國立臺灣大學獸醫專業學院獸醫學研究所 博士論文
Graduate Institute of Veterinary Medicine School of Veterinary Medicine
National Taiwan University Doctoral Dissertation
臺灣禽類副黏液病毒分離、鑑定與親緣分析
Isolation, Identification, and Phylogenetic Analyses of Avian Paramyxoviruses in Taiwan
劉玉彬 Yu-Pin Liu
指導教授:蔡向榮 博士
Advisor: Hsiang-Jung Tsai, D.V.M., Ph.D.
中華民國 110 年 1 月
January, 2021
誌謝
在這些年博士班研究生涯中,最由衷感謝指導教授蔡向榮老師在學術研究及 論文寫作的悉心指導與鼓勵,學生永銘於心。感謝周崇熙老師、連一洋老師、張 伯俊老師、張紹光老師、鄭明珠老師與謝明昆老師在學生口試提供寶貴建議與細 心審查斧正,使本篇論文能夠更臻完善,在此致上誠摯的謝忱。
感謝在禽場及野鳥濕地辛苦採樣的防治所及鳥會同仁,使得本論文的調查可 以更具體呈現臺灣禽類副黏液病毒的演化及遺傳多樣性;也萬分感謝禽病診斷實 驗室的夥伴們,在禽流感疫情爆發的這幾年中患難與共,同甘共苦的一起走過這 段艱辛歲月;對於發表文章提供鉅細靡遺之建議與釜正的李璠組長也致以最真 摯、最深的謝意。
最後感謝最心愛的家人,感謝你們的支持、陪伴與鼓勵,讓我能夠完成博士 學位,在我的心中,你們永遠是最重要的。
劉玉彬 謹誌
中文摘要
禽類副黏液病毒為 Paramyxoviridae 科的 Avulavirinae 亞科下病毒群,目前已 知共 22 型,且廣泛分布於世界眾多鳥種中。禽類副黏液病毒一型(亦稱為新城病 病毒)為家禽新城病的致病原,已廣佈於全球,能夠感染兩百種以上的鳥類。目 前已有許多即時反轉錄聚合酶反應針對新城病病毒不同標的基因偵測被研發出,
但檢測的敏感性與效力仍待改進。在本研究中,於 2009 年至 2020 年期間由臺灣 家禽、候鳥、輸入鳥類及本地留鳥檢體中分離禽類副黏液病毒,並進行病毒基因 體或融合蛋白基因全長定序及分析。同時建立核鞘蛋白即時反轉錄聚合酶反應技 術以檢測具高度變異性的新城病病毒核酸。本研究共分離 100 株禽類副黏液病毒 株,分屬於一型(52 株)、二型(1 株)、四型(20 株)、六型(7 株)、十二型(2
株)、二十一型(2 株)以及二十二型(16 株)等 7 型。核酸序列分析顯示除了 10
株第一類禽類副黏液病毒一型及 2 株四型病毒屬於北美株外,其餘所有病毒均歸 屬於歐亞株。禽類副黏液病毒一型的融合蛋白親緣分析顯示 52 株分離株中,50 株 屬於第一類的第一基因型或第二類的第一、六及七基因型病毒、1 株為第一類新增 的第二基因型及 1 株為無法分類之特異病毒;臺灣近十年的鴿子副黏液病毒流行 株為第二類第六基因型的 2.1.1.2.1 與 2.1.1.2.2 亞型;造成臺灣家禽新城病疫情的 病毒分離株均歸屬於第七基因型的 1.1 亞型。本研究在 2009 年的禽流感主動監測
檢體中分離到一株特殊的副黏液病毒株(APMV/dove/Taiwan/AHRI33/2009),經基
因體核酸序列比對,AHRI33 相近於第七型禽類副黏液病毒,但相似度僅 62.8%。
依據 RNA 依賴性 RNA 聚合酶基因親緣性分析,AHRI33 病毒株應新成立為禽類副 黏液病毒二十二型。本研究建立之核鞘蛋白即時反轉錄聚合酶反應檢測技術在不 同型別新城病病毒株的測試下,全部 23 株均成功檢出,而經美國農業部認證之基 質蛋白即時反轉錄聚合酶反應則未能檢出 8 株第一類及 2 株鴿子副黏液病毒株;
新檢測方法對於 35 種非新城病病毒均無陽性反應,顯示高度之特異性;實際應用
於臨床檢體時,與黃金標準的病毒分離檢測結果比對,其敏感性與特異性分別為 100%與 96.61%。綜合上述,本篇研究證實臺灣禽鳥至少帶有 7 種不同型別之禽類 副黏液病毒,而候鳥對於歐亞及北美間的跨洲際病毒傳播扮演重要之角色。
AHRI33 病毒應於 Avulavirinae 亞科 Metaavulavirus 屬中設立為 Avian metaavulavirus 22 新病毒種;臺灣家禽與野鳥至少帶有 5 種不同基因型之新城病病毒;其中
AHRI67 病毒株應於第一類下新成立第二基因型;而新建立的核鞘蛋白即時反轉錄 聚合酶反應技術具高敏感性、特異性,可檢出高度演化變異的新城病病毒。總結 本篇研究結果可更深入瞭解臺灣禽類副黏液病毒之天然宿主、演化及遺傳多樣性。
關鍵詞:
禽類副黏液病毒、新城病、融合蛋白、跨洲際傳播、即時反轉錄聚合酶反應
Abstract
Avian paramyxoviruses (APMVs) belonging to the subfamily Avulavirinae within the family Paramyxoviridae consist of twenty-two known species, and are constantly isolated from a wide variety of avian species around the world. Avian paramyxovirus 1, synonymous with Newcastle disease virus (NDV), is a worldwide viral agent that infects over 200 species of birds and responsible for outbreaks of Newcastle disease.
Although a number of real-time reverse transcription polymerase chain reaction
(rRT-PCR) assays have been developed for detecting different genes of NDV, diagnostic sensitivity and efficiency could be improved. In this study, the APMV isolates collected from poultry, migratory, imported and resident birds during 2009-2020 in Taiwan were genetically characterized by sequence analysis of the complete fusion protein gene or full-length genome. Furthermore, this study describes a nucleocapsid protein gene rRT-PCR screening assay based on TaqMan technology for the detection of divergent NDV strains and isolates. One hundred isolates belonging to seven species were identified as APMV-1 (n=52), APMV-2 (n=1), APMV-4 (n=20), APMV-6 (n=7), APMV-12 (n=2), APMV-21 (n=2) and APMV-22 (n=16). Genetic studies showed that the recovered APMVs isolates had highest homology with Eurasian isolates, except ten class I APMV-1 and two APMV-4 isolates related to North America strains. Our
phylogenetic analysis of complete fusion protein gene of the APMV-1 isolates revealed that 50 of the 52 Taiwanese isolates were closely related to APMV-1 of class I genotype 1 or class II genotypes I, VI or VII, one isolate belonged to a group that can be
classified as a novel genotype 2 within class I, and one isolate was grouped within class I viruses but formed a monophyletic lineage, with a genetic distance of 5.6% between them. Viruses placed in class II sub-genotype VI.2.1.1.2.1 and sub-genotype
VI.2.1.1.2.2 were the dominant pigeon paramyxovirus 1 circulating in the last decade in Taiwan. All the Newcastle disease outbreak-associated isolates belonged to class II sub-genotype VII.1.1, which was mainly responsible for the present epizootic of Newcastle disease in Taiwan. In 2009, the isolate APMV/dove/Taiwan/AHRI33/2009 was isolated from swabs of red collared doves during active surveillance of avian influenza in resident birds in Taiwan. Nucleotide sequence comparisons of the genome between each prototype of APMVs had shown AHRI33 to be more closely related to APMV-7 than to the others, with a sequence identity of 62.8%. Based on topology of the phylogenetic tree of RdRp genes and the branch length between the nearest node and the tip of the branch, AHRI33 met the criteria for designation as distinct species.
Using the NP-gene rRT-PCR assay, all 23 representative NDV strains of class I and II in the tested panel were detected, whereas eight class I and two class II NDV isolates cannot be detected by the USDA-validated matrix-gene assay. The new assay also has a high degree of specificity with no false-positive results of 35 non–NDV viruses. A total of 146 clinical specimens were also tested and the NP-gene assay gave high relative sensitivity (100%) and specificity (96.61%) when compared with virus isolation. We conclude that at least seven species of APMVs were obtained from multiple avian host species in Taiwan. Migratory birds may play an important role in intercontinental spread of APMVs between Eurasia and North America. The data suggest that the isolate
APMV/dove/Taiwan/AHRI33/2009 should be considered as the prototype strain of the new species Avian metaavulavirus 22 in the genus Metaavulavirus in the subfamily Avulavirinae. The current data confirm that at least five genotypes of APMV-1 circulate
in both domestic and wild birds throughout Taiwan. One genetically divergent group of APMV-1 should be considered as a novel genotype within class I. Furthermore, the developed NP-gene rRT-PCR assay offers a sensitive, specific and rapid assay for
detecting both class I and II NDV and could be used as part of a panel of diagnostic assays for this notifiable disease agent. Together, this study contributes to the
knowledge of the distribution, evolution, and genetic diversity of APMVs in Taiwan.
