
1.9.5 Förster resonance energy transfer ... 32
Chapter 2. Results and Discussion ... 35
2.1 Design of TGase inhibitors ... 35
2.2 Synthesis of TGase inhibitors ... 39
2.2.1 Synthesis of phosphoglycerate derivatives ... 41
2.2.2 Synthesis of biphenyl esters ... 42
2.2.3 Synthesis of 4-arylbenzamide derivatives ... 44
2.2.4 Synthesis of 4-arylbenzylamino derivatives ... 45
2.2.5 Coupling of phosphoglycerate with 4-arylbenzamide derivative . 48 2.2.6 Coupling of phosphoglycerate with 4-arylbenzylamino derivative ... 49
2.2.7 Synthesis of pyrophosphate derivatives ... 50
2.3 Biological activity ... 51
2.3.1 Transglycosylase activity assay ... 53
2.3.2 Minimum inhibitory concentration (MIC) assay ... 56
2.4 Conclusions ... 58
Chapter 3. Experimental Section ... 61
3.1 General part ... 61
3.2 General procedures of HPLC-based TGase fluorescence assay ... 62
3.3 General procedures of MIC assay ... 63
3.4 Synthetic procedures and characterization of compounds ... 64
References……….………..………..101
Appendix NMR spectra………...……….110
Index of Figures
Figure 1. (a) Gram-positive bacteria cell envelope; (b) Gram-negative bacteria cell
envelope ... 2
Figure 2. Biosynthetic pathway of bacterial cell wall peptidoglycan ... 5
Figure 3. Antibiotics development timeline ... 6
Figure 4. Chemical structures of penicillin G and vancomycin ... 8
Figure 5. (a) Interactions between vancomycin and lipid II terminal peptide; (b) Interactions between vancomycin and mutated lipid II terminal peptide. ... 9
Figure 6. Overall structure of S. aureus PBP2 ... 11
Figure 7. Crystal structure of the complex PBP1b and moenomycin ... 11
Figure 8. Characteristic hydrogen bonding interactions and electrostatic between moenomycin and E. coli TGase ... 12
Figure 9. Two possible polymerization directions for formation of cell wall glycan ... 13
Figure 10. Experimental design to verify the direction of chain elongation in transglycosylation ... 14
Figure 11. Proposed mechanism for lipid II polymerization by TGase. ... 15
Figure 12. Structures of nisin and moenomycin A ... 17
Figure 13. TGase inhibitors reported by Sofia and coworkers. ... 20
Figure 14. TGase inhibition assay using radioactive labeling lipid II ... 27
Figure 15. HPLC-based TGase inhibition assay using fluorescence labeling lipid II .... 28
Figure 16. Design for a fluorescent TGase substrate of Lipid II-Dansyl-C20 ... 29
Figure 17. Modification of the A ring of moenomycin A to a fluorescent derivative (Moe-NH2) ... 31
Figure 18. Screening for TGase inhibitors by fluorescence anisotropy assay using fluorescent moenomycin (F-Moe) ... 32
Figure 19. TGase activity assay by Förster resonance energy transfer... 33
Figure 20. The concept for design of potential TGase inhibitors ... 35
Figure 21. The TGase inhibitors synthesized in our laboratory... 38
Figure 22. Retrosynthetic scheme of TGase inhibitors bearing biphenyl derivatives .... 40
Figure 23. The designed TGase inhibitors ... 52
Figure 24. Transglycosylase activity assay ... 53
Figure 25. Comparison of structures 66‒69 and their possible interactions in TGase active pocket ... 55
Index of Schemes
Scheme 1. Synthesis of phosphoglycerate derivative 37 ... 42
Scheme 2. Synthesis of tetramethoxy biphenyl compound 43 ... 43
Scheme 3. Synthesis of tetrabenzyloxy compound 45 ... 44
Scheme 4. Synthesis of aldehyde compounds 50 and 51 ... 45
Scheme 5. Synthesis of Fmoc derivative 53 ... 46
Scheme 6. Synthesis of acid compounds 58 and 59 ... 48
Scheme 7. Synthesis of acid compound 61 ... 49
Scheme 8. Synthesis of acid compounds 66 and 67 ... 49
Scheme 9. Synthesis of monophosphate compound 72 ... 51
Scheme 10. Synthesis of pyrophosphate compounds 73 and 74 ... 51
Scheme 11. Proposed structures of potent TGase inhibitors ... 60
Index of Tables
Table 1. TGase inhibitors reported by Walker, Kahne and coworkers ... 19
Table 2. The reported TGase inhibitors by Walker, Vederas and coworkers ... 21
Table 3. TGase inhibitors reported by Wong, Ma, Cheng and coworkers ... 22
Table 4. TGase inhibitors with salicylanilide-based core structure ... 23
Table 5. TGase inhibitors reported by Wong, Cheng and coworkers ... 24
Table 6. TGase inhibitors reported by Zuegg and coworkers ... 25
Table 7. Antibacterial activity of the lactate and glycerate phosphate compounds synthesized in our laboratory ... 26
Table 8. Synthesis of amine compound 54 ... 47
Table 9. Transglycosylase activity assay... 54
Table 10. MIC assay ... 57
Abbreviations
Ac acetyl
Ala alanine
Arg arginine
Asp aspartate
Bn benzyl
DIBAL-H diisobutylaluminum hydride
DMF N,N-dimethylformamide
DMSO dimethylsulfoxide
EDCI 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
ESI electrospray ionization
Et ethyl
Fmoc 9-fluorenylmethoxycarbonyl
GlcNAc N-acetylglucosamine
Glu glutamate
HOBt hydroxybenzotriazole
HPLC high-performance liquid chromatography
HRMS high resolution mass spectroscopy
IR infrared
NBD 7-nitrobenz-2-oxa-1,3-diazol-4-yl
NMR nuclear magnetic resonance
mCPBA meta-chloroperoxybenzoic acid
Me methyl
MIC minimum inhibitory concentration
mp melting point
MS molecular sieve
MurNAc N-acetylmuramic acid
MW molecular weight
PDC pyridinium dichromate
PBPs penicillin-binding proteins
Rf retention factor
rt room temperature
TBAI tetrabutylammonium iodide
TGase transglycosylase
TG transglycosylation
TLC thin-layer chromatography
THF tetrahydrofuran
TPase transpeptidase
TP transpeptidation
UDP uridine diphosphate
Chapter 1. Introduction
1.1 Background
Bacteria are everywhere in nature. The communities of various bacteria can be tracked in ice, sediment, meltwater, acidic hot springs and even in radioactive waste.1 The existence of cell wall, which mammals lack, protects bacteria for their survival in such tough environment. No matter bacteria are malignant or beneficial: humans are always coexisting with them. Pathogenic bacteria had caused great menace to humans before the discovery of penicillin by Fleming in 1928. As time passed, there are increasing numbers of antibiotics developed for treatment of bacterial infections. However, the abuse of antibiotics leads to the rising of emerging multi-drug resistant bacteria which impose serious threat to human health. Developing new and effective antibiotics against resistant bacteria is urgently required.
1.2 Gram-positive bacteria and Gram-negative bacteria2
In 1884, Hans Christian Gram developed a staining procedure that could categorize a large proportion of bacteria into two groups. One group was called “Gram-positive bacteria” that were stained purple by crystal violet because they had thick peptidoglycan.
