國立臺灣大學理學院化學研究所 碩士論文
Department of Chemistry College of Science
National Taiwan University Master Thesis
設計與合成人類胸苷酸激酶之抑制劑 Design and Synthesis of
Inhibitors against Human Thymidylate Kinase
黃夢若 Meng-Ruo Huang
指導教授:方俊民 博士 Advisor: Jim-Min Fang, Ph.D.
中華民國 102 年 7 月
July, 2013
2
3
謝誌
時光飛逝,轉眼間研究所的兩年就這樣匆匆過了。想當初還只是個懵懵懂懂的 大學畢業生,一腳踏進台大這個國內首屈一指的學術殿堂,還真怕自己程度跟不上 大家。所幸在老師與學長姐們的協助下總算慢慢熬了過來,雖然過程歷經不少挫 折,現在回首望去,也許那些都是磨練我們具備更佳抗壓性的必經之路吧!真的非 常感謝指導教授方俊民老師在這兩年時間的諄諄教誨,每當我實驗遇到困難時,您 總是非常有耐心的替我想辦法解決問題,才讓我得以順利的將實驗完成。此外,也 很感謝您每年大方提供的出遊計畫和各種聚餐活動,讓我們在實驗之餘也能享受到 精神上的調劑。陽明大學的張智芬老師,謝謝您提供我們生物活性實驗協助以及相 關研究方向的建議,利用 TMPK 的抑制來對癌症進行治療真的是一項偉大的新發 現。同為陽明大學的許世宜老師,感謝您替我們做的分子動態模擬實驗,也謝謝您 親切的為我解答實驗不懂的地方。羅禮強老師,這麼晚才向您敲定口試時間深感抱 歉,謝謝您提出的論文改進方向,讓我能將論文整理得更加完整。另外,實驗部份 要 特 別 感 謝 詹 迺 立 老 師 替 我 們 做 蛋 白 質 的 結 晶 , 效 率 超 群 的 趙 玉 正 小 姐 的 ESI-HRMS 分析,以及親切的蘇仁寬先生在 NMR 上的協助,每次都要麻煩你測 overnight 真是非常不好意思。
除了老師之外,實驗室的同仁也幫了我許多忙。個性穩健的怡禎學長,生化專 家大溫學長,唱歌很有感情的公政學長,笑容親切的珈男學長,總是能提出很多問 題的農森學長,充滿親和力又有趣的銘祥學長,以及又會唱歌又會招呼學弟妹的紹 宏學長,希望你們在就業的路上都能平安順遂,財源滾滾。為人親切的俊霖學長,
感謝你在我需要打 HPLC 時冒著吸到 chloroform 的危險教我純化技術。幽默風趣的 志安哥哥,謝謝你在 meeting 時提出的建議,讓我獲益良多。生活作息超規律的北 濤學長,每次看到你認真做實驗的態度總是自嘆不如,相信付出一定會有代價的!
超級帥氣的正港台灣男兒小彬彬學長,雖然剛開始覺得你像流氓(好像本來就是),
不過熟了之後真的覺得你人超 nice!感謝你在我碩一時像保母一樣的將我拉拔長 大,讓我學到不少實驗知識和技巧,現在想起當初作伙「買涼的」都覺得格外懷念 呢!希望你能趕快找到合適對象,來個閃電結婚也不錯唷。外表看起來超年輕的育 禎學姐,雖然覺得你就這樣休學有點可惜,不過相信你也是做了一番評估才下定決 心,祝福你和親愛的白頭偕老喔!飛到荷蘭從此成為脫韁野馬一去不復返的乳焜同 學,很懷念當初和你一邊做報告一邊暢快聊天的感覺,希望你早日回國讓我們砲一 下。總是很搞笑的密友佩琪姊姊,我的手機跟我說沒有你玩它的日子有些落寞耶,
是否該回來露臉一下呢?希望你在台積能夠繼續奮鬥下去,不過錢要賺肝也要顧 喔!笑容靦腆的瑋霖學長,每次看你報進度都覺得你真的好認真,相信這樣的態度 到業界一定是備受肯定的,希望你感情事業都順順利利。盡責又會照顧學弟妹的蔡 MOMO,你的開朗個性總能讓實驗室活潑熱鬧許多,希望你能順利完成實驗,早 日畢業。善解人意的凱哥,每次被你逼問八卦都有點招架不住的感覺,不過你真的 人很 nice 又很風趣,祝你實驗順利。體貼又美麗的思涵姊姊,從剛進實驗室一直
4
到畢業都麻煩你很多事情,謝謝你總是不厭其煩的幫我,相信現在實驗上的辛苦一 定會有代價,再撐一下以後就是高薪貴婦了,期待被你丟紅色炸彈的那天,不過滷 肉飯桌可以加點配菜嗎?人超 nice 的鵬皓學長,雖然沒有跟你聊很多,不過幾次 接觸下來覺得你人很好,希望你能順利推上理想的學校。認真的哲哲學長,希望你 實驗一切順利,早日畢業。外表看似嚴肅內心卻很幽默的氣象專家俞瑞哥,感謝你 常常請我們吃美食,可惜我還是不爭氣的沒長胖,看樣子你乾脆列個美食清單給我 比較快,祝你實驗順利,TG 組的大家長要 hold 住團隊唷!親愛的阿王,謝謝你這 兩年來的陪伴,不論何時,你總能看出我的低潮並且陪我一起走過,今天之所以能 順利畢業有一半的功勞都要歸功於你,真的很感謝。雖然你的題目時常遇到瓶頸,
不過你的認真程度是大家有目共睹的,相信老天不會辜負這樣努力的你,要堅持下 去唷!有點冒失唱歌卻很好聽的子瑋,一直覺得你是個很樂觀開朗的人,但是太過 大而化之是優點也是缺點,你要自己好好衡量一下,然後實驗要加油喔,現在有學 妹陪你奮鬥更要好好努力,fighting!實驗超厲害的隆誌學長,感覺你真的是念博 班的料,要繼續加油下去!同為 TMPK 組的怡瑄學姊,有你加入之後讓我覺得自 己終於不是孤軍奮鬥,謝謝你教了我很多生物方面的知識,以後實驗就靠你了,有 問題歡迎與我聯絡。幽默的程健哥,很可惜你沒能繼續唸下去,不過現在有了愛情 的滋潤加上不錯的工作,應該算是愛情事業兩得意吧,先跟你說聲恭喜囉!
很有自己規劃的小宥宥,兩年下來受你不少幫助,希望你口試和就業順利,和 女朋友也能愛情長跑下去。認真又很有責任感的翊瑋,總覺得你常常給自己太大壓 力,要記得適度的放鬆才能保持身心健康,祝你未來一切順遂。活潑又有原則的小 單,剛開始覺得你酷酷的,久了才知道其實你是外冷內熱,雖然你很不想承認,但 我們終究是密友對吧,希望以後有機會還能分享彼此的心情跟想法唷!很有自己想 法的乃維,看得出你對自己的要求以及堅持,不過努力之餘還是要記得放鬆一下,
朝著自己的理想邁進吧!個性隨和又健談的宛蓁,感覺你也是蠻容易緊張的人,不 過相信以你的能力應該能在畢業前做出不少 final,要對自己有信心!開朗風趣的日 平,謝謝你以前常常在我忙不過來的時候幫我代班,實驗方面再加把勁就可以發 paper 了,加油,你可以的。自戀又多愁善感的年加,其實越跟你相處就越覺得你 人不錯又搞笑,做事也很盡責,只是發條要上緊點,還有個性要再硬一些,然後少 講些冷笑話,才不會常常句點,加油好嗎?常常放空卻又帶點喜感的昱婷,生物背 景來做有機合成真是辛苦你了,雖然覺得休學有點可惜,不過既然你有自己的轉換 目標就好好堅持下去吧!新進的碩一,苓瑋、哲瑄、仁垚、若綺、晏靈,以及助理 子禎,每次到實驗室都覺得你們很認真,以後實驗室就靠你們這些新血了,加油!