Keywords:
avian paramyxovirus; Newcastle disease; fusion protein; intercontinental dispersal;
real-time RT-PCR
Contents
口試委員審定書 --- i
誌謝 --- ii
中文摘要 --- iii
Abstract --- v
Contents --- viii
List of Figures --- xii
List of Tables --- xiii
Chapter 1 Introduction --- 1
1.1 Background of avian paramyxoviruses --- 1
1.2 Research aims --- 2
1.3 The layout and format of this dissertation --- 2
Chapter 2 Literature review --- 3
2.1 Avian paramyxovirus --- 3
2.1.1 Taxonomy of APMV --- 3
2.1.2 Virus characteristics --- 3
2.1.3 Members of APMV --- 4
2.1.4 Natural host of APMV --- 5
2.1.5 Pathogenicity of APMV --- 6
2.1.6 Classification system of APMV --- 7
2.2 Newcastle disease virus --- 8
2.2.1 Classification of APMV-1 --- 8
2.2.2 Host, virulence, and genetic diversity of APMV-1 --- 9
2.2.3 Molecular basis for pathogenicity --- 11
2.2.4 Detection of APMV-1 by real-time RT-PCR assay --- 12
2.3 Intercontinental dispersal of avian pathogens by migratory birds --- 13
2.4 APMV study in Taiwan --- 13
Chapter 3 Novel avian metaavulavirus isolated from birds of the family Columbidae in Taiwan --- 15
3.1 Introduction --- 15
3.2 Materials and methods --- 16
3.2.1 Sample collection and virus isolation --- 16
3.2.2 Electron microscopy --- 17
3.2.3 Determination of nucleotide sequence of full-length APMV genome -- 17
3.2.4 Intracerebral pathogenicity index --- 18
3.2.5 Serological characterization --- 18
3.2.6 Phylogenetic analysis --- 19
3.2.7 Nucleotide sequence accession number --- 20
3.3 Results --- 20
3.3.1 Sample collection and virus isolation --- 20
3.3.2 Electron microscopy --- 21
3.3.3 Intracerebral pathogenicity index --- 21
3.3.4 Serological characterization --- 21
3.3.5 Determination of nucleotide sequences of full-length APMV genome - 21 3.3.6 Sequence comparison and phylogenetic analysis --- 22
3.4 Discussion --- 23
Chapter 4 Phylogenetic analysis of avian paramyxoviruses 1 isolated in Taiwan from 2010 to 2018 and evidence for their intercontinental dispersal by migratory bird --- 40
4.1 Introduction --- 40
4.2 Materials and methods --- 42
4.2.1 Sample collection and virus isolation --- 42
4.2.2 RNA extraction and reverse transcription-polymerase chain reaction -- 43
4.2.3 Nucleotide sequencing of fusion protein gene and full-length genome - 43 4.2.4 Phylogenetic analysis --- 44
4.3 Results --- 45
4.3.1 Sample collection and virus isolation --- 45
4.3.2 GenBank accession numbers --- 45
4.3.3 Genetic analysis of class I APMV-1 --- 46
4.3.4 Genetic analysis of class II APMV-1 --- 46
4.4 Discussion --- 48 Chapter 5 A highly sensitive real-time reverse transcription polymerase chain
reaction for detecting nucleocapsid protein gene of both classes I and
II of Newcastle disease virus --- 63
5.1 Introduction --- 63
5.2 Materials and methods --- 64
5.2.1 Probe and primers design --- 64
5.2.2 Virus isolates and characterization --- 65
5.2.3 RNA extraction and rRT-PCR --- 66
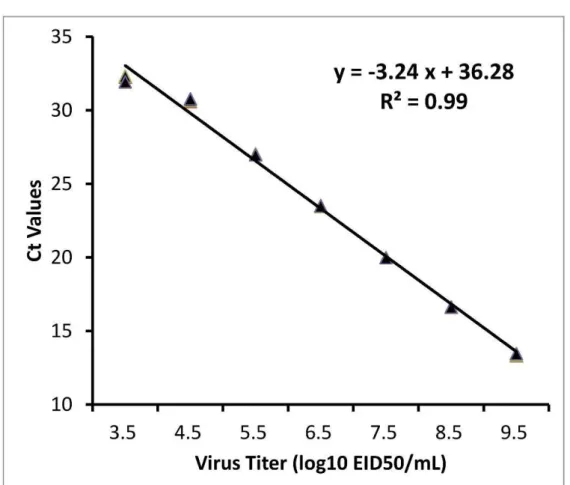
5.2.4 Limit of detection --- 66
5.2.5 Specificity testing --- 67
5.2.6 Comparison with virus isolation in clinical specimens --- 67
5.3 Results --- 68
5.3.1 Probe and primers design --- 68
5.3.2 Virus isolates and characterization --- 68
5.3.3 Limit of detection --- 68
5.3.4 Specificity testing --- 69
5.3.5 Comparison with virus isolation in clinical specimens --- 69
5.4 Discussion --- 69
Chapter 6 Genetic diversity of avian paramyxoviruses isolated from wild birds and domestic poultry in Taiwan between 2009 and 2020 --- 77
6.1 Introduction --- 77
6.2 Materials and methods --- 79
6.2.1 Sample collection and virus isolation --- 79
6.2.2 RNA extraction and seminested reverse transcription-polymerase chain reaction --- 79
6.2.3 Nucleotide sequencing of fusion protein gene and full-length genome - 80 6.2.4 Phylogenetic analysis --- 81