Another group was called “Gram-negative bacteria” that were not stained with crystal
violet because they had additional outer membrane. Gram-positive and Gram-negative
bacteria differ not only in the fundamental structures but also in their physiology and pathogenicity.
The cell envelope of Gram-positive bacteria with peptidoglycan of 20–40 nm thickness is anchored by long anionic polymers of glycolipid, called wall teichoic acids, which comprises a disaccharide linker and a polymer of phosphodiester-linked polyol repeat units (Figure 1). The cell wall also contains other subunits such as lipoproteins and lipoteichoic acids which are embedded in the head groups of cell membrane.
Figure 1. (a) Gram-positive bacteria cell envelope; (b) Gram-negative bacteria cell envelope3
Gram-negative cell envelope has a distinct feature of extra layer that is outer membrane comprising lipid bilayer. The outer membrane has lipopolysaccharides (LPS) on the outer leaflet and porin that allows nutrients to flow in and resists hydrophilic molecules larger than 700 Daltons. The space between inner and outer membrane is called periplasmic space which contains a thin peptidoglycan (7–10 nm). LPS molecule is
(a) (b)
nontoxic when it is incorporated in outer membrane. However, degradation of Gram- negative bacteria peptidoglycan releases lipid A that can evoke inflammatory response and cause hyperpyrexia even the septic shock. Therefore, harm of the released lipid A may be more serious than inflection.4
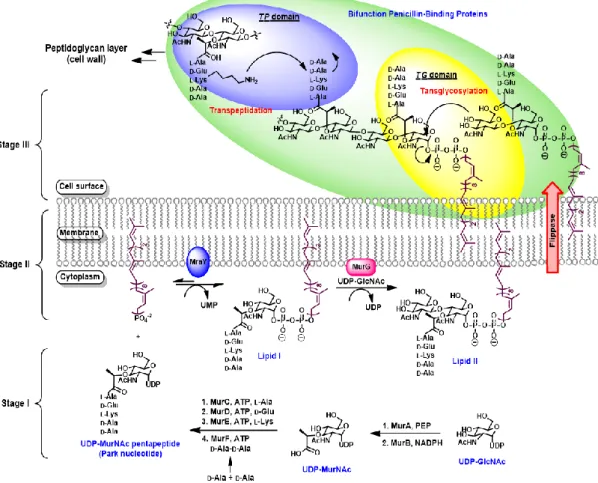
1.3 Biosynthesis of peptidoglycan
Bacteria cell walls are constituted by the mesh-like peptidoglycan, which is a heteropolymer to maintain the characteristic cell shape and protect cell integrity by resisting the internal osmotic pressure. Interruption of peptidoglycan biosynthesis will cause cell death. The bacterial cell wall peptidoglycan is formed by polymerization and cross-linked of lipid II, which consists of -,4-linked N-acetylglucosamine and N- acetylmuramic acid pentapeptide.
The biosynthesis of bacterial peptidoglycan is a complicated process. The biosynthetic pathway can be roughly categorized into three stages (Figure 2).5
First stage: formation of UDP-N-acetylmuramic acid pentapeptide (Park nucleotide)
In the first step, UDP-N-acetyl glucosamine (UDP-GlcNAc) is reacted with phosphoenol pyruvate (PEP) by catalysis of transferase MurA, and then reduced by reductase MurB to yield UDP-N-acetylmuramic acid (UDP-MurNAc). UDP-GlcNAc is not only an essential precursor for peptidoglycan but also the precursor for many other
cell wall macromolecules such as teichoic acids and lipopolysaccharides. The assembly of pentapeptide moiety onto UDP-MurNAc to produce UDP-MurNAc pentapeptide (also known as Park nucleotide) is accomplished by a series of enzymes designated as Mur synthetase including MurC, MurD, MurE and MurF, which are responsible for successive addition of L-alanine, D-glutamic acid, L-lysine or meso-diaminopimelic acid (depending on Gram-positive or Gram-negative bacteria), D-alanine and D-alanine.
Second stage: formation of lipid II
UDP-MurNAc pentapeptide is catalyzed by MraY to connect the membrane-bound acceptor undecaprenyl diphosphate (C55PP), forming lipid I along with release of UMP.
MurG subsequently catalyzes the transfer of N-acetylglucosamine from UDP-GlcNAc to the C4 hydroxyl group of lipid I in a -1,4-linkage to yield lipid II. Lipid II is then translocated to the outer membrane by the flippase MurJ, which is a polytopic innermembrane protein belong to the multidrug/oligo-saccharidyl-lipid/polysaccharide (MOP) exporter superfamily.6 Another potent flippase Amj (formerly YdaH) regulated in vivo by a stress-response pathwayin the absence of MurJ is found in Bacillus subtilis.7 Third stage: formation of peptidoglycan
At the final stage, lipid II binds to transglycosylase (TGase) of penicillin-binding proteins (PBPs) to undergo transglycosylation (TG) by attaching its C4 hydroxyl group
at the GlcNAc terminal to the polyprenyldiphosphate moiety at the anomeric center of MurNAc unit to produce immature elongated glycan chains by releasing C55PP to the interior of cell membrane. Then, transpeptidase (TPase) in PBPs catalyzes cross-linking of the nascent glycan chains by connecting the amine group of L-Lys to the penultimate
D-Ala of an adjacent strand with liberation of one D-Ala to form the firm and mesh-like peptidoglycan.
Figure 2. Biosynthetic pathway of bacterial cell wall peptidoglycan8
1.4 History of antibiotics
The injured people have been treated by using herbs or potions for thousands of years.
In ancient times people did not know that the infections were caused by bacteria. People could only fight bacteria by autoimmune system. The diseases caused by infection of bacteria could possibly be cured only until the discovery of antibiotics.
In 1928, Alexander Fleming accidently discovered penicillin (Figure 3). However, this discovery did not get attention until 1940 when Howard Walter Florey and Ernst Boris Chain successfully isolated penicillin and identified its efficacy in therapy of bacterial infectious diseases. Despite the success of penicillin, the demand of new antibacterial agents remained.
Figure 3. Antibiotics development timeline9,10
The continuing researches led to the discovery of many antibiotics such as sulfonamides, bacitracin, tetracycline, chloramphenicol, macrolide, glycopeptides,
and cycloserine in different scaffolds.9 However, there has been short of major classes of antibiotics since 1987, presumably because Big Pharma has reduced their researches in this area.10
According to the mechanism of action; different antibiotics can be categorized into five types: 9
i. Inhibition of cellular metabolism: sulfonamides and trimethoprim.
ii. Inhibition of bacterial cell wall synthesis: penicillins, carbapenems, and glycopeptides.
iii. Interactions with the plasma membrane: cycloserine and lipopeptides.
iv. Disruption of protein synthesis: tetracyclines, chloramphenicol, macrolides, streptogramins, lincosamides and oxazolidinones.
v. Inhibition of nucleic acid replication and transcription: rifamycins and quniolones.