專題生哲凡、旻泓,和曹一包,看到你們對自己未來的規劃就很替你們開心,希望 你們繼續朝夢想努力下去。菁菁、王婷、薇立,以及欣傳,希望你們不論是推甄和 考試都能一切順利,早日成為研究生,享受做實驗的樂趣。
最後要感謝我的家人,願意一路支持我走到現在並且給予關心,還要忍受我的 壞脾氣。沒有你們我就無法順利從這裡畢業,謝謝你們。同時也祝 JMF 的大家都 能實驗順利,日日有 data,月月發 paper,年年出專利!
I
摘要
癌症一直以來是造成國人死亡率居高不下的原因,如何有效對抗癌症因此成
為科學家致力研究的方向。目前醫療上廣泛應用的化療藥物小紅莓(doxorubicin) 雖具有良好抑制效果,然而其嚴重的副作用也使得這個藥物的發展受到限制。因此 找尋小分子抑制劑來敏化小紅莓對癌症的毒殺能力,進而降低其藥劑量便成為一個 開發方向。
根據高通量藥物篩選的結果,YMU1 被發現對人類胸苷酸激酶(hTMPK)具有良 好的抑制效果,同時在低劑量小紅莓的共同作用下,能選擇性對癌症細胞造成毒殺 作用。因此在本篇論文裡,我將 YMU1 結構中的硫原子置換成其他原子,進而比 較其結構與活性的關係。另外,為了改善 YMU1 的水溶性問題,設計一系列親水 性 YMU1 衍生物也是我們研究的方向。最後,由於二聚體(dimer)結構的化合物具 有比 YMU1 更好的抑制活性,我們合成不同的二聚體衍生物並測試其活性。
由目前合成的 30 個類似物來看,有 7 個衍生物已證實具有和 YMU1 相同,甚 至更好的抑制活性。此外,我們也透過這些衍生物歸納出結構與活性之關係,包含 硫原子確實在 YMU1 中扮演重要角色、環上的氫氧基能大幅提升 YMU1 衍生物之 抑制活性,以及二聚體結構普遍具有良好的活性等,希冀能有助於未來的人類胸苷 酸激酶抑制劑之開發。
II
II
III
III
Abstract
Cancer has become one of the most fatal diseases in recent 30 years. Anti-cancer drugs such as doxorubicin, which can inhibit the function of type II topoisomerase, have been used extensively. However, doxorubicin treatment causes many side effects including vomit, hair loss, myelosuppression and cardiotoxicity. Searching for new drugs to cure cancer with reduced side effects from doxorubicin has become an important task. After screening a series of compounds, YMU1 was found to inhibit the activity of human thymidylate kinase (hTMPK), which plays an important role in synthesis of DNA. YMU1 could sensitize cancer cells to chemotherapeutic agent such as doxorubicin. The combined use of YMU1 and doxorubicin substantially inhibits tumor growth and reduces the side effect of doxorubicin.
My research aims to modify the structure of YMU1 to find derivatives showing better inhibition against hTMPK. In order to test the importance of sulfur atom in YMU1,
IV
IV
the sulfur atom was replaced by other atoms and the inhibitory activities of such derivatives were compared with the sulfur-containing structure. To solve the low solubility of YMU1 in water, I also prepared YMU1 derivatives having high hydrophilicity good for animal experiments. Because dimeric compound has a potential to show better inhibition than YMU1, different cores of dimeric derivatives were synthesized.
Among the above-prepared derivatives, 7 compounds were proved to have either comparable or better inhibition than YMU1. We also made several conclusions for the relationship between structure and activity, including the crucial role of the sulfur atom in the heterocyclic core, the enhanced hTMPK inhibition with a hydroxyl group in the core structure, and better inhibition of the dimeric compounds. This study will be useful for the development of anticancer therapy.
V
V
Table of Contents
Abstract in Chinese………...I
Abstract in English………III
Table of Contents………...V
Index of Figures………...VIII
Index of Schemes………..………....XII
Index of Tables………..……….……….XIII
Abbreviations………..………XIV
Chapter 1. Introduction ... ..1
1.1. Brief introduction of cancer ... ..1
1.2. Inhibition of topoisomerase ... ..2
1.3. Mechanism of DNA repair ... ..5
1.4. Biosynthetic pathway of dTTP in cells ... ..7
1.5. Antimetabolite agents targeting dTTP formation ... ..8
1.6. Structure and physiology of TMPK ... 10
1.7. The relationship between the doxorubicin sensitization by TMPK inhibition to p53 status ... 13
1.8. Confirming the characteristic role of hTMPK during DNA repair ... 15
VI
VI
1.9. Screening inhibitors against hTMPK ... 21
Chapter 2. Results and Discussion ... 27
2.1.1. Structural feature of YMU1 ... 27
2.1.2. Molecular dynamic simulation of YMU1 with TMPK ... 28
2.1.3. Specific YMU1 derivatives proposed in this study ... 29
2.1.4. Luciferase-coupled TMPK assay ... 30
2.1.5 General synthetic scheme ... 32
2.2.1. Replacement of the sulfur atom in heterocyclic core... 32
2.2.2. Synthesis of piperazine linkers ... 34
2.2.3. Synthesis of YMU1 derivatives 13–17 ... 35
2.2.4. Structure–activity relationship of compounds 3–7, 9 and 13–18 ... 36
2.3.1. Hydrophilic YMU1 derivatives ... 38
2.3.2. Synthesis of hydrophilic derivatives ... 40
2.3.3. Structure–activity relationship of compounds 3, 20, 22–29, and 33–34 ... 46
2.4.1. Dimeric inhibitors ... 52
2.4.2. Synthetic schemes for dimeric inhibitors ... 52
2.4.3. SAR of dimeric compounds 35, 38–39, and 42 ... 55
2.5. Molecular docking of YMU1 derivatives ... 58
2.6. Conclusion ... 61
VII
VII
Chapter 3. Experimental Section ... 63
3.1. General part ... 63
3.2. Expression and purification of enzymes ... 64
3.3. Luciferase-coupled TMPK assay ... 64
3.4. NADH-coupled TMPK assay ... 65
3.5. Molecular docking ... 65
3.6. Synthetic procedures and characterization of compounds ... 66
References ... 99
Appendixes 1H and 13C NMR spectra ... 109
VIII
VIII
Index of Figures
Figure1. Chemical structures of drugs targeting topoisomerases I and II ... 3
Figure2. Mechanism of DNA homologous recombination ... 6
Figure3. Biosynthetic pathways of cellular dTTPs ... 7
Figure4. Chemical structures of drugs targeting thymidylate kinase (TS) ... 8
Figure5. Mechanism of TS inhibition by 5-FU... 9
Figure6. Ribbon structure of human TMPK (PDB ID: 1E2F) ... 11
Figure7. Movement of human TMPK structures ... 12
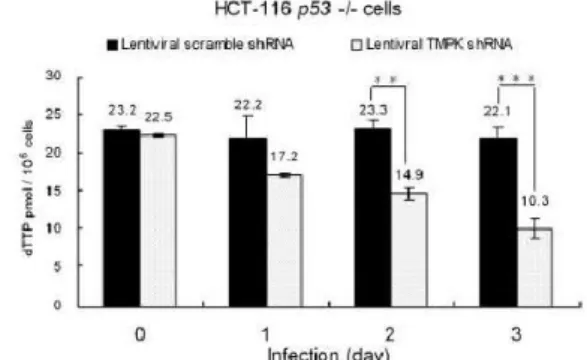
Figure 8. dTTP pool size measurement of lentiviral TMPKshRNA and scrambleshRNA in p53-proficient and p53-null HCT-116 cells ... 14
Figure 9. Comparison of TMPK and TS knockdown on doxorubicin-induced apoptosis in p53(+/+) and p53(-/-) HCT-116 cells ... 15
Figure 10. Combined treatment of doxorubicin and hTMPK inhibitor to trigger cell apoptosis ... 16
Figure 11. (a) γH2AX foci staining of MDA-MB231 cells with or without TMPK silencing after recovery from doxorubicin ... 18
(b) XRCC1 foci staining of LacZ or UNG lentivirus infected cells recovered from doxorubicin treatment for 24 h ... 18
IX
IX
Figure 12. (a) pCMV2-YFP-Nuc (vector) or pCMV2-YFP-Nuc-R1C (plasmid)
transfected into TMPK or TMPK knockdown cells ... 18 (b) Laser-micro-irradiation of Hela cells followed by immuno-
fluorescence staining ... 18 (c) γH2AX foci staining of MCF-7 cells with TMPK, R2 and
TMPK/R2 knockdown at the indicated time points ... 19 Figure 13. γH2AX foci staining of MCF-7, H184B5F5/M10, and MCF-10A cells
transfected with TMPK siRNA after DNA damage recovery ... 20 Figure 14. Flow cytometry of cells at the indicated time points after recovery from