6.3 Results --- 81
6.3.1 Sample collection and virus isolation --- 81
6.3.2 Genetic analysis of APMV-1 --- 82
6.3.3 Genetic analysis of APMV-2 --- 83
6.3.4 Genetic analysis of APMV-4 --- 84
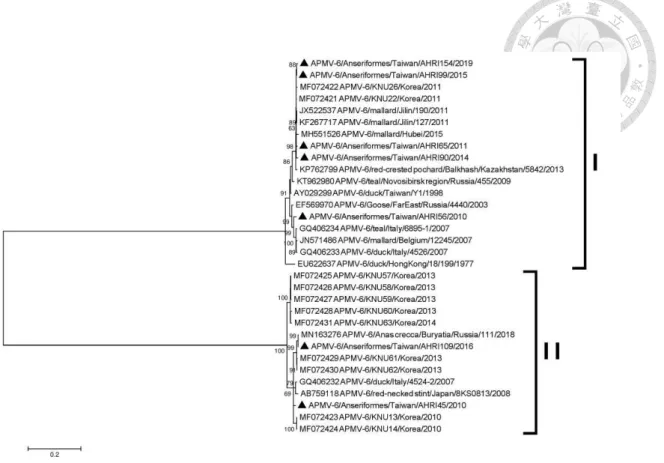
6.3.5 Genetic analysis of APMV-6 --- 84
6.3.6 Genetic analysis of APMV-12 --- 85
6.3.7 Genetic analysis of APMV-21 --- 86
6.3.8 Genetic analysis of putative APMV-22 --- 86
6.4 Discussion --- 87
Chapter 7 Conclusions --- 106
References --- 110
Original publications --- 123
List of Figures
Figure 3.1 Paramyxovirus virion found in allantoic fluid of embryonated chicken egg inoculated with swab sample extract from a red collared dove in
Taiwan --- 30 Figure 3.2 Schematic diagram of avian paramyxovirus AHRI33 isolate and
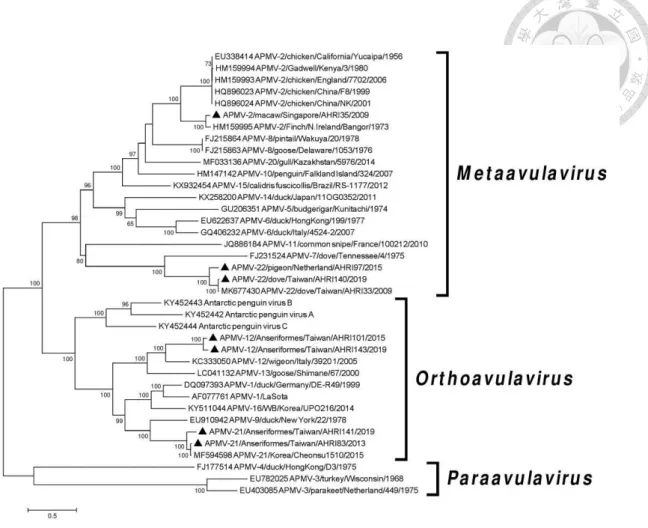
APMV-7 genome --- 31 Figure 3.3 Phylogenetic tree of the avian paramyxoviruses, based on comparison
of their full-length genomes --- 32 Figure 3.4 Phylogenetic tree of the fusion gene --- 34 Figure 3.5 Phylogenetic tree of the hemagglutinin-neuraminidase gene --- 36 Figure 3.6 Maximum Likelihood phylogenetic tree of the amino acid sequences
of RdRp gene --- 38 Figure 4.1 Phylogenetic tree based on the complete fusion protein gene
sequences of isolates of avian paramyxovirus 1 class I --- 55 Figure 4.2 Phylogenetic tree based on the complete fusion protein gene
sequences of isolates of avian paramyxovirus 1 class II genotype I --- 57 Figure 4.3 Phylogenetic tree based on the complete fusion protein gene
sequences of isolates of avian paramyxovirus 1 class II genotype VI - 59 Figure 4.4 Phylogenetic tree based on the complete fusion protein gene
sequences of isolates of avian paramyxovirus 1 class II genotype VII 61 Figure 5.1 Limit of detection of the nucleocapsid protein gene rRT-PCR assays. - 76 Figure 6.1 Phylogenetic tree based on the complete fusion protein gene
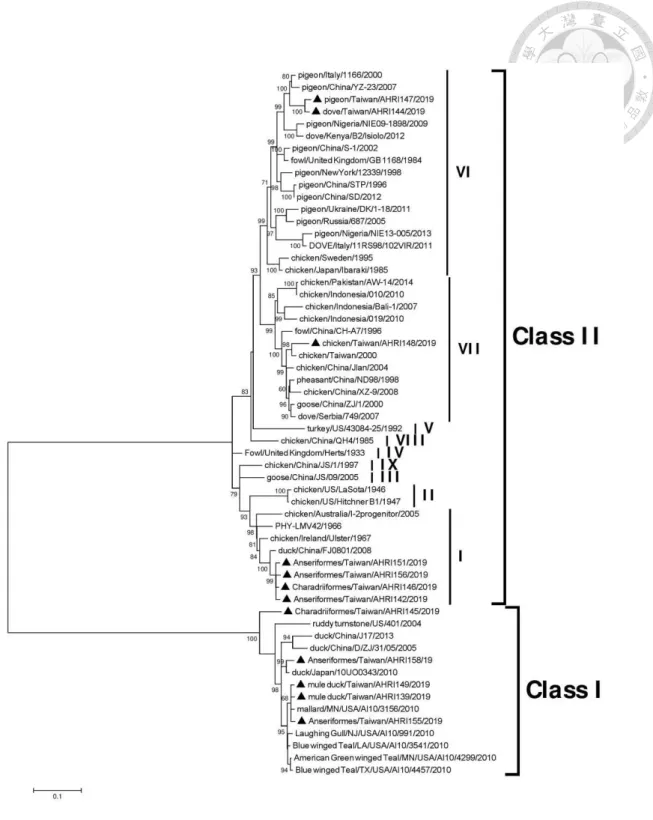
sequences of isolates of the avian paramyxovirus 1 --- 100 Figure 6.2 Phylogenetic tree based on the complete fusion protein gene
sequences of isolates of the avian paramyxoviruses --- 102 Figure 6.3 Phylogenetic tree based on the complete fusion protein gene
sequences of isolates of avian paramyxovirus 4 --- 103 Figure 6.4 Phylogenetic tree based on the complete fusion protein gene
sequences of isolates of avian paramyxovirus 6 --- 105
List of Tables
Table 3.1 Antigenicity between newly isolated AHRI33 virus and representative avian paramyxoviruses, measured by cross-hemagglutination
inhibition tests --- 27
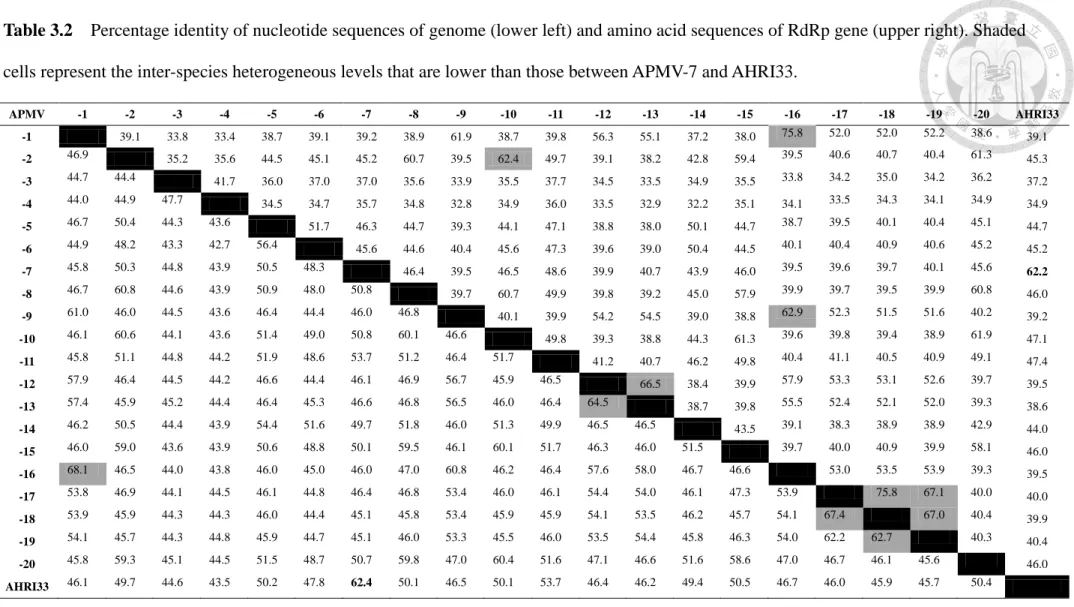
Table 3.2 Percentage identity of nucleotide sequences of genome and amino acid sequences of RdRp gene --- 28
Table 3.3 Percentage nucleotide and deduced amino acid sequence identities between the AHRI33 isolate and avian paramyxoviruses representing the species in the subfamily Avulavirinae --- 29
Table 4.1 List of RT-PCR primers --- 52
Table 4.2 Isolate details --- 53
Table 5.1 Nucleotide sequences of the primers and probe used in the NP-gene rRT-PCR and fusion gene RT-PCR assay --- 73
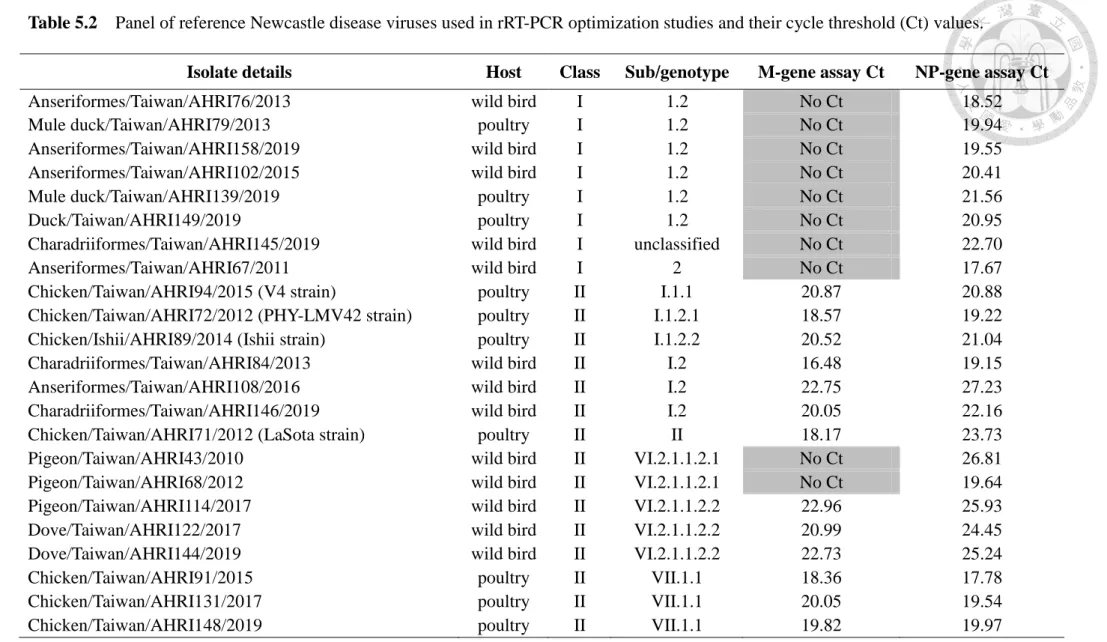
Table 5.2 Panel of reference Newcastle disease viruses used in rRT-PCR optimization studies and their cycle threshold values--- 74
Table 5.3 Two-by-two tables comparing the relative sensitivity and specificity of NP-gene rRT-PCR assay with gold standard --- 75
Table 6.1 List of RT-PCR primers --- 94
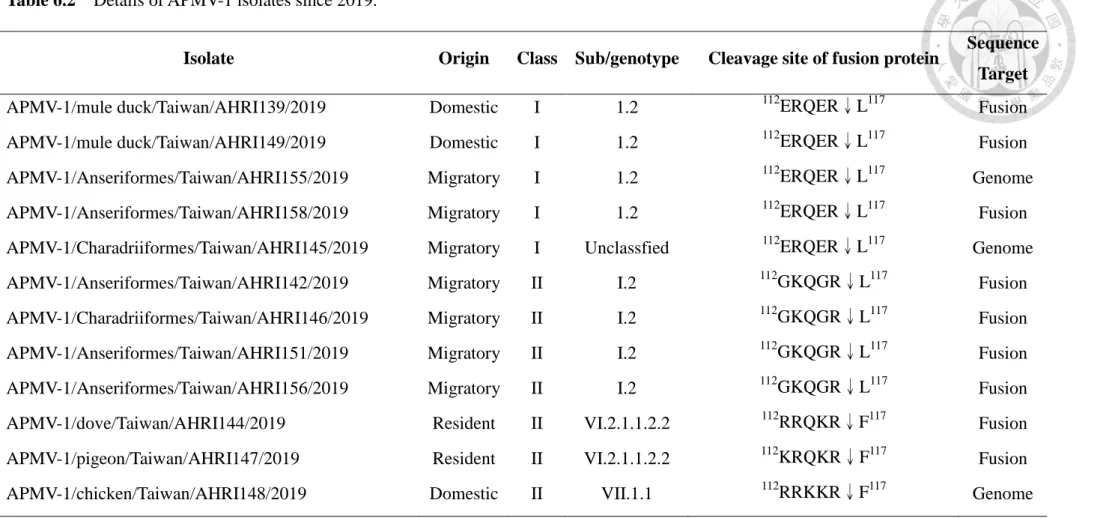
Table 6.2 Details of APMV-1 isolates since 2019 --- 97
Table 6.3. Details of non-APMV-1 isolates during 2009-2020 --- 98
Chapter 1 Introduction
1.1 Background of avian paramyxoviruses
Avian paramyxoviruses (APMVs) are one of the most important viruses and have been reported from a wide variety of avian species around the world (Gogoi et al., 2017).