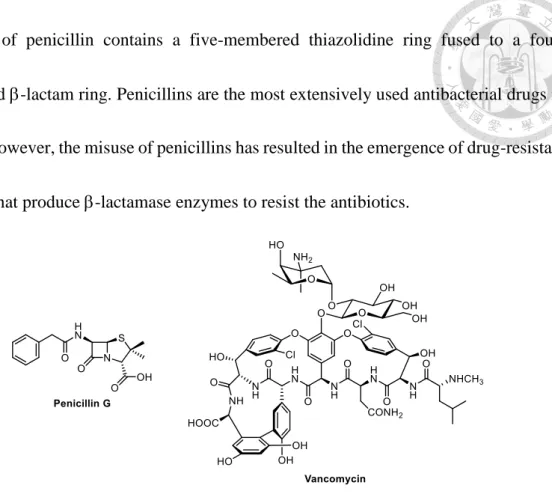
Mammal cells do not possess cell walls, so the optimal target for antibiotics is to inhibit bacterial cell wall synthesis without harm to treated individual. Penicillins and vancomycin (Figure 4) are two prominent antibiotics to inhibit the synthesis of peptidoglycan by obstructing transpeptidation (TP) in Gram-positive bacteria. The
structure of penicillin contains a five-membered thiazolidine ring fused to a four- membered -lactam ring. Penicillins are the most extensively used antibacterial drugs in
clinics. However, the misuse of penicillins has resulted in the emergence of drug-resistant bacteria that produce -lactamase enzymes to resist the antibiotics.
Figure 4. Chemical structures of penicillin G and vancomycin
The resistance of bacteria may be mediated by the following ways.11 i. Active efflux or reduced uptake antibiotics into the cells.
ii. Modification by enzyme to deactivate antibiotics.
iii. Alteration the target of the drug to diminish the binding of the antibiotics.
iv. Enormous generation of the antibiotic target.
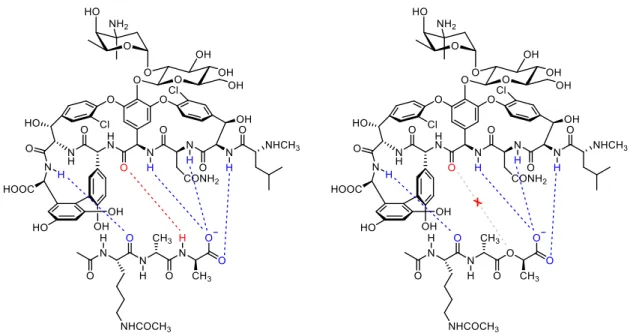
Vancomycin is the last resort for patients in clinical use to treat drug-resistant infections. The structure of vancomycin forms a pocket that can bind to D-Ala-D-Ala of lipid II by yielding five hydrogen bonds (Figure 5). The development of bacteria
resistance to vancomycin proceeds slowly, but the mutation of bacteria in hospital finally leads to the emergence of vancomycin-resistant Enterococci (VRE) and vancomycin- resistant Staphylococcus aureus (VRSA). The mutated bacteria reprogram the termini of lipid II peptide from D-Ala-D-Ala to D-Ala-D-Lac. The substitution of amide in Ala by ester in Lac not only loses one hydrogen bond to NH, but also renders repulsion by the lone-pair electrons of oxygens, thus causing 1000-fold decrease of binding affinity for vancomycin.12
(a) (b)
Figure 5. (a) Interactions between vancomycin and lipid II terminal peptide;
(b) Interactions between vancomycin and mutated lipid II terminal peptide12
1.5 Penicillin-binding proteins (PBPs) and structure of transglycosylase
Bacteria possess an adaptable number of penicillin-binding proteins (PBPs) that can catalyze the two last important steps, TG and TP, in peptidoglycan biosynthesis. There are high-molecular-weight (HMW) PBPs and low molecular weight (LMW) PBPs. HMW PBPs can be further categorized into class A and class B by the catalytic activity and structure of N-terminal domain. Class A PBPs are bifunctional because the C-terminal domain has TPase activity and the N-terminal domain possess TGase activity. In class B, the C-terminal domain also maintains TPase activity but the N-terminal domain is just unrelated to TG.13
Although TGase is highly conserved in all bacteria, it is laborious to isolate TGase due to the low content of PBPs is in the cell. In 2007, Strynadka et al. reported the crystal structures of S. aureus PBP2 and its complex with antibiotic moenomycin (Figure 6).14 The crystal structure revealed a bilobal fold with the TPase and TGase domains
partitioned by a -rich linker. The TGase fold is primarily -helical and is split into globular “head ” region and smaller “jaw” subdomains. The globular segment is composed of seven helices plus one region, and the jaw segment is constituted by
three helices closer to the membrane. In addition, the crystal structure of S. aureus PBP2-moenomycin complex indicated that the catalytic glutamic acid residues E114 and
E171 play important roles in the binding with moenomycin.15
Figure 6. Overall structure of S. aureus PBP214
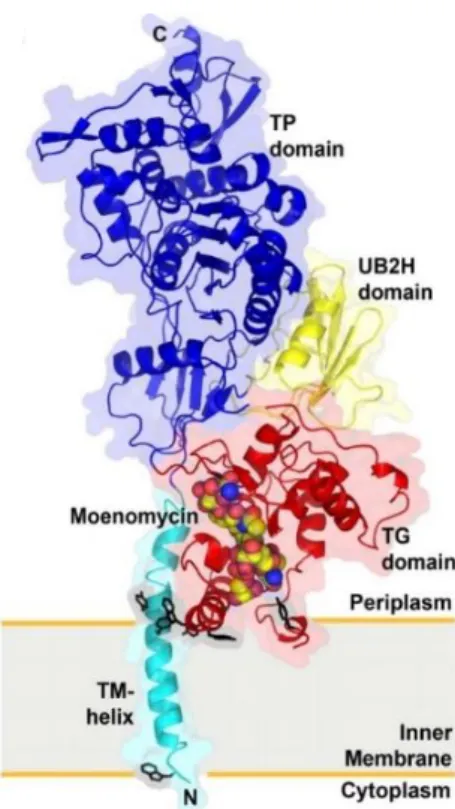
In 2009, Ma et al. reported the X-ray crystal structure of the complex of moenomycin with PBP1b from Escherichia coli (Figure 7). The PBP1b contains transmembrane (TM) helix, TGase, TPase and UB2H domain that consisted of about 100 amino acid residues.
Figure 7. Crystal structure of the complex PBP1b and moenomycin16 E114
E171
The binding affinity of E. coli PBP1b and moenomycin is TM domain dependent.