doxorubicin ... 20 Figure 15. Principles of NADH- and luciferase-coupled TMPK assays ... 22 Figure 16. Chemical structure of YMU1 ... 24 Figure 17. (a) H184B5F5/M10, MCF10A, MCF-7, and HCT-116 p53(-/-) cells
were treated with YMU1 or 5-FU at different concentration, and stained by crystal violet after 14 days ... 25 (b) γH2AX foci staining of cells after treated with vehicle or YMU1
along with doxorubicin exposure ... 25 Figure 18. (a) On treatment with vehicle or YMU1 for 72 h, followed by 4 h
doxorubicin exposure, cells were incubated for colony formation over
X
X
14 days ... 25
(b) Comparison of tumor growth in mice with combined treatment of YMU1 and doxorubicin or doxorubicin/YMU1 alone ... 25
Figure 19. Structure of YMU1 ... 27
Figure 20.Some structural features of YMU1 derivatives ... 28
Figure21. MD simulation of the mechanism of TMPK with or without YMU1 ... 29
Figure 22. Stabilzation of YMU1 in hTMPKvia the π-π stacking interaction with Arg16 ... 29
Figure 23. Specific YMU1 derivatives ... 30
Figure 24. Protocol of luciferase-coupled TMPK assay ... 31
Figure 25. Chemical structures of benzo[d]isothiazol-3(2H)-one (BT), compounds 3–7, 9, 13–18 and YMU1 ... 37
Figure 26. Chemical structures of compounds 3, 20, 22–29, 33 and 34 ... 47
Figure 27. Dosage-dependent TMPK inhibition of YMU1, compounds 22, 29 and 33. 50 Figure 28. The chemical structure of dimeric TMPK inhibitor 35 ... 52
Figure 29. Chemical structures of dimeric inhibitors 35, 38, 39, and 42 as well as suicide inhibitors 40 and 41 ... 55
Figure 30. Dosage-dependent inhibition of YMU1 and dimeric compounds 35, 38, 39 and 42 against TMPK ... 57
XI
XI
Figure 31. Selected compounds for molecular docking ... 58
Figure 32. Docking of compounds 22 and 42 with hTMPK... 60
Figure 33. Chemical structures of efficient hTMPK inhibitors ... 61
Figure 34. Structural features of YMU1 derivatives against hTMPK ... 62
XII
XII
Index of Schemes
Scheme1. General synthetic scheme for YMU1 derivatives ... 32
Scheme2. Synthesis of compound 12 ... 35
Scheme3. Synthesis of compounds 13–17 ... 35
Scheme4. Synthesis of compounds 20 and 22 ... 41
Scheme5. Synthesis of compounds 23 and 24 ... 42
Scheme6. Synthesis of compounds 25 and 26 ... 42
Scheme7. Synthesis of compounds 27 and 28 ... 43
Scheme8. Attempted synthesis of compound 29 ... 43
Scheme9. Synthesis of compound 29 ... 45
Scheme 10.Synthesis of derivatives 33 and 34 ... 46
Scheme 11.Synthesis of dimeric inhibitors 35, 38 and 39 ... 53
Scheme 12.Synthesis of compounds 40–42 ... 54
XIII
XIII
Index of Tables
Table 1. Comparison of sensitivity in NADH- and luciferase-coupled TMPK assay .. 23 Table 2. Reaction conditions for the synthesis of compound 16 ... 36 Table 3. Normalized TMPK inhibition of compounds BT, 3–7, 9, 13–18 and YMU1 . 37 Table 4. Attempted protection of compound 22 ... 44 Table 5. Synthesis of compound 34 ... 46 Table 6. cLog P values and TMPK inhibition of compounds 3, 20, 22–29, 33 and 34 48 Table 7. Dosage-dependent assays of YMU1, compounds 22, 29 and 33 against
TMPK ... 50 Table 8. Normalized TMPK inhibition of dimeric compounds 35 and 38–42 at 2 μM 55 Table 9. Dosage-dependent assays of YMU1 and dimeric compounds 35, 38, 39
and 42 against TMPK ... 56 Table 10. Ki values of compounds 22 and 42 for hTMPK ... 59
XIV
XIV
Abbreviations
5-FU 5-fluorouracil
Ac acetyl
ADP adenosine 5’-diphosphate
Ala alanine
AMP adenosine 5’-monophosphate
AppNHp adenosine 5’-(β,γ-imido)triphosphate lithium
Arg arginine
Asp aspartate
ATP adenosine 5’-triphosphate
Bn benzyl
Boc tert-butoxycarbonyl
BSA
BT
bovine serum albumin
benzo[d]isothiazol-3(2H)-one CH2THF 5,10-methylene tetrahydrofolate cLog P
CPT
calculated lipophilicity camptothecin
dADP deoxyadenosine diphosphate
dCDP deoxycytidine diphosphate
XV
XV
DIEA diisopropylethylamine
DMAP 4-dimethylaminopyridine
DMF N,N-dimethyl formamide
DMSO dimethyl sulfoxide
DNA deoxyribonucleic acid
dNTP deoxyribonecleotide
dTDP deoxythymidine diphosphate
dTMP
DTNB
deoxythymidine monophosphate 5,5’-dithiobis(2-nitrobenzoic acid)
dTTP deoxythymidine triphosphate
dUDP deoxyuridine diphosphate
dUMP deoxyuridine monophosphate
dUTP
EDTA
deoxyuridine triphosphate ethylenediaminetetraacetic acid
ESI electrospray ionization
Et ethyl
EtOAc ethyl acetate
EtOH ethanol
FdUMP fluorodeoxyuridine monophosphate
XVI
XVI
FdUTP fluorodeoxyuridine triphosphate
FITC fluorescein isothiocyanate
FUTP fluorouridine triphosphate
Glu glutamate
Gly glycine
HB hydrogen bond
HJ Holliday junction
HPLC high-performance liquid chromatography
HR homologous recombination
HRMS high-resolution mass spectrum
hTMPK human thymidylate kinase
IC50
IPTG
half maximal inhibitory concentration isopropyl-β-D-thiogalactopyranoside
IR infrared
Kiapp
apparent inhibition constant
MD molecular dynamics
Me methyl
MeOH methanol
mp melting point
XVII
XVII
MW molecular weight
NADH nicotinamide adenine dinucleotide
NEt3 triethylamine
NHEJ non-homologous end joining
NMP nucleotide monophosphate
NMR nuclear magnetic resonance
PBS phosphate buffered saline
Pd/C palladium on carbon
PEP
PG
Piv
PPi
PTSA
phosphoenol pyruvate protecting group pivaloyl
pyrophosphate
p-toluenesulfonic acid
RNA ribonucleic acid
RNR ribonucleotide reductase
rt room temperature
SAR structure–activity relationship
shRNA small hairpin RNA
siRNA small interfering RNA
XVIII
XVIII
SSBs single-strand breaks
TBAI
TBDMS
tetrabutylammonium iodide tert-butyldimethylsilyl
TFA trifluoroacetic acid
THF tetrahydrofuran
TK thymidine kinase
TLC thin-layer chromatography
TOP2 topoisomerase II
TS thymidylate synthase
UDG uracil-DNA-glycosylase
UMP uridine monophosphate
YFP yellow fluorescent protein
1
Chapter 1. Introduction
1.1. Brief introduction of cancer
Cancer has remained top spot among the ten leading causes of death in Taiwan for
31 years. It is estimated that one person died of cancer in an average of every 3 minutes and 25 seconds, indicating the severe damage to human beings. Cancers usually arise from genomic instability,1 making cells become uncontrolled in proliferation and differentiation. By genome evolution, cancer cells have potential to proceed metastasis to other locations in body through blood and lymphatic systems, endangering one’s health enormously.