To date, 21 different species of APMVs (APMV-1 to 21) have been reported and their complete genome sequences are available in GenBank. Avian paramyxoviruses belong to the subfamily Avulavirinae within the family Paramyxoviridae. APMV-1 (commonly termed Newcastle disease virus, NDV) is the most recognized species, but information on the distributions of other APMVs in domestic poultry and wild birds is limited.
These findings suggest that wild birds and domestic poultry might harbor previously unrecognized genetic diversity of APMVs, and the full extent of the distribution, evolution, and host species of APMVs has remained unexplored.
Newcastle disease (ND) is one of the most important diseases of poultry and caused by virulent strains of avian paramyxovirus 1 (APMV-1), also known as
Newcastle disease virus (NDV). Newcastle disease is an OIE (World Organization for Animal Health) notifiable disease that needs implementing control measures and trading restrictions to prevent the spread of the disease (OIE, 2012). Based on its genetic
characteristics, the APMV-1 was re-assigned into the new genus Orthoavulavirus within a new subfamily Avulavirinae of the family Paramyxoviridae by the International Committee on Taxonomy of Viruses in 2019 (Amarasinghe et al., 2019). The virus has high genetic diversity and the infection of NDV has been reported in a wide variety of avian species around the world (Dimitrov et al., 2016).
1.2 Research aims
In the present study, the APMVs isolates obtained from migratory birds and poultry in Taiwan were characterized by sequencing of complete fusion protein gene or
full-length genome sequences and were compared to those available in GenBank. Based on the results of the phylogenetic analyses, we aim to i) illustrate the genetic diversity of APMVs in various avian hosts and present new epidemiological information on APMVs in Taiwan; ii) present new information on the genetic evolution and
sub/genotype classification of NDV in Taiwan; iii) provide evidence for the potential intercontinental transmission of APMVs by migratory birds; iv) develop a rapid, sensitive, and reliable TaqMan rRT-PCR assay that could be used for detecting the NP gene from both class I and II NDV.
1.3 The layout and format of this dissertation
The dissertation comprises a series of studies that address different aspects of the evolution, current distribution and detection of different APMV species isolated from domestic poultry and wild birds in Taiwan. All the results chapters shown in (3 to 6) have already been published (3, 4 and 5) or have been prepared to submit to
international journals. These result chapters contain the same text and structure as published or submitted to the respective journals. The formatting and referencing have been altered to form a standard style for all chapters. In the final chapter, a summary and discussion of all findings are presented.
Chapter 2
Literature review
2.1 Avian paramyxovirus 2.1.1 Taxonomy of APMV
According to the taxonomy of the order Mononegavirales: updated in 2018, three genera, named Orthoavulavirus, Metaavulavirus, and Paraavulavirus has recently been created within a new subfamily Avulavirinae of the family Paramyxoviridae
(Amarasinghe et al., 2019). Avian paramyxoviruses (APMVs) belong to three genera, Metaavulavirus (APMV-2, -5, -6, -7, -8, -10, -11, -14, -15 and -20), Orthoavulavirus
(APMV-1, -9, -12, -13, -16, APV-A, APV-B, APV-C and -21) and Paraavulavirus (APMV-3 and -4). The subfamily Avulavirinae comprises of all APMVs that have been isolated from avian species.
2.1.2 Virus characteristics
The APMVs contain a non-segmented, negative-sense, single-stranded RNA genome that ranges from 14,904 to 17,412 nucleotides (nt) in length (Aziz-ul-Rahman et al., 2018). For most of the APMVs, the genome encodes at least six proteins: the nucleocapsid protein (NP), phosphoprotein (P), matrix protein (M), fusion protein (F), hemagglutinin-neuraminidase (HN), and large polymerase (L) except for APMV-6 that had an additional short hydrophobic (SH) region between F and HN genes (Chang et al., 2001). The order of the genes for these proteins in the genome is 3′
leader-N-P-M-F-HN-L-5′ trailer (Anderson andWang, 2011). The viral envelope is derived from the host cell membrane. The outer surface of the envelope consists of two
glycoproteins, namely F and HN proteins. The HN protein mediates attachment by binding to the sialic acid receptor, has neuraminidase activity and plays a role in fusion promotion where as F protein directs the membrane fusion. The F and HN proteins play a defined role in the fusion, attachment and release from the cells. The cleavage site of the F protein acts as key determinant of virulence and existence of
phenylalanine/leucine residue assists in classification of virulent and avirulent strains (Alexander, 2000). M protein lies below the envelope and is shown to play a major role in virus assembly and budding of NDV. The viral envelope encloses the
ribonucleocapsid core that is formed by the nucleoprotein, the phosphoprotein and large polymerase protein bound with viral genomic RNA (Anderson andWang, 2011). Viral transcription begins at single promoter at the 3′ leader end, and the genes are copied into individual mRNAs by a start-stop-restart mechanism guided by conserved gene-start (GS) and gene-end (GE) transcription signals that flank the individual genes. For the APMVs and other members of the subfamily Paramyxovirinae, the genome nucleotide length is an even multiple of six, known as ‘rule of six’ which is required for efficient RNA replication and the precise packaging of the polynucleotide in the nucleocapsid (Kolakofsky et al., 1998).
2.1.3 Members of APMV
Until recently, the known APMV species were restricted to APMV-1 to -9, which were isolated and characterized based on haemagglutination inhibition (HI) and neuraminidase inhibition (NI) assays in the 1970s (Alexander, 1987). The prototype virus, APMV-1 of genus Orthoavulavirus, also well known as Newcastle disease virus or NDV, is the most extensively studied virus in this group. NDV is a geographically widespread viral agent that infects wild and domestic avian hosts. Virulent strains of
APMV-1 have significant global economic impacts on domestic poultry production and are notifiable to the World Organization for Animal Health (OIE). APMV-2 to -9 had been isolated from chicken, ducks, turkeys, and wild birds all over the world prior to the 1980s (Alexander, 2000). Following the expansion of viral surveillance initiatives and improvements in sequencing technology, 12 novel species have been designated since 2010. Many of these novel APMVs were isolated as a result of influenza virus
surveillance programs. A virus isolated from rockhopper penguins was antigenically and genetically distinct from all known APMV-1 to -9 and considered to represent the prototype of a new species, APMV-10 (Miller et al., 2010). A further novel APMV-11 isolate was obtained in France from a common snipe in 2010 (Briand et al., 2012).
APMV-12 was isolated in Northern Italy in 2005 from an Eurasian widgeon (Terregino et al., 2013). In 2015 and 2016, three publications described a novel APMV-13 species found in three separate regions of Eurasia: Japan, Kazakhstan and Ukraine (Yamamoto et al., 2015; Karamendin et al., 2016; Goraichuk et al., 2016). In 2017, seven novel APMV species were announced: APMV-14 from ducks in Japan (Thampaisarn et al., 2017), APMV-15 from shorebirds in Brazil (Thomazelli et al., 2017), and APMV-16 from ducks in Korea (Lee et al., 2017). Three novel species, Antarctic penguin virus A, B, and C (APV-A, -B, and -C), were simultaneously isolated from Antarctic penguins (Neira et al., 2017), and APMV-20 from gulls in Kazakhstan (Karamendin et al., 2017).
In 2018, one novel APMV species, APMV-21, was isolated from wild birds in South Korea (Jeong et al., 2018). These findings suggest that wild birds maintain a previously unrecognized genetic diversity of APMVs. Till date, 21 species of APMVs (APMV-1 to 21) are reported and their complete genome sequences are available in GenBank.