The TM domain comprises a single long helix, which may simply stabilize the protein- membrane interaction and restrict the orientation between PBP1b and moenomycin (or lipid II).16
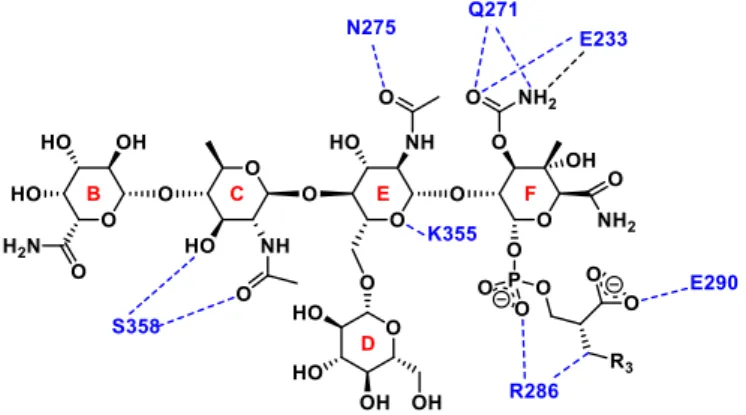
Some characteristic hydrogen bonding interactions between moenomycin and E. coli TGase are shown in Figure 8.16 The sugar parts of moenomycin is considered to mimic the sugar moieties of lipid IV. The E and F rings are suggested to act as the glycosyl donor to have stronger binding affinity with TGase than the B and C rings, which are considered the glycosyl acceptor. Besides, there are also electrostatic interactions between TGase and phosphoglycerol chain of moenomycin.16
Figure 8. Characteristic hydrogen bonding interactions and electrostatic between
moenomycin and E. coli TGase16
1.6 Mechanism of transglycosylation
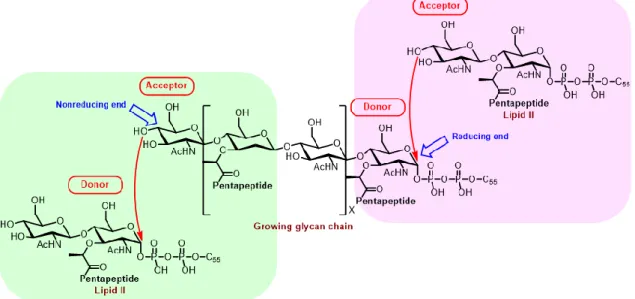
Whether lipid II plays the role of TG donor or acceptor has remained extensively debated for many years. One argument is that lipid II acts as a glycosyl acceptor with the lipid-pyrophosphate retained by addition of new disaccharide units to the reducing end of the elongating polymer (Figure 9, right panel). The other proposal is that lipid II plays the role of glycosyl donor with the lipid-pyrophosphate lost by joining new disaccharide units to the nonreducing end of the growing glycan chain (Figure 9, left panel).17
Figure 9. Two possible polymerization directions for formation of cell wall glycan17
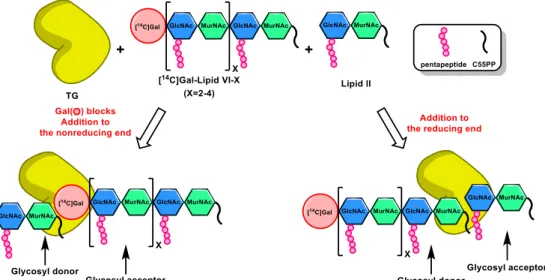
In 2007, Kahne, Walker and their coworkers designed an experimental strategy to verify the direction of chain elongation (Figure 10). The synthesized lipid IV was incubated with lipid II with E. coli PBP1A to form TGase substrates lipid VI–X. The nonreducing ends of these longer substrates bear a [14C]-Gal, which is introduced to
GlcNAc by catalysis of -1,4-galactosyltransferase (GalT), so that the TG reaction would
not take place at the nonreducing end. To test this assumption the Gal-blocked lipid IV– X were incubated with lipid II, respectively, in the presence of four different TGases obtained
from A. aeolicus PBP1A, S. aureus PBP2, E. coli PBP1A, and E. coli PBP1B. The gel
electrophoresis assay showed that the longer glycan chains were formed. These experimental results support that lipid II acts as the glycosyl acceptor and the growing glycan acts as the glycosyl donor for TG reaction at the reducing end.17
Figure 10. Experimental design to verify the direction of chain elongation in
transglycosylation17
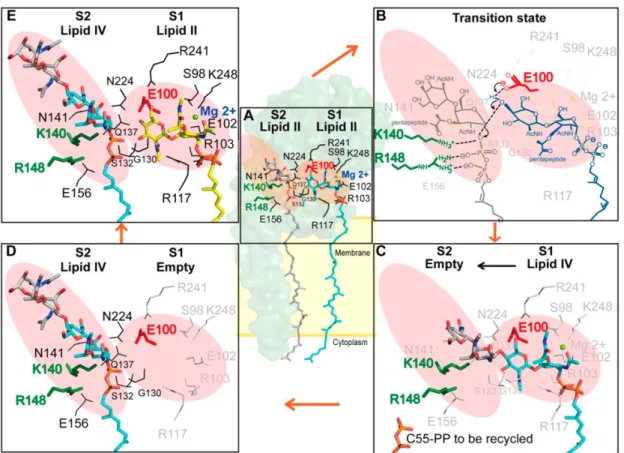
In 2012, Ma et al. reported the X-ray diffraction data of S. aureus PBP in complexation with a lipid II analog and S. aureus PBP in complexation with moenomycin.
Accordingly, a schematic model for the TG reaction was proposed (Figure 11). The substrate-binding site can be separated into two parts; the glycosyl acceptor site (S1) and
glycosyl donor site (S2). The 4-OH group of the acceptor in S1 site is deprotonated by E100 residue, allowed by a simultaneous reaction at the C1 of the -linked MurNAc donor in an SN2–like mechanism to give the -1,4-linked product. In addition, the K140 and R148 assisted the leaving of the pyrophosphate group by direct protonation of phosphate or by stabilizing the pyrophosphate via a divalent cation. After lipid II is transferred to the glycan chain, the elongated product will shift to the glycosyl donor site (S2), and a new lipid II molecule can reside in the glycosyl acceptor site for the next TG reaction.18
Figure 11. Proposed mechanism for lipid II polymerization by TGase18
1.7 Development of transglycosylase inhibitors
TPase and TGase are two important enzymes that catalyze the formation of three- dimension backbone of peptidoglycan. The TPase inhibitors such as vancomycin and penicillin have been used in clinical treatment for decades. With the more and more resistant cases of TPase inhibitors, scientists now shift the goal to this appealing target for several reasons:
i. TGase is located on the external of cell membrane, so inhibitors can reach the target without entering cytoplasm.
ii. Polysaccharide backbone retains intact in wild-type and resistant bacterial strains.
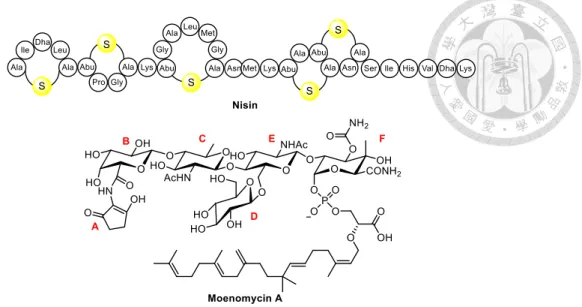
iii. TGase is unique in prokaryote and does not exists in eukaryotic counterparts.11 Natural products that inhibit TG can be divided into two types: 1) substrate binder which binds to lipid II, and 2) enzyme binder that directly inhibits TGase.11 The most salient TGase substrate binder is a lantibiotics, nisin (Figure 12) which coordinates the pyrophosphate moiety of lipid II by the N-terminal backbone amides to form a cage structure to interfere with the biosynthesis of peptidoglycan. Moenomycin A, a member of moenomycin family, is the most prominent TGase binder. It shows better affinity to TGase than the growing glycan chain, so moenomycin A can directly interact with the active site of TGase to inhibit the polymerization of peptidoglycan.5d
Figure 12. Structures of nisin and moenomycin A
Moenomycin A is composed of three components, including a pentasaccharide, a phosphoglycerate and a C25 moenocinol lipid tail which is attached to C1 of the F ring via phosphoglycerate. The minimal inhibitory concentration (MIC) of moenomycin A against several Gram-positive bacteria ranges from 1 to 100 ng/mL. However, the poor pharmacokinetic properties render moenomycin A inadequate for treatment to humans.