Oncogenes and tumor-suppressor genes have been confirmed to correlate with the production of cancers.2 The former are genes that has the potential to cause cancer, while the latter are, as its name, the genes which help to restrain formation of cancers. Once the function of tumor-suppressor genes such as Tp53 is disrupted, genomic instability drives cancer cell formation.3
Substances causing genomic mutation are called carcinogens, and can be roughly divided into three categories: chemical carcinogens, physical carcinogens, and biological carcinogens. Chemical carcinogens include organic and inorganic compounds, physical carcinogens contain radiation, especially ultraviolet and X-ray exposure, and biological
2
2
carcinogens comprise viruses and other microorganisms.
1.2. Inhibition of topoisomerase
Each human cell contains DNA with approximately 1.8 m long, winding around the nucleus tightly.4 Due to the double-helix structure of DNA, it is crucial for DNA to unwind its double strands in order to obtain information from genes. Topoisomeraseis an extremely important enzyme which contains a conserved tyrosine residue to react with phosphodiester bond in DNA, forming phosphotyrosine to cut the double strands.
Once the break has formed, DNA can be untangled or unwound, followed by reconnecting DNA backbone to alter the topology.5
Topoisomerase can be divided into two types according to the number of strands cut in one round of performance.6 Type I topoisomerase cleaves one strand of DNA, making the other strand rotate around to release the stress from supercoiled and reanneal the break site. On the other hand, type II topoisomerase cleaves both strands in DNA double helix; thus, the unbroken DNA duplex can pass through the breakage. After religation, DNA loop may be able to increase or decrease the tension.
Since topoisomerase is essential for DNA functionality, several drugs and
treatments are designed to target this enzyme (Figure 1). For example, camptothecin (CPT) and topotecan are widely used in clinical treatment against topoisomerase I.7
3
3
Chemotherapeutic agents targeting topoisomerase II (TOP2) are classified into two categories. The first class of compounds, which trap the enzyme intermediate, a TOP2–DNA covalent complex, resulting in permanent DNA lesion are called TOP2 poisons. The second class of compounds, which diminish enzymatic activity of TOP2 and lead to cell death pathway, are called TOP2 inhibitors. Most of the clinically active drugs are TOP2 poisons, including etoposide, mitoxantrone, and doxorubicin.8
Figure 1. Chemical structures of drugs targeting topoisomerases I and II.
4
4
Doxorubicin (adriamycin) is an antibiotic agent that blocks the synthesis of DNA by intercalating into DNA double strands, and further inhibits the function of TOP2 from religation. Cancer cells with rapid proliferation and high levels of TOP2 expression are most sensitive to doxorubicin. Doxorubicin is widely used for treating leukemias as well as cancers in breast, ovaries, multiple myeloma, and others.4, 9 However, the radical oxygen species (ROS) generated by interaction of doxorubicin with irons may result in severe side effects, such as cardiotoxicity,9 acute infection of bowels,10 and secondary malignancies.11 Moreover, cancer cells showing resistance to doxorubicin has emerged by alteration of NADPH metabolism,12 variation in cellular glutathione levels, and regulation of gene expression and TOP2 activity.13 Finding new drugs in combination with low dosage of doxorubicin to trigger cell apoptosis is a promising approach to cancer therapy.
5
5
1.3. Mechanism of DNA repair
To regulate the cellular concentrations of four deoxyribonucleoside triphosphates
(dNTPs) is critical for DNA synthesis during replication and repair. Once the levels of dNTP pool are too high or too low, DNA may be prone to mutation. Ribonucleotide reductase (RNR), which catalyzes the reduction of ribonucleotides into the corresponding deoxyribonucleotides (dADP, dUDP, dCDP, and dGDP) through radical reaction, is composed of two different dimeric subunits often called R1 and R2. These two subunits are regulated by different cell cycles, with RNR controlled by the level of R2.14 Besides, R2 may be able to collaborate with multiple oncogenes for malignant transformation and tumorigenesis.15 R2 is often overexpressed in tumor cells,16 suggesting that R2 mediates the activity of DNA repair.
Double-strand breaks (DSB) can be repaired through non-homologous end joining (NHEJ) or homologous recombination (HR).17 NHEJ ligates the break ends directly without the assistance of homologous template, and mainly predominates in the G1 phase.18 Compared with NHEJ, HR repair takes place during S/G2 phases, and requires a long homologous strand (usually sister chromatids) to lead repair.18, 19 After DSB occurs, HR repair starts by resection at the 5’ end on either side of the break, providing 3’ overhangs of single strand DNA (Figure 2). Strand invasion then proceeds on a homologous template by 3’ overhang strand to form a D-loop junction. Afterwards,
6
6
non-crossover and crossover products can be generated from synthesis-dependent strand annealing (SDSA) pathway or double-strand break repair (DSBR) pathway that forms an intermediate harboring two Holliday junctions (HJ).19 HR requires more than 10000 dNTPs for incorporation into DNA strands during DSB invasion.20 Therefore, RNR that mediates the recruitment of dNTPs, with R2 expressing through S and G2/M phases,16 plays a significant role for HR repair.
Figure 2. Mechanism of DNA homologous recombination.19
7
7
1.4. Biosynthetic pathway of dTTP in cells21
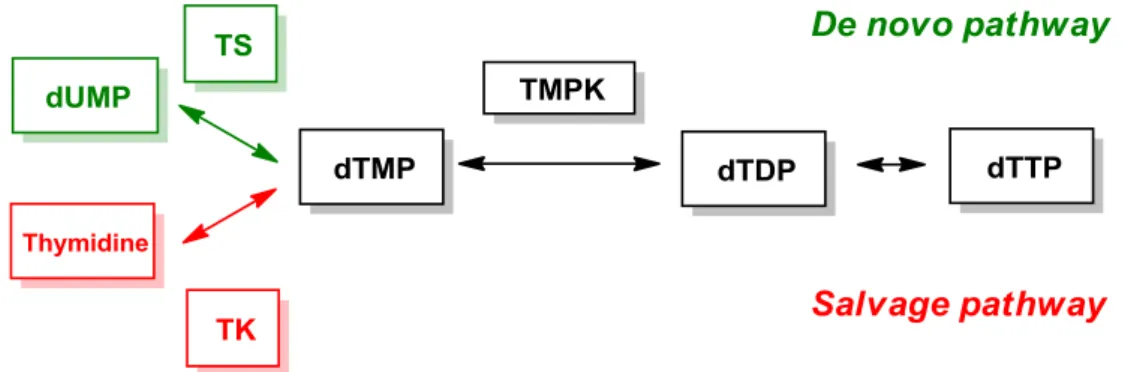
Among the four dNTPs in cells, dTTP is the only deoxyribonucleotide synthesized without RNR. The biosynthesis of dTTP can be divided into two pathways (Figure 3). In the de novo pathway, dUMP is catalyzed by rate-limiting enzyme thymidylate synthase (TS) to transform into dTMP. Thymidylate kinase (TMPK) then modifies dTMP to dTDP, followed by pyrophosphorylation through nucleotide diphosphate kinase to generate dTTP. For salvage pathway, dTMP is synthesized from thymidine with the help of thymidine kinase (TK). The subsequent phosphorylation steps are just the same as that in de novo pathway. Owing to the unique biosynthetic mechanism of dTTP, several cancer chemotherapeutic drugs are designed to focus on the inhibition of dTTP formation during DNA repair.