2.1.4 Natural host of APMV
APMV-1 (commonly termed Newcastle disease virus, NDV) has a wide host range and is able to infect over 240 species of birds (Kaleta andBaldauf, 1988). In contrast, the information about the host ranges and the distribution of the other APMVs species in wild birds and domestic poultry is limited. The natural hosts for APMV-2 are turkeys, psittacines, and passerines, but chickens and rails act as incidental host (Alexander, 2000; Zhang et al., 2007). Apart from prototype strain Yucaipa, APMV-2 strains Bangor, England and Kenya are also characterized. APMV-2 strains Bangor, England and Kenya were isolated from a finch, chicken and gadwall duck, respectively. Apparently during a routine quarantine evaluation, APMV-2 strain Bangor was isolated and on its
characterization, both biologically and serologically, it was observed that it might depict a different serotype or as a subgroup within serotype (Subbiah et al., 2010). APMV-3 and -7 have been isolated from captive caged birds and domestic poultry including chickens and turkeys; APMV-4 have mostly been isolated from waterfowl belonging to the order Anseriformes; APMV-5 have been isolated mostly from budgerigars; APMV-6 was first isolated from a healthy duck at a domestic duck farm in the year 1977 from Hong Kong; APMV-12 was first isolated from a cloacal swab of an infected Eurasian wigeon in Italy during the influenza surveillance programme in 2005 (Terregino et al., 2013) and was no other strains of APMV-12 have been reported till date; and the other APMVs appear to be more restricted to wild birds including ducks, geese, gulls and penguins, but very little is known regarding the disease potential and the epidemiology of these APMVs (Aziz-ul-Rahman et al., 2018).
2.1.5 Pathogenicity of APMV
APMV-1 is the best characterized virus among other APMVs due to the severity of disease caused by this virus in chickens. APMV-1 is also historically known as
Newcastle disease virus (NDV) because of its origin from the seaport town of
Newcastle upon Tyne in England. APMV-1 are categorized as lentogenic, mesogenic, and velogenic depending on clinical signs in chickens and the cleavage site amino acid sequence of fusion protein (Miller and Koch, 2013). Depending on its predilection site, velogenic NDV can be either viscerotropic or neurotropic (Alexander, 2000). Other APMVs are also associated with varying degrees of pathogenicity in chickens and other avian species. Most of these APMVs appear to be present in natural reservoirs of
specific feral avian species, although other host species are usually susceptible. Only APMV-2 and APMV-3 viruses have made a significant disease and economic impact on poultry production. Both types of viruses cause respiratory disease and egg production losses which may be severe when exacerbated by other infections or environmental stresses. No report demonstrated natural infections of chickens with APMV-3 viruses (Alexander, 2000). APMV-2 infection can cause a decreased yield and decreased hatchability in turkeys (Bankowski et al., 1981). APMV-3 was isolated from turkeys which are showing signs of nasal discharge, coughing and swelling of the infra-orbital sinus (Kumar et al., 2010). APMV-6 seems to be apathogenic to chickens but can cause mild respiratory disease and a drop in egg production in turkeys (Alexander, 2000). The pathogenicity and the economic significance of other APMVs are not well understood.
2.1.6 Classification system of APMV
APMVs have been placed in different groups, or serotypes, based on their
antigenic relation, and nine different groups were defined in this way (Alexander et al., 1983). However, there are multiple problems associated with the use of serology, including the inability to classify some APMVs by comparing them to the sera of the nine defined APMVs alone (Alexander et al., 1989). In addition, cross-reactivity
between different serotypes have been documented for many years (Terregino et al., 2013). Because of obstacles imposed by the cross-reactivity as mentioned above and the limitation of available reference antiserum against each APMV serotype, analysis of complete genome sequences should be conducted to identify new APMV serotypes as suggested previously (Miller et al., 2010). The analysis of complete genome nucleotide identity is a simple method for identification of novel APMV, and up to 25% divergence may be suggestive of novel virus or strain (Terregino et al., 2013). This is evident in pairwise comparison of whole genome sequences of all APMVs where a substantial divergence was observed. The divergence in percentage nucleotide identity among inter-APMV can also be a marker of novel strain and may highlight the evolutionary distance of APMV. In this context, the complete genomes of all APMVs showed a high nucleotide divergence except in APMV-1, -9, -12, -16, -17, -18 and -19, which revealed a high nucleotide identity (70.6%-88.4%) compared to rest of APMVs (Aziz-ul-Rahman et al., 2018). Based on a sequence comparison of the RNA-dependent RNA polymerases (RdRps) of the viruses, APMVs are divided into twenty species according to the
taxonomy of the order Mononegavirales updated in 2019 (ICTV, 2019). In the family Paramyxoviridae, the ICTV Study Group has decided that, since a number of other
criteria are no longer applicable or have been applied inconsistently (Rima et al., 2018), the classification of the viruses should now be based on a sequence comparison of the RdRp gene. The primary criterion is the tree topology however using such trees for classification may require defining taxon-specific cut-off criteria.
2.2 Newcastle disease virus 2.2.1 Classification of APMV-1
Newcastle disease virus was assigned into the new genus Orthoavulavirus within
the subfamily Avulavirinae of the family Paramyxoviridae by the International
Committee on Taxonomy of Viruses in 2018 (Amarasinghe et al., 2018). The complete F gene sequence is considered as the main target for molecular epidemiological
investigations and genotyping of APMV-1. Based on phylogenetic analyses of fusion protein gene, NDV has been divided into two clades, class I and class II (Czeglédi et al., 2006). An unified and objective classification system of APMV-1 was proposed in 2012 based on the coding sequences of the complete fusion protein gene (Diel et al., 2012), and this system and nomenclature criteria for APMV-1 were revised and updated by a global consortium in 2019 (Dimitrov et al., 2019). This system utilized the complete F gene coding sequences and incorporated a number of objective criteria for classification of NDV, including: i) New genotypes are created based on the phylogenetic tree
topology (need to cluster into monophyletic branches) using the Maximum Likelihood method and the general time-reversible (GTR) model with gamma distribution; ii) Different genotypes have an average distance per site above 10%; iii) The bootstrap value at the genotype and sub-genotype defining node is 70% or above (≥70%); and iv) New genotypes are created only when four or more independent isolates, without a direct epidemiologic link (i.e. distinct outbreaks), are available. The use of Dimitrov et al. (2019) system and criteria led to the classification of class I isolates belong to a single ‘genotype’ (genotype 1) whereas class II isolates are divided into 21 genotypes.
The classification system of APMV-1 then was applied to other species of APMVs to provide a more rational and scientific genotyping method for epidemiological studies (Yin et al., 2017; Chen et al., 2018; Tseren-Ochir et al., 2018).
2.2.2 Host, virulence, and genetic diversity of APMV-1
Almost all of the class I viruses are lentogenic strains and have been isolated
primarily from waterfowl of the family Anatidae worldwide and occasionally from poultry in live bird markets (Kim et al., 2007). Class II viruses are genetically and phenotypically more diverse, frequently isolated from poultry with occasional spillovers into wild birds, and exhibit a wider range of virulence. Waterfowl, cormorants, and pigeons are natural reservoirs of all APMV-1 pathotypes, except viscerotropic velogenic viruses for which natural reservoirs have not been identified. APMV-1 isolates of class II, genotype I consist of lentogenic strains and have been widely recovered from a diversity of wild and domestic waterfowl. Genotypes I and II within class II include isolates of high and low virulence, the latter often being used as vaccines. Viruses of genotypes III and IX that emerged decades ago are now isolated rarely, but may be found in domestic and wild birds in China. Containing only virulent viruses and responsible for the majority of recent outbreaks in poultry and wild birds, viruses from genotypes V, VI, and VII, are highly mobile and have been isolated on different
continents. A pigeon-adapted variant of genotype VI NDV, often termed pigeon paramyxovirus 1 (PPMV-1), is commonly isolated from columbids and can cause ND-like infectious disease in wild and domestic birds. Unlike any other NDV genotype, genotype VI viruses have been isolated in all continents, except Antarctica. Their
potential to infect chickens has been demonstrated, but only rarely, and they appear to be highly adapted to some Columbiform birds (Aldous et al., 2014). Genotype VII is another large and genetically diverse group of viruses. Class II genotype VII viruses might emerge in the Far East in the1990s and subsequently spread to Europe, South Africa, South America, and Asia, including Taiwan (Dimitrov et al., 2016). All genotype VII viruses are predicted to be virulent based on deduced amino acid motifs and many have been shown to be velogenic via pathotyping. Genotype VII viruses have been associated with recurrent poultry outbreaks in Eastern Europe, the Middle East, and
Asia and sporadic events in Africa and South America, making this large and diverse group of viruses of significant global economic importance (Miller et al., 2015).
Conversely, virulent viruses of genotypes XI (Madagascar), XIII (mainly Southwest Asia), XVI (North America) and XIV-XXI appear to have a more limited geographic distribution and have been isolated predominantly from poultry (Dimitrov et al., 2016).