Moenomycins have been legally added to animal feed as the growth promoters under the
trademarks Flavomycin and Flavophospholipol. The issue of using antibiotic additives in animal nutrition has been debated for a long time, and an ultimate decision is made in 2006 to forbid the use of antibiotic additives on all animal feed in entire European Union.19
Welzel and co-workers have reported the structure activity relationships of moenomycin A derivatives with TGase inhibition. It is shown that the lipid chain of
moenomycin A first anchors to the cytoplasmic membrane and the sugar part
subsequently binds to the donor binding site of TGase to compete with the growing glycan chain rather than lipid II. The -[1,2] junction between the E and F rings of moenomycin differs from the -[1,4] connection of growing glycan chain, and thus prevents the 4-OH
of lipid II GlcNAc from attacking the phosphate group of the F sugar.5d, 20 In contrast, rings A and D of moenomycin A, which resemble the pentapeptide of MurNAc, project from the planar polysaccharide scaffold and thus make fewer contacts to TGase.15
The degradation studies of moenomycin have shown that the rings E and F are crucial for activity and the modifications of the substituents on these rings decrease the biological activity. Particularly, the carbamoyl group at C3, the hydroxyl group at C4 and the carbamoyl groups at C3 and C5 of ring F, the acetyl group at C2 of ring E as well as the carboxyl group of phosphoglycerate are the pivotal functional groups that are exposed to the surface of TGase.20, 21
In 2010, Walker, Kahne and coworkers have reported that the carboxylate group of phosphoglycerate and the isoprenyl lipid in moenomycin A are important to TGase activity (Table 1). The inhibitory activity of moenomycin A decreases when the carboxylate group is replaced by carboxamide group, acylsulfonamide group or hydrogen atom. This results indicate that the carboxylate group binds to target by ionic interactions
with the positively charged side chains of enzyme, and the interactions help to dispose the lipid chain to attach the hydrophobic position of active site to cell membrane. In addition, when the isoprenyl chain is altered by neryl chain, the inhibitory activity also decreases. It indicates that the C10 chain is too short to maintain the biological activity and the C25 chain is crucial to interact with the hydrophobic cell membrane.22
Table 1. TGase inhibitors reported by Walker, Kahne and coworkers22
Compound
IC50 (M) MIC (g mL-1)
S. aureus PBP2 E. faecalis PBP2A S. aureus E. faecalis
Moenomycin 0.002 ± 0.001 0.035 ± 0.007 0.13 0.063
1 0.340 ± 0.209 0.203 ± 0.101 > 250 > 250
2 56 ± 14 57 ± 14 > 250 > 250
3 68 ± 26 88 ± 40 > 250 > 250
Due to the low quantities and difficult isolation of lipid II in nature, the development of artificial TGase inhibitors progresses slowly.23 In 1999, Sofia et al. have applied a
solid-phase combinatorial approach to construct a series of disaccharides mimicking the E and F rings of moenomycin A (Figure 13). The resin containing photocleavable linker is removed after the derivatization of disaccharides. A library of 1300 disaccharides is prepared and compounds 4‒9 are shown to inhibit bacterial growth. Therefore compounds
4‒9 were resynthesized and purified to show the IC50 values of 8‒10 g/mL for TGase and 3.12‒12.5 g/mL of MIC values.
Figure 13. TGase inhibitors reported by Sofia and coworkers24
In 2004, Walker, Vederas and coworkers have reported a series of mono- and di- saccharides C-phosphonates containing curtailed alkyl chains to mimic the active portion of moenomycin A and lipid II (Table 2). The activity assay against E. coli PBP1b is performed based on the radioactive labeling. Compounds 10a and 10b which are prepared to test the distance between the noncleavable C-phosphonate and the sugar, are shown to have 17% and 0% inhibition at 100 M. In addition, compound 11a‒11c with extended
linkage which are prepared to examine the effect of the first sugar of moenomycin, show inhibition of 25%, 10% and 25% at 100 M, respectively.These results suggest that the
introduction of properly extended linkage between mono- or di-saccharides and phosphate are needed for TGase inhibition.25
Table 2. The reported TGase inhibitors by Walker, Vederas and coworkers25
Compound (100 M)
Inhibition (%) E. coli PBP1b
10a 17
10b ‒
11a 25
11b 10
11c 25
In 2008, Wong, Ma, Cheng and coworkers have used a fluorescence anisotropy (FA)- based assay as a new strategy to search TGase inhibitors. The high-throughput screening (HTS) of 57,000 molecules reveals compounds 12‒14, that have IC50 values of 34.0, 3.7 and 9.3 M, respectively (Table 3). The structures of these compounds all contain numerous planar hydrophobic groups and negative charge properties, which are
speculated to compete the pyrophosphate binding site with the fluorescent moenomycin derivatives.26
Table 3. TGase inhibitors reported by Wong, Ma, Cheng and coworkers26
Compound IC50 (M) MIC (M)
B. subtilis E. faecalis S. aureus S. pneumoniae
Moenomycin 0.36 0.33 0.04 < 0.01 0.33
12 34.0 0.25 0.25 1.0 4.0
13 3.7 0.25 1.0 4.0 ‒
14 9.3 4.0 > 4.0 > 4.0 > 4.0
In 2010, Wong and coworkers have again conducted an HTS of nearly 2 M compound library, and further narrowed down to 252 hit compounds. These hits are tested by FA assay for TGase inhibition, and then verified by TGase-catalyzed lipid II polymerization.
Among all the synthesized compounds, 15‒18 with the salicylanilide-based core structure (Table 4) have the best inhibitory activity at 100 M against TGase. It is concluded that
the third aryl group on the anilide is necessary for TGase inhibition.27
Table 4. TGase inhibitors with salicylanilide-based core structure27
Compound IC90 of TGase (M) MIC (g/mL) S. aureus MRSA
15 50 0.5 0.25
16 ≤ 25 4 2
17 ≤ 25 1 1
18 ≤ 25 8 4
In 2010, Wong, Cheng and coworkers have reported the small molecules with azasugars-based structure, which is conjugated with a truncated pyrophosphate lipid chain and lipophilic carboxylic acids (Table 5). The azasugars are designed to mimic the charge and shape of the oxonium ion transition state during the TG reaction. In order to enhance the stability of the inhibitors, the O-glycoside was replaced by C-glycoside to connect the azasugar and phosphate. The potential hit compounds are identified by the
FA-based assay. The best inhibitor 19 shows 100% inhibition at 1 mM and 80% inhibition at 100 M against E. coli PBP1b. In comparison, the pentapeptide-containing lipid I and
lipid II-based inhibitors (22 and 23), which are synthesized by walker and Vederas et al.
show worse inhibition than compound 19.28
Table 5. TGase inhibitors reported by Wong, Cheng and coworkers28
Compound
Inhibition%
1 mM 100 M
Moenomycin 100 100
19 100 > 80
20 69 11
21 73 15
22 41 13
23 77 42
In 2015, Zuegg and coworkers have reported the novel compounds with non-planar pyranose scaffold and benzimidazole moiety to replace the phosphoglycerate group in moenomycin (Table 6), in order to reduce the molecular weight and hydrophobicity.