Figure 3. Biosynthetic pathways of cellular dTTPs.22
8
8
1.5. Antimetabolite agents targeting dTTP formation
Among the enzymes involving in biosynthesis of dTTP, thymidylate synthase (TS)
is a potential target for dTTP inhibition.23 It has been reported that several anticancer drugs including 5-fluorouracil (5-FU)23, 24 and raltitrexed (Tomudex),25 a folate analog, block the function of TS (Figure 4), thereby diminishing the production of dTMP and dTTP. Consequently, imbalanced recruitment of dNTPs mediated by RNR will induce DNA breakage during replication or repair to cause cells death.
Figure 4. Chemical structures of drugs targeting thymidylate kinase (TS).
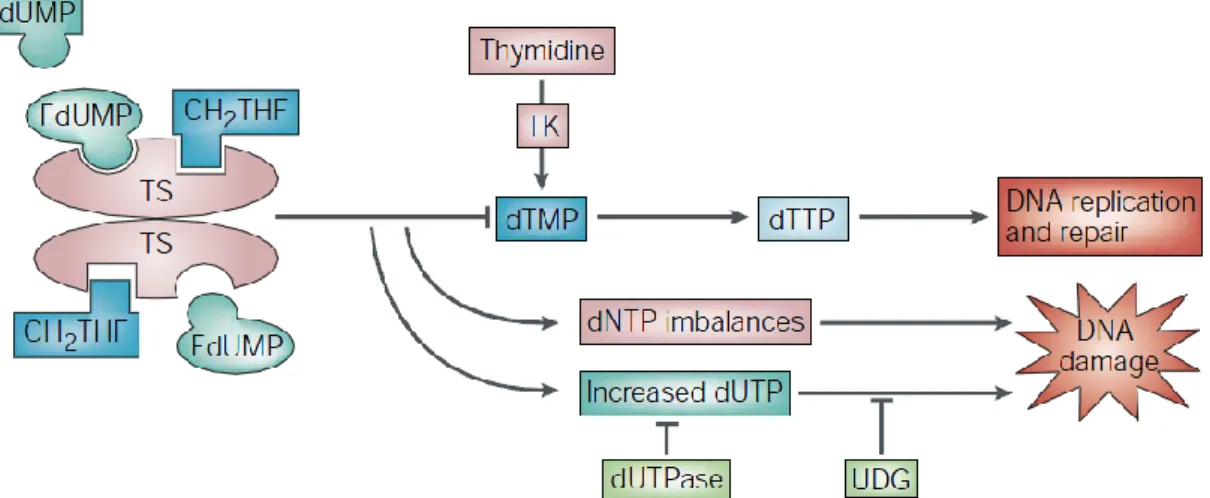
5-Fluorouracil (5-FU) is an analog of uracil with hydrogen atom at the C-5 position
being replaced by fluorine. Once entering into cells, 5-FU can camouflage as uracil to convert into several active metabolites including fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine triphosphate (FdUTP) and fluorouridine triphosphate (FUTP) (Figure 5). FdUMP binds irreversibly to TS that catalyzes the transformation of dUMP into dTMP with the help of the one-carbon methyl donor 5,10-methylenetetrahydrofolate (CH2THF), thereby blocking the binding site of normal
9
9
dUMP.24 The formation of dTMP in de novo pathway is inhibited, causing thymineless death. Moreover, inhibition of TS may result in accumulation of dUMP, and enhance the level of dUTP in cells. DNA polymerase is unable to distinguish between dUTP and dTTP,26 therefore, excessive amount of dUTP and FdUTP can be misincorporated into DNA. Such a futile process of misincorporation will eventually lead to DNA damage-induced cell death.
Figure 5. Mechanism of TS inhibition by 5-FU.24
The treatment of 5-FU for cancer chemotherapy has been used more than 40 years.
However, many of the therapies that use 5-FU alone or in combination with other agents show drug resistance. For instance, p53 disruption makes human colorectal cancer cells less sensitive to 5-FU.27 TS deficiency only sensitizes the p53- deficient cells to doxorubicin to a limit extent, probably due to the compensation of salvage pathway
10
10
mediated by TK.28 Since tumor cells are frequently mutated in p53,27 finding new anticancer drugs to inhibit the formation of dTTP without dependence on the expression of p53 will be particularly important.
1.6. Structure and physiology of TMPK
Because targeting TS may be complemented by TK through salvage pathway,
thereby diminishing the inhibition efficiency against TS,24 the enzymes participating in both dTTP biosynthetic pathways will be more reliable targets for drug development.
For example, TMPK is a good target because it stands at the junction of de novo and salvage pathways to catalyze the transfer of phosphate group from ATP to dTMP in the
presence of Mg2+.29
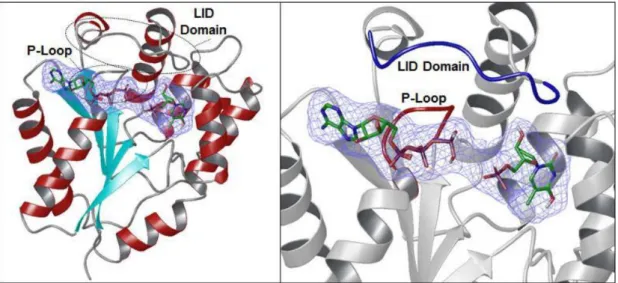
Thymidylate kinase (TMPK) is a member of nucleoside monophosphate (NMP) kinase family.30 The structure is usually a homodimer with each subunit composed of a central five-stranded β-sheet surrounded by α-helices(Figure 6).31 The enzymes are divided into three parts termed CORE domain, NMP binding site, and ligand-induced degradation (LID, residues 135-150) domain (Figure 6).31, 32 CORE domain is highly conserved among NMP kinases and contains the central parallel β-sheet as well as a phosphate binding loop (P-loop, residues 13-17) that controls the position of phosphate donor (ATP) for substrate recognition and catalysis. NMP binding region is largely
11
11
helical among most of the NMP kinases except guanylate monophosphate kinase. LID domain is flexible and covers a phosphate donor site partially for phosphate transfer.31, 32, 33
Figure 6. Ribbon structure of human TMPK (PDB ID: 1E2F). Left panel: central five-stranded β-sheet (light blue) surrounded by α-helices (red). Right panel: The P-loop (red) and LID domain (blue).31
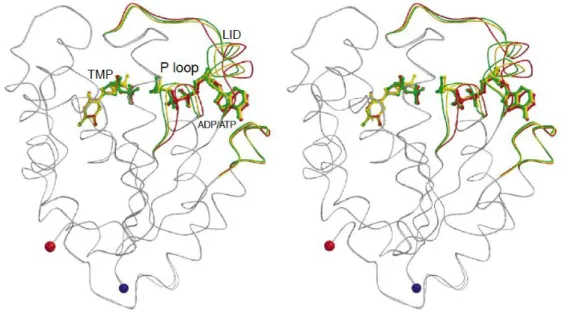
When both the nucleotide binding sites are occupied, the active site of TMPK will exist as a globally closed conformation.34, 35 Several conformational changes of adenine-base binding loop (residue 178-188 for human TMPK), P-loop, and LID loop in the globally closed conformation of TMPK have been confirmed to correlate with the phosphorylation activity. The structure of TMPK in the presence of TMP and ADP is termed P-loop open form, and the partially closed form is observed in combination with TMP and AppNHp (an ATP analog). When TDP and ADP bind to TMPK, P-loop is
12
12
situated in a fully closed state.32, 34, 35 Among the catalytic process, the conformation of TMPK will change from open into closed form according to the movement of P-loop, and bring the phosphoryl donor close to the acceptor.31, 35 It is noted that the distance between Arg16 (P-loop) and the β,γ-phosphate bridging atom of ATP in the partially and fully closed conformation is closer than that in the open form. This is important for stabilizing the negative charge occurring during the phosphoryl transfer. Besides, the side chain of Asp15 that is essential in TMPK catalysis is too far away to interact with the 3’-hydroxyl group of TMP or the Arg97 residue in the open form.35 Thus, the partially closed and fully closed conformations with P-loop may be structurally relevant to the catalytically active conformation, whereas the P-loop open form is an inactive state.34, 35
Figure 7. Movement of human TMPK structures: P-loop open (red), partially closed (yellow), and fully closed conformation (green) in response to binding of substrates.