2.2.3 Molecular basis for pathogenicity
Although all NDV isolates belong to a single serotype, there is great genetic variability among different strains. The cleavability of protein F is the main determinant for viral virulence, but other genes such as HN and P are also believed to influence pathogenicity (deLeeuw et al., 2005). During replication, APMV-1 particles are
produced with a precursor fusion glycoprotein, F0, which has to be cleaved to Fl and F2 for the virus particles to be infectious. This post translation cleavage is mediated by host cell proteases. Most APMV-1 viruses that are pathogenic for chickens have the sequence
112R/K-R-Q/K/R-K/R-R↓F117 at their F protein cleavage site, whereas the lentogenic APMV-1 have sequences of 112G/E-K/R-Q-G/E-R↓L117 (Choi et al., 2010). Therefore, predicted virulence based upon deduced amino acid motifs at the fusion cleavage site has been incorporated into the definition of notifiable APMV-1 strains by the OIE. The OIE definition for reporting an outbreak of ND is: Newcastle disease is defined as an infection of birds caused by a virus of APMV-1 that meets one of the following criteria for virulence: i) The virus has an intracerebral pathogenicity index (ICPI) in day-old chicks of 0.7 or higher (2.0 is maximum); ii) Multiple basic amino acids have been demonstrated in the virus at the C-terminus of the F2 protein and phenylalanine (F) at residue 117, which is the N-terminus of the F1 protein. The term ‘multiple basic amino
acids’ refers to at least three arginine (R) or lysine (K) residues between residues 113 and 116 (OIE, 2012).
2.2.4 Detection of APMV-1 by real-time RT-PCR assay
With its efficiency and high throughput, real-time reverse transcription polymerase chain reaction (rRT-PCR) has become one of the most widely applied assays for NDV diagnosis and surveillance (Hoffmann et al., 2009). At present, the U.S. Department of Agriculture (USDA) validated M-gene rRT-PCR assay described by Wise et al. (2004) is widely used in North America, Europe, and Taiwan for detecting most NDVs, mainly class II isolates. However, even within the more conserved genes of APMV-1 targeted in screening rRT-PCR assays, there is still considerably high genetic variation within APMV-1 to challenge primer/probe combinations to detect all contemporaneously circulating strains with sufficient diagnostic specificity/sensitivity. Recent data revealing significant heterogeneity in the genomes of APMV-1 suggests that some highly divergent viruses may not be detected (Khan et al., 2010). A large number of class I APMV-1 have been identified in samples recovered from waterfowl, shorebirds, and from poultry in live bird markets; however, the M-gene assay failed to detect the majority (73%) of these viruses (Kim et al., 2007). To minimize the false negatives produced by the USDA-validated M-gene assay, rRT-PCR assays targeting other NDV genomic regions were developed and evaluated. These assays include TaqMan rRT-PCR assays detecting the M gene (Hines et al., 2012), L gene (Fuller et al., 2010; Sutton et al., 2019), both L gene and M gene (Kim et al., 2008), along with many assays targeting the F gene, which also enable sequencing or pathotyping (Fuller et al., 2009; Yacoub et al., 2012). However, these assays cannot detect all NDV strains and a multiple testing approach may be needed for detecting the index case (OIE, 2012).
2.3 Intercontinental dispersal of avian pathogens by migratory birds
There is a growing body of literature supporting a routine exchange of infectious agents between regions by migratory birds and the potential for identifying patterns of dispersal that may be useful to predict the spread of emerging avian pathogens. Based on the global phylodynamic analysis, a study provides evidence for East Asia
representing a critically important node for the global dispersion of APMV-1 (Hicks et al., 2019). In a study of fusion protein gene sequences of two sub-genotypes of class II APMV-1 strains isolated from wild birds in Eurasia and North America, evidence of intercontinental dispersal by wild birds has been found (Ramey et al., 2013). The finding that APMV-1 sequences originating from isolates derived from waterfowl sampled in Alaska and a Delaware Bay shorebird were phylogenetically nested within mixed clades with samples from Eurasia is similar to findings of intercontinental gene flow in low-pathogenic avian influenza virus isolates (Koehler et al., 2008). The phylogentic study for global APMV-4 isolates presented limited evidence for historical viral movement between continents (Reeves et al., 2016). Phylogenetic network analysis also supported the introduction of Asia-origin clade 2.3.4.4 H5N8 avian
influenza viruses into North America via intercontinental associations of waterfowl (Lee et al., 2015). Collectively, these findings suggest that migratory birds may play a
potential role in the global spread of kinds of avian infectious agents.
2.4 APMV study in Taiwan
At least three major epidemics of ND have occurred in Taiwan, the first in 1969, the second in 1984, and the third in 1995 ( Lu et al., 1986; Yang et al., 1997). Results obtained from a study of Taiwanese NDV isolates led Yang et al. (1997) to suggest that
the 1969 outbreak of ND in Taiwan was caused by the genotype III virus, whereas the 1984 and 1995 outbreaks were caused by the genotype VII viruses. In Taiwan, despite that intensive vaccination programs against ND have been implemented for decades, NDV still has caused sporadic outbreaks among poultry flocks up to now. The antigenic and genetic characterization of velogenic NDV in Taiwan’s poultry population has been studied previously (Tsai et al., 2004; Lien et al., 2007), and all of the 20 isolates (Lien et al., 2007) and 30 isolates (Ke et al., 2010) collected from ND outbreaks in Taiwan from 2003 to 2006 and from 2002 to 2008, respectively, were assigned to former class II genotype VIIe, with an exception of a VIIc isolate by phylogenetic analysis of partial fusion protein gene sequences. However, the full extent of the distribution, evolution, and host species of APMV-1 circulating in domestic and wild birds has remained unexplored. In Taiwan, an APMV-6 strain was isolated from domestic ducks which appeared to be healthy in Taiwan in the year 1998, as a result of influenza surveillance program. The complete genome of an APMV-6 strain isolated from ducks was reported in 2001 (Chang et al., 2001).
Chapter 3
Novel avian metaavulavirus isolated from birds of the family Columbidae in Taiwan
3.1 Introduction
The International Committee on Taxonomy of Viruses has recently created and renamed three new genera, Orthoavulavirus, Metaavulavirus, and Paraavulavirus, within a new subfamily Avulavirinae of the family Paramyxoviridae (Amarasinghe et al., 2019). Viruses in the subfamily Avulavirinae, commonly known as avian
paramyxoviruses (APMVs, used hereafter for the purposes of this chapter), have been reported from a wide variety of avian species around the world (Gogoi et al., 2017).
APMVs contain a non-segmented negative sense single-stranded RNA genome ranging from 15 to 17 kb in length (Aziz-Ul-Rahman et al., 2018). According to the taxonomy of the order Mononegavirales updated in 2019, APMVs are divided into twenty species based on a sequence comparison of the RNA-dependent RNA polymerases (RdRps) of the viruses (ICTV, 2019). APMV-1 (commonly termed Newcastle disease virus, NDV) is the most recognized species, but information on the distributions of all other APMVs in domestic poultry and wild birds is limited.
Until recently, the known APMV species were restricted to APMV-1 to -9, which were isolated and characterized in the 1970s (Alexander, 1987). Following the
expansion of viral surveillance initiatives and improvements in sequencing technology, 11 novel species have been designated since 2010. A virus isolated from rockhopper penguins was antigenically and genetically distinct from all known APMV-1 to -9 and considered to represent the prototype of a new species, APMV-10 (P. J.Miller et al.,
2010). A further novel APMV-11 isolate was obtained in France from a common snipe in 2010 (Briand et al., 2012). APMV-12 was isolated in Northern Italy in 2005 from an Eurasian widgeon (Terregino et al., 2013). In 2015 and 2016, three publications
described a novel APMV-13 species found in three separate regions of Eurasia: Japan, Kazakhstan and Ukraine (Yamamoto et al., 2015; Karamendin et al., 2016; Goraichuk et al., 2016). In 2017, seven novel APMV species were announced: APMV-14 from ducks in Japan (Thampaisarn et al., 2017), APMV-15 from shorebirds in Brazil (Thomazelli et al., 2017), and APMV-16 from ducks in Korea (Lee et al., 2017). Three novel species, Antarctic penguin virus A, B, and C (APV-A, -B, and -C), were simultaneously isolated from Antarctic penguins (Neira et al., 2017), and APMV-20 from gulls in Kazakhstan (Karamendin et al., 2017). These findings suggest that wild birds maintain a previously unrecognized genetic diversity of APMVs. In Taiwan, the complete genome of an APMV-6 strain isolated from ducks was reported in 2001 (Chang et al., 2001). In the present article, we present the antigenic and genetic characterization of a novel APMV isolated from doves in Taiwan, which represents a previously unknown species.
3.2 Materials and methods
3.2.1 Sample collection and virus isolation
The samples of this study were collected from domestic poultry and wild birds in Taiwan as part of an avian influenza (AI) surveillance program. The tracheal/cloacal swabs and tissue samples of the trachea, lung, liver, spleen, heart, and kidney were inoculated into the allantoic cavities of 9- to 11-day-old specific-pathogen-free (SPF) embryonated chicken eggs (Animal Drugs Inspection Branch, Animal Health Research Institute, Miaoli, Taiwan) and then incubated at 37℃ for 72 h. The allantoic fluid from each inoculated embryo was collected after two passages in eggs and individually
examined for hemagglutination assay (HA). The HA-positive allantoic fluid was tested by AIV (Spackman et al., 2002) and NDV (Wise et al., 2004) real-time reverse
transcription polymerase chain reaction (rRT-PCR). Samples that tested negative were subjected to further analyses.