Compounds 24 and 25 possess good in vitro antibacterial activity against several Gram- positive bacteria. In addition, the in vitro docking experiment suggests that the two compounds are able to bind at both the donor and acceptor sites. The in vivo experiments
show that both compounds can be tolerated in mice up to a dose of 100 mg/kg, and have good metabolic stability in rats.29
Table 6. TGase inhibitors reported by Zuegg and coworkers29
Compound inhibition
MIC (g/mL)
S. aureus E. faecium E. faecalis S. pneumoniae E. coli
24 200 g/mL 4 16 4 4 > 64
25 200 g/mL 4 2 8 8-16 > 64
1.8Research of transglycosylase inhibitors in our laboratory
In 2010, Wang, J-T. has designed a series of TGase inhibitors that utilize azasugar to mimic the charge and structure of the oxonium ion transition state during the TG reaction and introduce a modified phosphoglycerate part of moenomycin (Table 7). The synthesized compounds 26 and 27 show poor inhibition against TGase.8 In 2011, Chang, J-H. also applied the same concept to mimic the TGase transition state during the TG reaction by using special sugar-DANA, GlcNAc or aryl functional groups to mimic the sugar part of lipid II and using phosphoglycerate derivatives to mimic the pyrophosphate part of lipid II. Furthermore, she has extended the amine linker in different lengths from
the phosphate of phosphoglycerate. Compound 28 has good inhibition against TGase at 100 M.30 In 2013, Cheng, H-J. designed a biphenyl group, which is extended from
phosphoglycerate, to imitate the saccharide part of lipid II. Compound 29 has the best inhibitory activity against TGase at 500 M and the MIC is 50 M.31 In 2014, Tsai, W-
C. has replace the phosphate to phosphonate in order to enhance the stability of TGase inhibitor and to decrease the rate of hydrolysis. Compound 30 shows moderate TGase inhibitory activity and antibacterial activity.32
Table 7. Antibacterial activity of the lactate and glycerate phosphate compounds
synthesized in our laboratory
Compound Inhibition
MIC (M) S. aureus
26 > 1 mM ‒
27 > 1 mM ‒
28 > 100 M 200
29 ≈ 500 mM 50
30 > 100 M 100
1.9 Analytic methods for assessment of transglycosylase activity 1.9.1 Radioactive labeling
The earliest TGase assay– radioactive labeling devised by Suzuki,33 van Heijenoort,34 Goldman35 and coworkers (Figure 14). The [14C]-radiolabel UDP-GlcNAc or UDP- MurNAc-pentapeptde undergoes the enzymatic TG to produce [14C]-lipid II. The radioactivity of lipid II and glycan chains are subjected to paper chromatography analysis or gel electrophoresis assay, and the radioactivity is counted to determine the activity of TGase or to identify the TGase inhibitors. This method is improper for detailed enzymology, the throughput is low and cannot be conducted in a common laboratory as manipulations of radiolabeled substances is hard to conduct in a normal laboratory.
Figure 14. TGase inhibition assay using radioactive labeling lipid II
1.9.2 Fluorescence labeling for HPLC
The fluorescence labeling HPLC-based method for TG assay is first reported by Schwartz et al. in 2001. Lipid II is incubated with PBP1b and the primary amine of lysine in unreacted lipid II is derivatized with fluorescamine at different reaction time. The
Peptidoglycan
Lipid II
Lipid II
signal of fluorescent labeling declines with the longer polymerization time, and the HPLC analysis will determine the activity of TGase.36
In 2002, Schwartz et al. has further devised a continuous fluorescence assay to measure the activity of TGase (Figure 15). The fluorescent dansyl-lipid II is incubated with PBP1b. TGase catalyzes the polymerization of dansyl-lipid II to form peptidoglycan, which was cleaved by muramidase at the 1,4-glycosidic linkages to produce the dansyl- tagged peptidoglycan monomers (F-PGM). In the presence of TGase inhibitor, the dansyl-lipid II substrate remains and appears at a retention time different from that of F- PGM on HPLC analysis.37
Figure 15. HPLC-based TGase inhibition assay using fluorescence labeling lipid II37
In 2002, Fang, Cheng and coworkers have designed a new HPLC-based fluorescence labeling method.38 The design concept of lipid II derivative was based on Walker and coworkers who have previously shown that the lipid chain of Lipid II-C35P containing four cis double bonds near pyrophosphate plays an important role in recognition with TGase.39 Hence, the four cis double bonds are maintained while the diprenyl moiety in
the lipid chain is replaced by the fluorescent dansyl group (Figure 16). Compared to the previous method,37 Lipid II-Dansyl-C20 has the fluorescent tag on the lipid chain rather than the lysine residue of the pentapeptide chain. The depletion of fluorescent Lipid II- Dansyl-C20 at different reaction times is detected by HPLC, indicating it is a substrate of TGase. The advantages of this method are using a curtailed lipid chain to improve solubility and having the fluorescent tag remote from the TG site. Furthermore, an extra step for using muramidase to hydrolyze glycan chain is unnecessary.
Figure 16. Design for a fluorescent TGase substrate of Lipid II-Dansyl-C20
1.9.3 Surface plasmon resonance
In 2002, Welzel et al. have developed an assay based on competitive surface plasmon resonance (SPR) to evaluate the affinity of TGase with moenomycin A analogues.40 The total internal reflection between incident slide and thin metal layer leads to different resonance angle when a polarized light strikes the surface of biochip with different compounds. According to their previous research, the A ring of moenomycin A is less influential in binding with TGase. Hence, the A ring is selectively modified to produce a moenomycin A derivative Moe-NH2 (Figure 17).41 Moe-NH2 is immobilized on the sensor chip surface by amide bond formation of the primary amine group with the N- hydroxysuccinimide on the chip. The bacterial PBP1b is then injected at different concentrations to the Moe-NH2 sensor chip. The binding response with PBP1b is concentration dependent. The competition experiment is carried out by incubation with inhibitor at different concentrates to compete with Moe-NH2 in PBP1b binding. The higher SPR response indicates the lower binding of inhibitor with PBP1b, and hence lower antibiotic activity.
Figure 17. Modification of the A ring of moenomycin A to a fluorescent derivative
(Moe-NH2)41 1.9.4 Fluorescence anisotropy
Although there are some methods to search for TGase inhibitors including radioactive assay and SPR, the low-throughput is still a major disadvantage. Therefore, Wong, Ma, Cheng and coworkers developed a high-throughput assay using fluorescence anisotropy- based method in 2008.26 Moenomycin A is modified by fluorophore to prepare the fluorescein-labeled probe F-Moe. The steady-state affinity (KD) values of moenomycin A (4.4 ×10‒7 M‒1 ) and F-Moe (5.2 × 10‒7 M‒1) are very similar. F-Moe is preincubated with Helicobacter pylori PBP1a, and then an inhibitor is added. If the inhibitor has
stronger binding affinity than F-Moe toward TGase, the signal of anisotropy will decrease.
Therefore, the TGase inhibitory activity can be determined by the signal change of anisotropy due to the competitive TGase binding with inhibitor against F-Moe (Figure 18).
Figure 18. Screening for TGase inhibitors by fluorescence anisotropy assay using
fluorescent moenomycin (F-Moe) 1.9.5 Förster resonance energy transfer
The slow development of TGase inhibitors is partly due to the shortage of continuous,
quantitative, and high-throughput assay.42 Hence, Cheng, Wong and coworkers have devised a new continuous fluorometric assay based on Förster resonance energy transfer
(FRET) to provide an efficient and convenient high-throughput screening of potential TGase inhibitors. The lipid II analogue is prepared with modification of a dimethylamino- azobenzenesulfonyl quencher in the lipid chain and a coumarin fluorophore in the
pentapeptide chain. During the process of TGase-catalyzed polymerization, the quencher- appended lipid-pyrophosphate moiety would be released, and the formed oligomer and polymer will emit fluorescence. The oligomer and polymer are digested by N- acetylmuramidase to liberate peptidoglycan monomer that shows enhanced fluorescence of coumarin at 460 nm (Figure 19).