13
13
P-loop, LID loop, and NMP binding site that change during catalytic process are colored.35
Lavie and coworkers36 classified TMPKs into two classes based on the position of basic residues (mostly Arg) in the active site. Type I TMPKs are mainly from eukaryotes (e.g. human and yeast). A basic residue occurs at position 10 in the P-loop that can interact with ATP; however the LID domain lacks this residue. In contrast, type II TMPKs are mainly from prokaryotes (e.g. E. coli). The basic residue is situated in the LID region (Arg153) rather than P-loop, which has a replacement of glycine.30,31, 36 By understanding the classification and mechanism of TMPK, one can better control the activity of enzyme for further drug discovery.
1.7. The relationship between the doxorubicin sensitization by TMPK inhibition to
p53 status
In order to confirm the relationship between human TMPK and p53 status, Chang and coworkers have generated lentivirus shRNA for hTMPK knockdown, and assessed the effect on dTTP pools in either isogenic p53 (+/+) or p53 (-/-) HCT-116 colon cancer cells.37 After infected by lentivirus for 3 days, the steady state level of dTTP diminished gradually up to 50% in TMPK silenced cells compared with normal cancer cells (scramble shRNA) in both p53 (+/+) and p53 (-/-) types, with the dTTP amount of p53
14
14
(+/+) cells higher then p53 (-/-) cells, indicating the essential role of TMPK in dTTP formation (Figure 8).
Additionally, the combination of TMPK silencing and genotoxic agents was also examined. As shown in Figure 9A, neither low dosage (0.5 μmol/L) of doxorubicin nor TMPK knockdown alone showed apparent effect on apoptotic induction. Nevertheless, TMPK silencing followed by doxorubicin treatment led to cell death (Annexin V-positive) in both p53 (+/+) and p53 (-/-) cells. On the other hand, 1 μmol/L administration of doxorubicin after TS deletion led to apoptosis with p53 (+/+) rather than p53 (-/-) cells (Figure 9B). These evidences revealed that low-dose doxorubicin in combination with TMPK knockdown was p53 independent, and was more effective than that with TS depletion (p53 dependent) to induce apoptosis of p53 deficient cells.
Figure 8. dTTP pool size measurement of lentiviral TMPKshRNA and scrambleshRNA in p53-proficient and p53-null HCT-116 cells. (*, P = 0.06; **, P = 0.01; ***, P < 0.001 based on a two-tailed Student’s t test.)
15
15
Figure 9. Comparison of TMPK and TS knockdown on doxorubicin-induced apoptosis in p53(+/+) and p53(-/-) HCT-116 cells by FITC-labeled Annexin V apoptosis assay.
1.8. Confirming the characteristic role of hTMPK during DNA repair
Based on the aforementioned information, it is hypothesized that reducing the
formation of dTTPs by inhibiting TMPK function will sensitize tumor cells to doxorubicin. As the DNA breakage occurs after doxorubicin treatment, DNA repair will start by incorporating dNTPs into DSB site. Since HR requires more than 10000 dNTPs
16
16
to repair each DSB, the supply of dNTPs mediated by RNR is highly demanded.
Blocking TMPK function reduces the level of dTTP at damage site, and causes the imbalanced incorporation of four dNTPs during HR repair. Since DNA polymerase is unable to differentiate between dUTP and dTTP, surpluses amount of dUTP will misincorporate into DNA and trigger cell death finally (Figure 10).
R2, which is a subunit of RNR, is cell-cycle regulated and usually expresses during G2/M phases. Malignant tumor cells are cell-cycle checkpoint defected,38 and thus show high G2/M-phase population after recovery from DNA damage, during which HR repair takes place along with R2 overexpression. On the other hand, the intact checkpoint functionality in normal cells reduces the population during S phase, and triggers cell quiescence into G0 phase, leading to low expression of R2 and less dUDP formation thereafter. Hence, TMPK knockdown will sensitize DNA damage only in tumor cells rather than normal cells.39
Figure 10. Combined treatment of doxorubicin and hTMPK inhibitor to trigger cell apoptosis.22
17
17
To confirm the hypotheses, Hu and Chang39 first investigated the contribution of TMPK during DNA repair. γH2AX, the indicator of double-strand breaks, was higher in the TMPK knockdown MDA-MB231 breast cancer cells compared with control cells after recovery from a low-dosage doxorubicin treatment for 24 h, demonstrating that TMPK is essential for DNA repair in cancer cells (Figure 11a). Thereafter, uracil misincorporation test was conducted by inhibiting uracil N-glycosylase (UNG), an enzyme which removes erroneous uracil from DNA and forms single-strand breaks (SSBs) subsequently. After 24 h recovery from DNA damage, XRCC1 foci (SSBs marker) was reduced apparently by UNG knockdown in TMPK-knockdown cells (Figure 11b).
In order to make sure RNR will generate dUDP for incorporation during DNA repair in the absence of TMPK, the recruitment of RNR to DNA damage site was blocked by yellow fluorescent protein (YFP) fusion. The results showed that decline of RNR recruitment abolished the effect of TMPK knockdown during recovery from doxorubicin treatment (Figure 12a). Besides, TMPK and RNR were proved to co-localize and bind to the damage site of DNA, where is the place needed for dNTPs balance (Figure 12b). Furthermore, reducing the expression of R2 generated DNA repair in spite of TMPK silencing in tumor cells (Figure 12c). These evidences indicated that DNA repair required the cooperation of TMPK and R2 in RNR at the damage site.
18
18
TMPK is essential to DNA repair by preventing dUTP misincorporation.
(a) (b)
Figure 11. (a) γH2AX foci staining of MDA-MB231 cells with or without TMPK silencing after recovery from doxorubicin. (b) XRCC1 foci staining of LacZ or UNG lentivirus infected cells recovered from doxorubicin treatment for 24 h.
(a) (b)
19
19
(c)
Figure 12. (a) pCMV2-YFP-Nuc (vector) or pCMV2-YFP-Nuc-R1C (plasmid) transfected into TMPK or TMPK knockdown cells. (b) Laser-micro-irradiation of Hela cells followed by immunofluorescence staining. (c) γH2AX foci staining of MCF-7 cells with TMPK, R2 and TMPK/R2 knockdown at the indicated time points.
Given that TMPK knockdown will trigger apoptosis of tumor cells with low-dosage doxorubicin treatment, it is worthwhile to investigate the selectivity between cancer and normal cells. Breast cancer cell line MCF-7 as well as nontumorigenic H184B5F5/M10 and MCF10A were tested for the influence of TMPK silence during DNA repair. After recovery from DNA damage, γH2AX foci remained in MCF-7 cells but not H184B5F5/M10 or MCF10A cell lines, indicating the nontoxicity of TMPK knockdown toward normal cells (Figure 13). To further understand the reason for selectivity, Chang and coworkers used flow cytometry analysis to study the cell-cycle population of cell lines after doxorubicin treatment. H184B5F5/M10 and MCF10A cells showed higher population in G0/G1 phases compared with MCF-7 and MDA-MB231 cancer cells, which possessed larger S and G2/M phase populations after DNA damage (Figure 14), demonstrating lower integrity of checkpoint function in these cancer cells.