3.2.2 Electron microscopy
HA-positive allantoic fluid which tested negative for AIV and NDV by rRT-PCR was centrifuged at 3000 × g for 20 min at 4℃. The supernatant was separated and then subjected to ultracentrifugation at 100,000 × g for 5 min. The pellet was resuspended in 25 μL of distilled water and stained with 2% phosphotungstic acid in a copper grid (Cohen, 1992) and then examined using a JEOL JEM-1400 electron microscope (JEOL Ltd., Tokyo, Japan).
3.2.3 Determination of nucleotide sequence of full-length APMV genome Viral RNA from HA-positive allantoic fluid which tested negative for AIV and NDV by rRT-PCR was isolated using the MagNA Pure Compact Nucleic Acid Isolation Kit I (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions. Amplification of the APMV RNA was performed using
Rubulavirus-Avulavirus genus subgroup-specific RT-PCR (Tong et al., 2008) with
SuperScript III One-Step RT-PCR kit (Invitrogen, Carlsbad, California, USA). To determine the nucleotide sequence of the full-length genome, a combination of random primers and primer walking was used to generate PCR amplicons covering the genome (except for the 3’ and 5’ ends). The sequences of both ends of the genome were
amplified using the rapid amplification of cDNA ends method (RACE) (Qiu et al., 2009). According to the assembling contig of primer-walking and RACE amplicons, we
redesigned 17 primer sets to amplify 17 overlapping segments covering the entire genome. All amplified products were separated on 2% agarose gel and purified using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). These products were cloned with the TOPO TA Cloning kit (Invitrogen) using the standard protocol, and inserted cDNA segments were amplified using M13 forward and reverse primers
provided with the kit. The PCR products with expected lengths were sequenced with the 3700XL DNA analyzer (Applied Biosystems, Life Technologies, Carlsbad, California, USA) by a commercial sequencing service (Mission Biotech, Taipei, Taiwan).
3.2.4 Intracerebral pathogenicity index (ICPI)
To determine the virulence of an APMV isolate, the ICPI test for NDV (OIE, 2012) was employed in day-old SPF chicks (Animal Drugs Inspection Branch, Animal Health Research Institute).
3.2.5 Serological characterization
In view of the difficulties of obtaining a whole set of APMV-1 to APMV-9 reference antisera and antigen, the new APMV isolate was submitted to the OIE/FAO and National Reference Laboratory for Newcastle Disease and Avian Influenza, Istituto Zooprofilattico, Italy. Hemagglutination inhibition (HI) tests (OIE, 2012) were
performed using antigens and chicken polyclonal antisera of representative APMV-1 to APMV-9 (except APMV-5). The antiserum against the new APMV isolate used in reverse HI assay was produced in adult SPF leghorn chickens by subcutaneously inoculating 0.2 mL of inactivated, adjuvanted virus emulsion twice with a 2-week interval, and the antiserum was collected two weeks after the second inoculation. The reverse HI assays were conducted for characterizing new the APMV isolate with
APMV-7 reference antigen and antiserum, which were kindly shared by the Italian OIE reference laboratory. The antigenic relationship between the two viruses was analyzed with the following formula (Archetti and Horsfall, 1950): R = (r1 × r2)1/2 where r1 is the ratio of the heterologous HI titer divided by the homologous HI titer for virus 1, and r2 is the ratio of the heterologous HI titer divided by the homologous HI titer for virus 2.
3.2.6 Phylogenetic analysis
The sequences of the full-length genome, fusion (F), and
hemagglutinin-neuraminidase gene (HN) of the isolated APMV were aligned with the sequences of APMV-1 to APMV-20 representative viruses retrieved from the GenBank database using ClustalW in Molecular Evolutionary Genetics Analysis version 7, or MEGA7 (Kumar et al., 2016). For the construction of the phylogenetic trees, the evolutionary history was inferred using the Maximum Likelihood method based on the general time reversible model with discrete gamma distribution and invariant sites (Nei and Kumar, 2000) by 1000 bootstrap replicates.
The phylogenetic analyses based on complete amino acid sequences of
RNA-dependent RNA polymerase (RdRp or L gene) of the viruses were conducted as previously described (ICTV, 2019). The alignment prepared with MEGA7 software with a gap-opening penalty of 5 and a gap extension penalty of 1. The evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model (Jones et al., 1992). Initial tree for the heuristic search were obtained
automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. Evolutionary analyses were conducted in MEGA7 (Kumar et al., 2016).
3.2.7 Nucleotide sequence accession number
The complete genome sequences of APMV/dove/Taiwan/AHRI33/2009 and three AHRI33-like isolates are available from GenBank under the accession numbers
MK677430-MK677433. GenBank accession numbers for the complete coding region of the fusion genes for the other ten AHRI33-like isolates sequenced as part of this study are as follows: MK677434–MK677443.
3.3 Results
3.3.1 Sample collection and virus isolation
As a result of the avian influenza surveillance program on resident birds in Taiwan, a novel virus, AHRI33, showing hemagglutination activity was isolated from a
clinically healthy red collared dove (Streptopelia tranquebarica) in 2009. The AHRI33 virus propagated well in embryonated chicken eggs, and the harvested infective
allantoic fluid had an HA titer of 32. Moreover, we also obtained additional 13 AHRI33-like virus isolates from resident doves and pigeons in two different time periods. In 2009-2010, a research project for surveillance of AIV in resident birds was conducted. Swab samples were mainly collected from sparrows, doves, pigeons, and Chinese bulbuls, the dominant species of resident birds in Taiwan, and 11 AHRI33-like strains were isolated only from red collared doves. Since the 2015 outbreak of the clade 2.3.4.4 H5Nx highly pathogenic avian influenza (HPAI) viruses in Taiwan, dead birds found in public places have been sent to AHRI for detection of AIV. Two AHRI33-like strains were isolated. Among them, AHRI104 was obtained from a dead oriental turtle dove (Streptopelia orientalis) in 2016 and AHRI128 from a dead pigeon (Columba livia) in 2017.
3.3.2 Electron microscopy
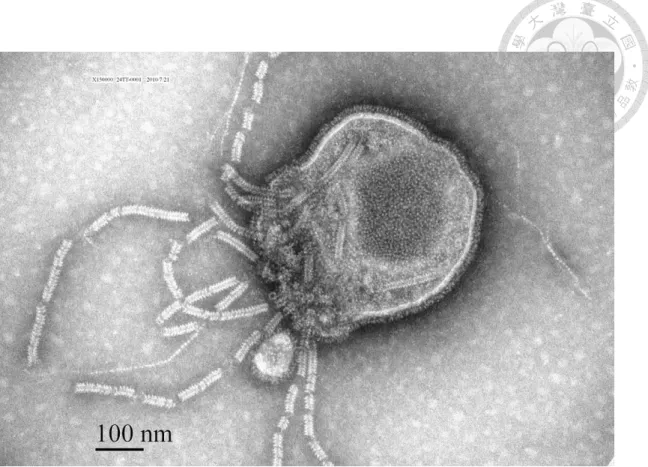
The fine structure of the virion showed typical characteristics of a paramyxovirus.
The virion was pleomorphic, roughly spherical, and varied from 150 to 500 nm in diameter. Herringbone-like nucleocapsids were encased in a fragile envelope coated with approximately 10 nm long spikes (Fig. 3.1).
3.3.3 Intracerebral pathogenicity index
After inoculation of the AHRI33 virus into the cerebrums of one-day-old chickens, the obtained ICPI value was zero, indicating no clinical signs. This result suggested that the AHRI33 isolate was avirulent to chickens.
3.3.4 Serological characterization
As shown in Table 1, the titers of antisera against each representative APMV species were higher with the homologous virus. The AHRI33 isolate only showed titers of 1:4 to 1:16 with the reference antisera against APMV-1, -2, -3, -4, -6, -8, and -9, but it was antigenically similar to APMV-7 on the basis of HI typization (4-fold difference in HI titers). The R-value between AHRI33 and APMV-7 was then calculated based on the HI titers obtained from the cross-HI assay, and the R-value of 0.125 indicated antigenic similarity between these viruses, although the R-value was less than would be expected between viruses of the same serogroup.
3.3.5 Determination of nucleotide sequences of full-length APMV genome
The complete genome sequence of the AHRI33 isolate was characterized using a Sanger sequencing approach. Assembly of the sequences produced a contig of 16,914
nucleotides (nt), making it close in size to APMV-5 (GU206351, 17,262 nt, 348 nt longer than that of AHRI33), APMV-11 (NC_025407, 17,412 nt, 498 nt longer), and APMV-3/Netherlands (EU403085, 16,272 nt, 642 nt shorter). Other APMV genome sequences range in length from 14,880 nt for APMV-2/F8 (HQ896023) to 17,412 nt for APMV-11.