Figure 19. TGase activity assay by Förster resonance energy transfer42
Although, there are methods reported to detect TGase activities, some shortcomings remain to be resolved. The radioactive labeling method with thin-layer chromatography is useful for enzymatic characterization of TGase but laborious to acquire quantitative results. Though SPR can afford quantitative analysis, it is only used to analyze small
group of compounds because this assay is low-to-medium throughput and time consuming. Neither the HPLC-based method can be applied for high-throughput analysis.
The fluorescence anisotropy-based assay is limited to identify the compounds that can compete F-Moe in TGase binding. The fluorescent compounds may interfere with F-Moe to give false results. It is still not an easy work to synthesize fluorescent lipid II analogs for TGase assays.
Chapter 2. Results and Discussion
2.1 Design of TGase inhibitors
The design of TGase inhibitors is primarily based on the skeleton of oxonium transition state during the lipid II TG. Besides, the structure of moenomycin is taken into consideration for modification of these TGase inhibitors (Figure 20).
Figure 20. The concept for design of potential TGase inhibitors
We designed a series of potential TGase inhibitors that contain a biphenyl moiety with different substituents to mimic the saccharide part of lipid II. A phosphoglycerate moiety which is a characteristic group of moenomycin bearing a curtailed aliphatic chain
mentioned moieties are connected by amide or amine linker. On the other hand, the dimeric pyrophosphate ester bearing different functional groups were also designed to mimic the pyrophosphate part of lipid II.
Using a properly extended linkage between the biphenyl derivatives and phosphoglycerate is inspired by the work of Walker and Vederas. The phosphoglycerate derivatives 10a‒11c were based on the characteristic lipid chain of moenomycin (Table 2).25 Compounds 11a‒11c with a properly extended linker between the saccharide and phosphorous atom have higher inhibitory activity than those compounds (10a‒10b) without linker.
The salicylanilide-based compounds 15‒18 (Table 4) found by high–throughput screening (HTS) with FA-based assay showed moderate TGase inhibitory activity.26 The structures of compounds 15‒18 were comprised by three to four planar aromatic rings.
Hence, we speculated that the existence of aromatic ring may lead to the inhibition of TGase activity. In addition, the salicylanilide core structure may provide hydrogen bonding with TGase. Accordingly, our designed TGase inhibitors would contain amides
and aromatic rings that are further modified by hydroxyl groups to increase the hydrogen bonding interaction and - stacking interactions.
The complex structure of moenomycin and E. coli PBP1b has revealed significant
amino acid residues in the TGase activity site.16 We speculated that not only glutamate 233 acts to deprotonate the 4-OH group of GlcNAc, but also asparagine 275 contains an
amide side chain to play an important role in binding with moenomycin (Figure 8). In 2009, Nakanishi et al. have assessed amide- interactions in the formamide-benzene
model system. They calculated all the possibilities of interaction between formamide and benzene including NH/, C=O/ and stacked geometries.43 The results suggested that the
energy between formamide and benzene after interaction would decrease energy.
Consequently, the favored amide-benzene interaction can support our concept of introducing aromatic rings to the TGase inhibitors. Moreover, the electrostatic interactions between phosphate and arginine 286 are also prominent.
Chang, J-H. in our lab has also applied the similar inhibitor design to imitate the transition state during TG reaction.30 In her design, DANA, GlcNAc and aryl functional group mimic the sugar part of lipid II, and phosphoglycerate derivatives mimic the pyrophosphate part of lipid II. However, the designed inhibitors only have decreased inhibitory activity against TGase, presumably because the inhibitors did not have suitable positively charged substituents to mimic the oxonium transition state during TG.
In 2012, Cheng, H-J. in our labs has connected different aromatic rings to the phophoglycerate moiety by amine linker to mimic the saccharide part of lipid II.31 Among
all the synthesized phosphoglycerate derivatives, the compounds bearing biphenyl substituent group have the best inhibitory activity. In 2014, Tsai, W-C. in our lab has changed the phosphate in TGase inhibitor 29 to phosphonate 30 to increase the stability and to decrease the rate of hydrolysis (Figure 21).32 However, phosphonate 30 shows lower inhibitory activity than phosphate compound 29. This results indicates that the oxygen atom of phosphate may interact with amino acid residues in TGase active site.
Figure 21. The TGase inhibitors synthesized in our laboratory31, 32
For the reasons outlined above, we take compound 29, a phosphoglycerate derivative, as the prototype and introduce an amine linker by reductive amination to connect the biphenyl moiety that may bear methoxy, benzyl or hydroxyl groups. The amino group would be protonated to positively charged aminium ion at physiological pH to mimic the
oxonium transition state during TG. The biphenyl containing methoxy, benzyl or hydroxyl groups was used to investigate the interactions such as hydrophobic,-
stacking and hydrogen-bond interactions between the inhibitors and TGase. In comparison, the analogous compounds with amide linker will also be synthesized to increase the hydrogen bondings toward TGase. In addition, the dimeric pyrophosphate
derivatives will be prepared to mimic the diphosphate part of lipid II. Along this line, more potent TGase inhibitors may be developed by the structural modification in various aspects.
2.2 Synthesis of TGase inhibitors
We designed and synthesized a series of potential TGase inhibitors that comprised three parts: a biphenyl moiety with different substituents, a phosphoglycerate moiety bearing curtailed aliphatic chain, an amide or amine linker to connect the two above- mentioned moieties. According to the retrosynthetic analysis, the biphenyl moieties were conjugated with phosphoglycerate derivatives by a phosphate coupling reaction. The benzamide derivatives underwent amide bond formation with the biphenyl compounds containing ester functional group and 3-amino-1-propanol under solvent-free condition.
On the other hand, the benzylamino derivatives were formed by reductive amination with the biphenyl compounds bearing aldehyde functional group and 3-amino-1-propanol (Figure 22).
Figure 22. Retrosynthetic scheme of TGase inhibitors bearing biphenyl derivatives
Moreover, we also designed and synthesized pyrophosphate derivatives that are simply coupled by above-mentioned monophosphate. The ester functional group of monophosphate was further modified by amide functional group to enhance the chelation ability to magnesium.
2.2.1 Synthesis of phosphoglycerate derivatives
Our synthetic route began with the preparation of the phosphoglycerate derivative (Scheme 1). The synthetic scheme of ethyl ester 35 has been reported by Heck43 and Vederas45. We used D-mannitol as the starting material, which was treated with benzaldehyde for regioselective protection of the 1,3- and 4,6-hydroxyl groups to afford the dibenzylidene mannitol derivative 31. The remaining two hydroxyl groups underwent alkylation reactions with octyl bromide to produce compound 32. The benzylidene groups of compound 32 were removed by hydrogenolysis in the presence of palladium hydroxide to afford diol compound 33. On treatment of 33 with sodium periodate, the resulting aldehyde intermediate was further oxidized by sodium chlorite to give an acid compound 34. This reaction was conducted in the presence of 2-methyl-2-butene to trap the evolving
chlorine gas. Acid compound 34 underwent a Fischer esterification to produce ethyl ester 35. We then followed the same synthetic scheme that was previously developed in our lab
to afford phosphate compound 37.30 Accordingly, compound 35 was reacted with 1H- tetrazole and dibenzyl (N,N-diisopropyl)phosphoramidite at 0 oC to form a phosphite intermediate, which was oxidized by mCPBA in situ to compound 36 bearing a moiety of pentavalent phosphonate diester. After debenzylation by hydrogenolysis using pallidum on charcoal as the catalyst, the desired phosphate compound 37 was obtained.