20
20
Figure 13. γH2AX foci staining of MCF-7, H184B5F5/M10, and MCF-10A cells transfected with TMPK siRNA after DNA damage recovery.
Figure 14. Flow cytometry of cells at the indicated time points after recovery from doxorubicin.
21
21
1.9. Screening inhibitors against hTMPK
Based on the significant characteristics of TMPK during DNA repair in tumor cells, searching for small molecules targeting TMPK will be a promising direction. Two methods have been developed for TMPK enzymatic assay.31, 40 The first method uses isotope labeled dTMP as the substrate. After incubation with TMPK, dTMP acquires a phosphate group from ATP, and the ratio between dTMP and dTDP are evaluated by the radiative intensities for calculation of enzymatic activity. However, this method requires long time and is not suitable for high-throughput screening even though it is accurate by using only one enzyme. Another wildly used method is NADH-coupled enzymatic assay.
This method comprises three enzymes, TMPK, pyruvate kinase, and lactate dehydrogenase (Figure 15). TMPK phosphorylates dTMP into dTDP along with the generation of ADP. The ADP then turns back to ATP by reacting rapidly with phosphoenolpyruvate (PEP) in the presence of pyruvate kinase. Pyruvate formed from PEP turns into lactate by lactate dehydrogenase with the cofactor NADH oxidized into NAD+. Since NADH shows absorbance at 340 nm, the activity of TMPK can be inferred from measurement of the absorbance at 340 nm. Two additional enzymes are required in this assay, and the sensitivity is limited by the low extinction coefficient of NADH. In addition, the accuracy of this assay may be interfered with any substrate absorbs near the region of 340 nm.
22
22
To circumvent the disadvantage of NADH-coupled enzymatic assay, Chang and coworkers40 develop a new method termed luciferase-coupled TMPK assay, which has been used for screening hTMPK inhibitors. Luciferase catalyzes the oxidation of luciferin in the presence of ATP and generates luminescence. Hence, inhibiting TMPK will decrease the amount of ATP for luciferase. According to the intensity of luminescence, ATP remains after TMPK consumption can be determined accordingly.
Compared with the conventional assays, this method measures luminescence intensity which is more sensitive and rapid in determining the activity of hTMPK without the interference of substrate absorption (Table 1).
NADH-coupled enzymatic assay
Luciferase-coupled TMPK assay
Figure 15. Principles of NADH- and luciferase-coupled TMPK assays.
23
23
Table 1. Comparison of sensitivity in NADH- and luciferase-coupled TMPK assays.40 hTMPK protein (μg) Luciferase-coupled assay NADH-coupled assay
0.01 3.8 ± 0.2 a Not detectable
0.05 18.2 ± 0.9 a Not detectable
0.10 36.5 ± 1.1 a 33.8 ± 7.4 a
1.00 89.3 ± 4.7 a 85.7 ± 6.1 a
0.50 177.3 ± 6.4 a 166.1 ± 4.0 a
1.00 373.4 ± 5.1 a 387.5 ± 9.7 a
a.Mean value (pmol of dTDP formation /min) of five independent experiments. Different concentrations (0.01 to 1 μg) of purified hTMPK protein were tested. In the luciferase-coupled assay, TMPK activity (ΔRLU/min) was converted to the amount of dTDP formation per minute. In the NADH-coupled assay, TMPK activity (ΔA340nm/min) was estimated by the following equation: dTDP formation (pmol/min 100ul-1) = ΔA340nm 6.22 × 105 / reaction time (min).
Using luciferase-coupled TMPK assay for high-throughput screening, a library of 21,120 small molecules was screened to find YMU1 as a potent compound (Figure 16), with IC50 0.061 ± 0.02 μM and Ki 0.22 ± 0.03 μM against TMPK.39 As shown in Figure 17a, YMU1 itself did not give rise to genome toxicity in cells, unlike 5-FU which caused cell death without selectivity between normal and tumor cells. However, pretreatment of YMU1 followed by treatment with low-dosage doxorubicin demonstrated apparent DNA damage in tumor cells rather than normal cells (Figure 17b), indicating that YMU1 impaired DNA repair as effective as TMPK silencing.
To further verify that the combined use of doxorubicin and YMU1 was also verified to inhibit the growth of tumor cells, HCT-116 and MCF-7 cells were test and all showed
24
24
growth suppression (Figure 18a). Moreover, the size of tumor in mice treated with both YMU1 and doxorubicin was significantly smaller than that individually treated with YMU1 or doxorubicin (Figure 18b). Taken together, these results indicate that YMU1 has high potential in targeting human TMPK for tumor suppression. Therefore, YMU1 is a promising lead compound for development in cancer chemotherapy.
Figure 16. Chemical structure of YMU1.
25
25
(a) (b)
Figure 17. (a) 5000 cells of H184B5F5/M10, 3000 cells of MCF10A, MCF-7, and HCT-116 p53(-/-) were treated with YMU1 or 5-FU at different concentration, and stained by crystal violet after 14 days. (b) γH2AX foci staining of cells after treated with vehicle or YMU1 along with doxorubicin exposure.
(a) (b)
Figure 18. (a) On treatment with vehicle or YMU1 for 72 h, followed by 4 h doxorubicin exposure, cells were incubated for colony formation over 14 days.
(b) Comparison of tumor growth in mice with combined treatment of YMU1 and doxorubicin or doxorubicin/YMU1 alone for two weeks.
26
26
27
27
Chapter 2. Results and Discussion
2.1.1. Structural feature of YMU1
Chemical structure of YMU1 can be divided into three parts: The heterocyclic core,
linker containing a six member ring, and the end group R (Figure 19). Based on the derivatives synthesized previously by Yeh,22 several structure features are considered to maintain the inhibitory activity against hTMPK, including the role of sulfur atom in core structure, the importance of N-alkylation with linker over O-alkylated analogs, the appropriate length of linker with a six member ring, and the alteration of the end group R. (Figure 20).
Figure 19. Structure of YMU1.
28
28
Figure 20. Some structural features of YMU1 derivatives.22
2.1.2. Molecular dynamic simulation of YMU1 with TMPK
By means of molecular dynamic (MD) simulations of YMU1 with the crystal structure of TMPK, the inhibition mechanism can be further investigated. As shown in Figure 21, P-loop and LID region are connected by several residues including Arg16, Gly144, Ala145, Glu152, Asp15, Arg143, Ala17, and Ala180 via hydrogen-bond network. When YMU1 is incubated, the connection between LID and P-loop is disrupted, and the LID region is pushed outward. Arg16 in P-loop is supposed to interact with the heterocyclic core part of YMU1 by a π-π stacking, which can stabilize the disposition of YMU1 (Figure 22). In addition, YMU1 is able to dock into the ATP binding site and thus prevents Mg2+ from interacting with Asp15, a residue that is crucial for the activity of TMPK. O-alkylation derivatives cannot prevent this interaction, thus show poor inhibition against TMPK.39 Moreover, the two carbonyl groups of YMU1 can chelate
29
29
one Mg2+ ion, therefore, a long linker will be disfavored. Further modification of YMU1 can be performed more practically with the assistance of the simulation results.
Figure 21. MD simulation of the mechanism of TMPK with or without YMU1.
Hydrogen bonds were indicated with dash lines and YMU1 was shown as bright blue structure.
Figure 22. Stabilzation of YMU1 in hTMPK via the π-π stacking interaction with Arg16.
2.1.3. Specific YMU1 derivatives proposed in this study
Although the previous study22 indicated that the end group R in YMU1 made no
30
30
apparent difference in TMPK inhibition, I still plan to synthesize the dimeric compounds having the R group containing another heterocyclic core structure to test their inhibitory activity against TMPK. Besides, I further modified the structure of YMU1 to investigate the importance of the sulfur atom in the heterocyclic core, and to enhance the hydrophilicity by changing substituents in the core structure and the R group.