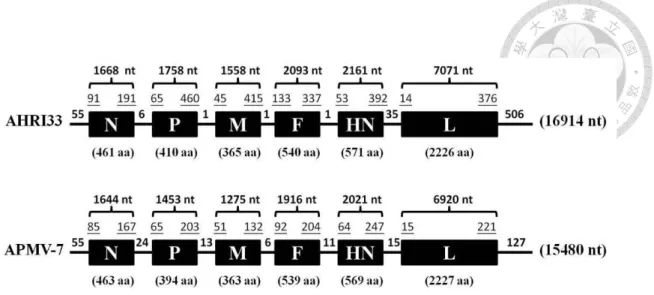
Six genes, nucleocapsid (N), phosphoprotein (P), matrix (M), fusion (F),
hemagglutinin-neuraminidase (HN), and large polymerase (L), were identified in the genome of the AHRI33 virus, showing a typical gene arrangement of APMVs
3'-leader-NP-P-M-F-HN-L-trailer-5' (Fig. 3.2), which predicted six viral proteins: NP, 461 amino acids (aa); P, 410 aa; M, 365 aa; F, 540 aa; HN, 571 aa, and L, 2226 aa. The 3’ leader sequence of the AHRI33 isolate was 55 nt in length, as conserved among most APMV species (Anderson andWang, 2011). The length of the trailer at the 5’ end was 506 nt. The first 13 nt of the leader sequence and the last 13 nt of the 5’ trailer sequence showed complete complement. The conserved sequences for the gene start (GS) and gene end (GE) of the AHRI33 virus were CCCCCNNUN and AAUNNU6-7, respectively.
The lengths of the intergenic region sequences ranged from 1 to 35 nt. The deduced amino acid sequence of the F gene cleavage site was STQQER/LFG, which lacked multiple basic residues at the C-terminus of the F2 protein and a phenylalanine residue at the N-terminus of the F1 protein.
3.3.6 Sequence comparison and phylogenetic analysis
Comparison of the AHRI33 full-length genome with those of known APMV species revealed that AHRI33 was more closely related to APMV-7 (62.4% nucleotide identity) than to the other APMVs (43.5-53.7%) (Table 3.2). The highest inter-serotypic identity was 68.1% between APMV-1 and -16, and the lowest was 42.7% between
APMV-4 and -6.
The nucleotide sequence identities of each AHRI33 gene to the corresponding genes of other APMVs ranged from 39.3% (P) to 68.0% (N) (Table 3.3). All six genes of AHRI33 were most similar to those of APMV-7, with identities of 55.7% to 68.0%.
The amino acid identities were consistent with the corresponding nucleotide identities (Table 3.3).
Phylogenetic trees were constructed based on alignment of the full-length genome of AHRI33 (Fig. 3.3), the F gene (Fig. 3.4), and the HN gene (Fig. 3.5) with
corresponding genes of representative APMV species. Phylogenetic analysis based on the full-length genome sequences (Fig. 3.3) revealed that the four AHRI33-like isolates were grouped along with APMV-7 and -11 but were distinguishable from these two species. Within this group, AHRI33-like isolates appeared to be more closely related to APMV-7 than to APMV-11. Phylogenetic analysis based on F gene sequences (Fig. 3.4) gave similar results. The other 13 AHRI33-like viruses isolated in Taiwan were highly similar to AHRI33, with 98.9% to 99.9% nucleotide identities of the fusion gene. All of them were assigned to the same cluster in the phylogenetic analyses (Fig. 3.4).
Phylogenetic analysis of amino acid sequences of the RdRp revealed that all APMVs clustered together in a group designated as subfamily Avulavirinae, when compared with the closely related Mumps virus as an outgroup (Fig. 3.6). Three main clades designated as genera Orthoavulavirus, Metaavulavirus, and Paraavulavirus were observed within the subfamily Avulavirinae. The AHRI33-like isolates characterized in this study nested within genus Metaavulavirus, and the branch length between the nearest node and the tip of the branch is 0.28.
3.4 Discussion
This study demonstrated that a unique APMV strain has been long-term circulating in resident doves and pigeons in Taiwan. In addition to the novel AHRI33 virus, 13 AHRI33-like APMVs were isolated from three different species of the family Columbidae in 2009-2017. Although the samples taken in the avian influenza
surveillance program covered many species of domestic poultry, migratory birds, resident birds, and imported birds, the novel APMV strain was isolated from members of the family Columbidae, implying that pigeons and doves may be one of the natural hosts of AHRI33-like viruses. Future surveillance of wild birds in Taiwan may help to better elucidate the host distribution of AHRI33-like viruses.
Sequencing of the viral genome of the AHRI33 isolate revealed characteristic APMV coding regions and the non-coding terminal sequences (e.g., the 55 nucleotide non-coding leader sequence at the 3' end that is present in all APMVs) (Fig. 3.2). The genome length of 16,914 nt is compatible with the “rule of six”, which plays an
important role in the replication of APMVs (Kolakofsky et al., 1998), and it is the third longest among the twenty species of APMVs reported to date, shorter than only those of APMV-11 and APMV-5. The schematic diagram of AHRI33 and the protovirus
APMV-7 genome made clear the significant disparities in length of the complete genome, all six of genes, the intergenic regions, and the 5’-trailer region (Fig. 3.2). The length difference of 1,434 nt between AHRI33 and APMV-7 (15,480 bp) is much greater than the largest difference, 120 nt, between intra-species of APMV-2/Yucaipa and APMV-2/Bangor. The phylogenetic relationship with APMV-7 is consistent throughout the genome, forming a monophyletic group, suggesting that these viruses share a more recent common ancestor than do other lineages (Fig. 3.3). The deduced amino acid sequence of the F-gene cleavage site was STQQER/LFG, which was significantly different from that of APMV-7/Tennessee (LPSSR/FAG) and all other
APMVs. This motif lacks multiple basic residues and phenylalanine at the N-terminus of the F1 protein, a characteristic that typically corresponds with non-pathogenic variants, which is concordant with the results of the ICPI test.
Traditionally, APMVs were classified based on their antigenic differences, and nine serotypes were defined by HI assay in the 1970s (Alexander, 1987). In the present study, HI assay revealed weak cross-reactivity between APMV-7 and AHRI33 (R=0.125), and there were extremely low relationships between AHRI33 and representatives of the other species. HI cross-reactivity is not rare between different species of APMV (e.g., between APMV-1 and APMV-12 (Terregino et al., 2013), and APMV-9 and APMV-16 (Lee et al., 2017)), and lack of HA activity observed in APMV-5 (Samuel et al., 2010) and one novel APMV-6 (Chen et al., 2018), and all this makes classification into serotypes problematic.
In contrast, Terregino et al. (2013) proposed a classification based on nucleotide sequence identities of the whole genome as one simple method. According to this classification method based on genome identities, AHRI33 is closest to APMV-7, at 62.4%, which less than those between APMV-1 and -16 (68.1%), APMV-A and -B (67.4%), APMV-12 and -13 (64.5%), and APMV-B and -C (62.7%) (Table 3.2). These results of genetic analyses indicate that the AHRI33 isolate evolved from a common ancestor of APMV-7 and -11 and is now a distinct branch of the APMV groups.
In the last proposal for taxonomy changes of the family Paramyxoviridae, the ICTV Study Group has decided that the classification should be based on a sequence comparison of the RdRps of the viruses (ICTV, 2019). Based on the phylogenetic tree topology (clustered into monophyletic branch within the clade of the genus
Metaavulavirus) and the branch length measured in the number of substitutions per site above 0.03, the AHRI33-like isolates met the criteria for designation as distinct species.
To sum up, we identified new APMVs from the birds of family Columbidae in Taiwan from 2009 to 2017. The new APMV isolates are more closely related to
APMV-7 based on estimates of nucleotide identities of the full-length genome; however, these heterogeneous levels are comparable to, or even greater than, those of several inter-species distances separating other accepted species. This, together with the analysis according to new RdRp phylogeny-based classification system, suggests that the newly-isolated APMV should be considered as a novel species and the prototype strain of a new APMV-22 group, with the full name
APMV-22/dove/Taiwan/AHRI33/2009.
Table 3.1 Antigenicity between newly isolated AHRI33 virus and representative avian paramyxoviruses (APMVs), measured by
cross-hemagglutination inhibition tests. The following representative viruses were used as antigens and to prepare homologous chicken antisera:
APMV-1/Ulster/2C/70, APMV-2/chicken/California/Yucaipa/56, APMV-3/parakeet/Netherlands/449/75, APMV-4/duck/Hong Kong/D3/75, APMV-6/duck/Hong Kong/199/77, APMV-7/dove/Tennessee/4/75, APMV-8/goose/Delaware/1053/75, and APMV-9/duck/New York/22/78.
Virus APMV-1 antiserum
APMV-2 antiserum
APMV-3 antiserum
APMV-4 antiserum
APMV-6 antiserum
APMV-7 antiserum
APMV-8 antiserum
APMV-9 antiserum
AHRI33 antiserum
APMV-1 512a
APMV-2 256
APMV-3 1024
APMV-4 1024
APMV-6 512
APMV-7 4096 8
APMV-8 1024
APMV-9 256
AHRI33 16 4 16 16 4 1024 16 4 128
a HI titres are expressed as the reciprocal of the highest dilution causing inhibition of 4 HA units of virus.