Scheme 1. Synthesis of phosphoglycerate derivative 37
2.2.2 Synthesis of biphenyl esters
To prepare the biphenyl compounds, 1-bromo-3,5-dimethoxylbenzene was treated with n-butyllithium and trimethyl borate to produce a boronic acid compound 39 (Eq.
1).46
The boronic acid compound 39 was then treated with commercially available 4- bromo-3,5-dimethoxy benzoic acid under the catalysis of palladium acetate in DMF (Eq.
2). Unfortunately, the yield of this reaction was low. It was suggested that the carboxylate group of 4-bromo-3,5-dimethoxy benzoic acid may react with potassium carbonate and the amount of potassium carbonate was insufficient for metathesis in the Suzuki coupling reaction (Eq. 2).
To improve yield, 4-bromo-3,5-dimethoxy benzoic acid was subjected to esterification in acidic condition to give methyl ester 42 (Scheme 2). Compounds 39 and 42 proceeded with Suzuki coupling reactions to produce tetramethoxy biphenyl
compound 43 in an excellent yield (99%).
Scheme 2. Synthesis of tetramethoxy biphenyl compound 43
In order to introduce benzyloxy substituents on the biphenyl moieties, the tetramethoxy substituents in the biphenyl compound 43 was converted to hydroxyl groups with boron tribromide, giving methyl ester 44 in 80% yield (Scheme 3). The hydroxyl
groups in ester 44 were then deprotonated by sodium hydride and underwent SN2 reaction with benzyl bromide to produce tetrakis(benzyloxy) compound 45.
Scheme 3. Synthesis of tetrabenzyloxy compound 45
2.2.3 Synthesis of 4-arylbenzamide derivatives
Ester 43 was treated with 3-amino-1-propanol (1.3 equiv) to afford amide 46 in 49 % yield by the promotion of lithium bromide at room temperature. Alternatively, compound 43 was reacted with excess 3-amino-1-propanol (20 equiv) at 160 oC to produce arylbenzamide compound 46 in 89% yield (Eq. 3).
By a similar procedure, the tetrakis(benzyloxyl) compound 45 was converted to amide 47 in 90% yield (Eq. 4).
2.2.4 Synthesis of 4-arylbenzylamino derivatives
Ester compounds 43 and 45 were reduced by diisobutylaluminium hydride to give alcohol compounds 48 and 49, which were subsequently oxidized by pyridinium dichromate to afford aldehydes 50 and 51, respectively (Scheme 4).
Scheme 4. Synthesis of aldehyde compounds 50 and 51
The reductive amination reaction of aldehyde 50 with 3-amino-1-propanol was achieved by the procedure previously performed in our lab31 using sodium triacetoxyborohydride as the reducing agent to produce an excellent yield of amine compound 52, which was then protected as the Fmoc derivative 53 by acylation with 9- fluorenylmethoxycarbonyl chloride (Scheme 5).
Scheme 5. Synthesis of Fmoc derivative 53
Surprisingly, the reductive amination of aldehyde 51 did not work under the similar reaction conditions (Table 8, entry 1). The reaction might be impeded by the steric effect of the benzyloxy substituents. By elevation of the reaction temperature to reflux, the desired product was only detected in trace amount (entry 2). By using NaBH4 in the presence of concentrated hydrogen chloride, aldehyde 51 underwent reductive amination to give 57 % of the product 54 (entry 3). Using concentrated sulfuric acid, a stronger acid than HCl, resulted in a lower yield (35 %, entry 4). In entry 5, addition of magnesium sulfate as a dehydration agent in combination with NaBH4 and H2SO4 did not improve the yield. Using anhydrous methanol that was presumably to stabilize the imine intermediate still not obviously increased the yield (entry 6). In entry 7, the solvent was changed back to 1,2-dichloroethane and acetic acid was added to maintain weak acidic environment suitable for imine formation. The yield of 54 was appreciably increased to
71%.
Table 8. Synthesis of amine compound 54
Entry Reducing agent
Additive Solvent Temp
(oC)
Time (h)
Yield
1 NaBH(OAc)3 none ClCH2CH2Cl rt 28 N.R.a
2 NaBH(OAc)3 none ClCH2CH2Cl Reflux 24 Trace
3 NaBH4 conc. HCl THF rt 46 57%
4 NaBH4 conc. H2SO4 THF rt 48 35%
5 NaBH4 conc. H2SO4, MgSO4
THF rt 5 37%
6 NaBH4 conc. H2SO4, anhydrous
MeOH
THF rt 20 38%
7 NaBH(OAc)3 CH3COOH ClCH2CH2Cl rt 3 71%
a. N.R. is no reaction
The amine compound 54 was then treated with 9-fluorenylmethoxycarbonyl chloride to afford Fmoc protected compound 55 (Eq. 5).
2.2.5 Coupling of phosphoglycerate with 4-arylbenzamide derivative
The hydroxyl group of compounds 46 and 47 were activated by trichloroacetonitrile and reacted with the phosphate compound 37 to produce the desired phosphate esters 56 and 57, respectively (Scheme 6). The ester moiety in compounds 56 and 57 were further hydrolyzed to carboxylic acid 58 and 59, respectively, to mimic the phosphoglycerate lipid part of moenomycin.
Scheme 6. Synthesis of acid compounds 58 and 59
On the other hand, the tetrakis(benzyloxy) compound 57 underwent hydrogenolysis by using the catalyst of palladium on charcoal to produce tetrahydroxyl compound 60 (Scheme 7). Finally, compound 60 was hydrolyzed by sodium hydroxide to afford acid compound 61.
Scheme 7. Synthesis of acid compound 61
2.2.6 Coupling of phosphoglycerate with 4-arylbenzylamino derivative
The hydroxyl group of 4-arylbenzylamino compound 53 and 55 was also activated by trichloroacetonitrile and reacted with phosphate compound 37 to produce phosphate esters 62 and 63, respectively (Scheme 8).
Scheme 8. Synthesis of acid compounds 66 and 67
Piperidine was used to remove the Fmoc protecting group in 62 and 63, giving 64 and 65 and the subsequent saponification yielded the acid compounds 66 and 67.
Additionally, compounds 65 and 67 were further hydrogenized by pallidum on charcoal and pallidum hydroxide to afford tetrahydroxyl 68 and 69 (eq.6).
2.2.7 Synthesis of pyrophosphate derivatives
The phosphate monoester 72 was prepared by Wong, Y-C. in our lab (Scheme 9). The acid compound 34 underwent amide bond formation by the assistance of 1-ethyl-3-(3- dimethylaminopropyl)carbodiimide (EDCI) and hydroxybenzotriazole (HOBt) to produce amide 70. Then the amide compound 70 was reacted with 1H-tetrazole and dibenzyl (N,N-diisopropyl)phosphoramidite at 0 oC to form a phosphite intermediate, which was oxidized by mCPBA in situ to compound 71. Debenzylation of 71 by using the catalyst of palladium on charcoal afforded phosphate monoester 72.