Figure 23. Specific YMU1 derivatives.
2.1.4. Luciferase-coupled TMPK assay
After setting the research target, the next step is to identify the inhibitory activity of YMU1 derivatives against hTMPK in cancer cells with the combined use of low-dosage doxorubicin. In order to screen inhibitors more efficiently and accurately, Chang and coworkers have established a convenient protocol of luciferase-coupled TMPK assay (Figure 24).40 Each test compound was dissolved in DMSO to a concentration of 5 mM,
31
31
and then diluted into different concentrations for tests. For example, a drug (10 μM) was transferred to a 96-well plate containing 10 μL of 0.5 μg TMPK for 10-min preincubation. Then, 30 μL of buffer solution containing 100 mM Tris-HCl (pH 7.5), 100 mM KCl, 10 mM MgCl2, 50 μM ATP and 100 μM dTMP were added to initiate the reaction. After 10 min, 200 μL of 100 μM 5,5’-dithiobis(2-nitrobenzoic acid) (DTNB) was added to stop the reaction. The sample solution (10 μL) was transferred to a 96-well plate, followed by adding 90 μL of luciferase assay buffer containing 50 mM Gly (pH 7.0), 0.1 μg luciferase, 0.5 mM EDTA, 50 μM luciferin, 5 mM MgCl2 and 0.1 % bovine serum albumin (BSA) into each well to measure the luminescence.
Figure 24. Protocol of luciferase-coupled TMPK assay.22
32
32
2.1.5. General synthetic scheme
The general synthetic strategy for YMU1 derivatives is shown in Scheme 1. Using piperazine as the starting material, the substitution reaction with 1 equiv of alkyl halide (R3Cl) would generate a mono-substituted piperazine A. The substitution reaction of chloroacetyl chloride would be performed via addition–elimination mechanism to form disubstituted piperazine derivative B. Further substitution of the chlorine atom with iodine atom would be conducted by using sodium iodide as a nucleophile in acetone.
Finally, diverse heterocyclic compounds D would be introduced by coupling with the iodo-substituted C to give a series of the desired YMU1 derivatives E.
Scheme 1. General synthetic scheme for YMU1 derivatives.
2.2.1. Replacement of the sulfur atom in heterocyclic core
Heterocyclic core 2 was synthesized from the commercially available 2-chloro-4,6-dimethlylnicotinonitrile (1) and thiourea in n-butanol at 118 oC. The
33
33
oxidative cyclization reaction was then performed in conc. H2SO4 at 100 oC for 4 h to afford compound 3 (Eq. 1).
In the first part, we aimed to test the significance of the sulfur atom in heterocyclic core for TMPK inhibition. Yeh has changed the sulfur atom of YMU1 into sulfone, and found the derivative did not show inhibition against hTMPK. We further replaced the sulfur atom with oxygen, nitrogen, carbon, carbonyl, and N-benzyl group to investigate the role of sulfur atom in the heterocyclic core.
We first focused on constructing the heterocyclic core having an N-benzyl group.
The commercially available 1H-indazol-3(2H)-one (4) was treated with benzyl chloride and sodium hydroxide in H2O at 70 oC for 2 h. The precipitates were collected and purified to afford compound 5 (Eq. 2).41
34
34
Using cyclohexane as the reducing agent, reduction of phthalimide (6) was promoted by aluminum chloride to afford phthalimidine 742 as the carbon-substituted YMU1 derivative (Eq. 3).
The oxygen-containing heterocyclic core 943 was synthesized from salicylhydroxamic acid (8) with carbonyl diimidazole in THF at 70 oC (Eq. 4).
2.2.2. Synthesis of piperazine linkers
The commercially available piperazine was treated with Boc-anhydride (1 equiv) in the presence of a base DIEA to form the mono-Boc-protected piperazine 10 (Scheme 2).
The reaction of 2-chloroacetyl chloride with 10 provided compound 11, which underwent a substitution reaction with sodium iodide in acetone to give the iodo compound 12.
35
35
Scheme 2. Synthesis of compound 12.22
2.2.3. Synthesis of YMU1 derivatives 13–17
The heterocyclic compounds 5–7 and 9 underwent substitution reactions with the iodo compound 12 in the presence of appropriate base to give the YMU1 derivatives 13 and 15–17, respectively (Scheme 3). Removal of the benzyl group from 13 was carried out by hydrogenation over Pd/C in MeOH to give compound 14.
Scheme 3. Synthesis of compounds 13–17.
Noteworthily, the substitution reaction of phthalimidine 7 with 12 could not be realized by using DIEA as shown in the general synthetic procedure. We thus tested different bases, including NaH, LiH and Cs2CO3, and solvents, including DMF, THF and CH3CN, for this transformation (Table 2). The reaction using Cs2CO3 in CH3CN at
36
36
50 oC showed the highest yield (entry 4).
Table 2. Reaction conditions for the synthesis of compound 16.
Entry Conditions Results
1 DIEA, CH2Cl2, rt, 5 d No reaction
2 NaH, DMF, rt, 5 d 16 (41%)
3 LiH, THF, reflux, 3 d 16 (16%) 4 Cs2CO3, CH3CN, 50 oC, 36 h 16 (49%)
2.2.4. Structure–activity relationship of compounds 3–7, 9 and 13–18
After synthesizing a variety of derivatives by replacement of the sulfur atom in YMU1, we started to study the relationship between the structure and inhibitory activity.
As shown in Figure 25, benzo[d]isothiazol-3(2H)-one (BT) and 3 was used to compare with compounds 4–7 and 9 having heterocyclic cores, whereas compound 18 was used to compare with compounds 13–17.
37
37
Figure 25. Chemical structures of benzo[d]isothiazol-3(2H)-one (BT), compounds 3–7, 9, 13–18 and YMU1.
Table 3. Normalized TMPK inhibition of compounds BT, 3–7, 9, 13–18 and YMU1.44
Compound Inhibition (%)a Compound Inhibition (%)a
BT 51.8 13 8.4 ± 2.5b
3 19.6 14 0.6 ± 1.5b
4 –3.3 15 18.7 ± 3.4b
5 –4.5 16 2.2 ± 5.4b
6 13.1 17 10.6 ± 7.6b
7 –6.0 18 91.3 ± 5.7b
9 –3.3 YMU1 70.0 ± 7.0b
a.Normalized TMPK inhibition of the test compound compared with YMU1 as 70%
inhibition at 2 μM.
b.Data of quadruplicate experiments.
38
38
Table 3 shows the inhibitory activity of compounds at 2 μM using luciferase-coupled TMPK assay as the preliminary test. The normalized inhibition value was calculated according to a formula:
(inhibitory activity of compound) ÷ (inhibitory activity of YMU1) × 100%.
Though BT and 3 still possessed moderate TMPK inhibition, compounds 4–7 and 9 having different heterocyclic cores without sulfur atom revealed poor activity against TMPK. Moreover, compounds 13–17 without sulfur atom were also inactive to TMPK, whereas the sulfur-containing derivative 18 was highly active with 91% normalized inhibition against TMPK. These results confirmed that the sulfur atom placed in the heterocyclic core of YMU1 indeed played an important role in the TMPK inhibition activity. Once the sulfur atom is replaced by other atom or functional group, the efficiency of inhibition against TMPK will decrease dramatically.
2.3.1. Hydrophilic YMU1 derivatives
Lipophilicity refers to the ability of chemical compound to partition between organic and polar aqueous phases (two immiscible solvents).45 Molecules with high capacity to form hydrogen bonds are more hydrophilic and tend to dissolve in high polar substances such as water. On the other hand, compounds having less hydrogen bonds often interact within themselves with London dispersion force, and are more favorable