pKNOT v.2: the protein KNOT web server
Yan-Long Lai
1, Chih-Chieh Chen
1and Jenn-Kang Hwang
1,2,*
1
Institute of Bioinformatics and Systems Biology, National Chiao Tung University and 2Center for Bioinformatics Research, National Chiao Tung University, Hsinchu 30068, Taiwan, Republic of China
Received February 17, 2012; Revised May 12, 2012; Accepted May 26, 2012
ABSTRACT
Knotted proteins have recently received lots of at-tention due to their interesting topological novelty as well as its puzzling folding mechanisms. We pre-viously published a pKNOT server, which provides a structural database of knotted proteins, analysis tools for detecting and analyzing knotted regions from structures as well as a Java-based 3D graphics viewer for visualizing knotted structures. However, there lacks a convenient platform performing similar tasks directly from ‘protein se-quences’. In the current version of the web server, referred to as pKNOT v.2, we implement a homology modeling tool such that the server can now accept protein sequences in addition to 3D structures or Protein Data Bank (PDB) IDs and return knot analysis. In addition, we have updated the database of knotted proteins from the current PDB with a combination of automatic and manual pro-cedure. We believe that the updated pKNOT server with its extended functionalities will provide better service to biologists interested in the research of knotted proteins. The pKNOT v.2 is available from http://pknot.life.nctu.edu.tw/.
INTRODUCTION
Knotted proteins are interesting not only in their extraor-dinary topologies (1,2) but also in their intriguing folding mechanisms (3–6). There are currently four types of knots identified in the protein structures in Protein Data Bank (PDB): the trefoil knot (or 31knot), the figure-eight knot
(or 41knot), the 52knot and the Stevedore’s knot
(or 61knot). Knotted proteins present a knotted problem
to both experimental and theoretical biologists: how does a peptide chain thread through the loops to form multiple crossing (up to six crossings) knots? Recent experiments showed that some dimeric knotted proteins appear to have a similar folding mechanism as that of unknotted
proteins (7), and that knotted proteins can exist in a knotted conformation even in their chemical unfolded states (8). Computer experiments have been performed to simulate possible folding mechanisms of knotted proteins (4,5). Protein’s knots are implicated in substrate binding and enzyme activity (9). For example, the knot of N-acetylornithine transcarbamylase is part of the active site (10), while the knot TrmD tRNA methyltransferase is shown to be important for substrate binding and cata-lytic activity (11).
Currently, there are two web servers available for analysis of knotted protein—pKNOT (12) (http://pknot .life.nctu.edu.tw) and KNOTS (13) (http://knots.mit.edu). Both web servers share similar functionalities: they provide a database of knotted protein structures; they can analyze structures for possible knots and they provide a 3D mo-lecular viewer for users to visualize and to manipulate the orientations of the knotted structures. In addition, pKNOT provides information about the smallest possible peptide chain that can form a knot structure and generates movie files of knot detection processes for pedagogical purpose (12). These web servers provide structure-based analysis for uses, but they cannot accept query sequences for knot analysis.
Herein, we implemented a structure modeling module based on (PS)2(14,15), which uses a consensus strategy in both template selection and target-template alignment to model 3D structures from homologous sequences. In this way, the updated pKNOT server can accept query se-quences, build its 3D structure, analyze its structure for possible knots and, if found, return their knot types and information such as the knot core and the knot depth (12) of the knots identified in the structures.
MATERIALS AND METHODS
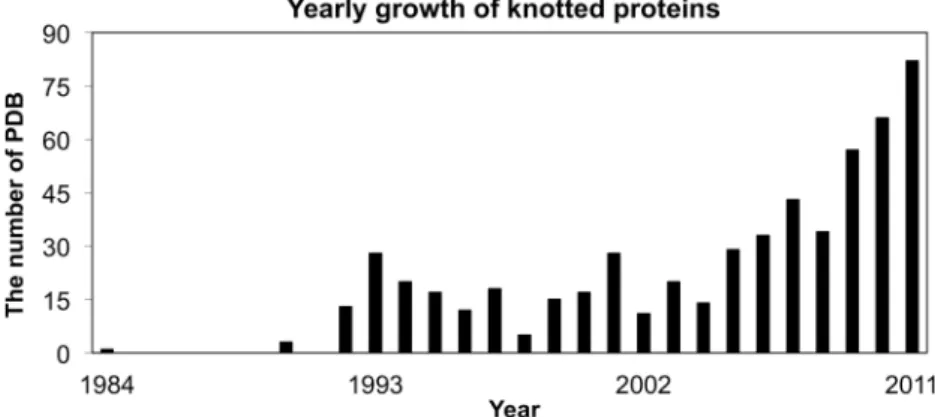
As the number of solved 3D protein structures increases, so is the number of knotted proteins, as shown in Figure 1. As reported previously (12,16,17), missing residues in 3D structures or non-standard PDB formats may cause mis-identification of knots in automatic approaches. To ensure the accuracy of our database of knotted proteins, we used
*To whom correspondence should be addressed. Tel: +886 3 513 1337; Fax: +886 3 572 9288; Email: [email protected]
W228–W231 Nucleic Acids Research, 2012, Vol. 40, Web Server issue Published online 12 June 2012 doi:10.1093/nar/gks592
ß The Author(s) 2012. Published by Oxford University Press.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/ by-nc/3.0), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
at National Chiao Tung University Library on April 28, 2014
http://nar.oxfordjournals.org/
a hybrid approach: we first scan the PDB using automatic procedures to generate a smaller set of knotted proteins (12), and then examine the smaller set by manual inspec-tion to remove structures with dubious knots.
Homology modeling and knot detection
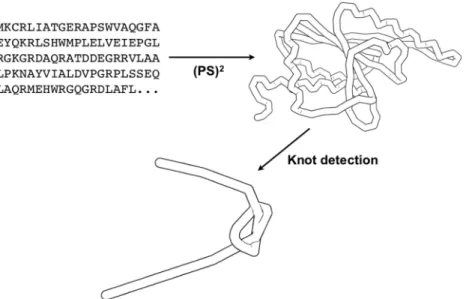
Since the protein families of knotted proteins are limited, it is feasible to build reliable homologous structures from protein sequences. 3D structures are modeled from protein sequences using (PS)2(14,15) previously developed by our laboratory. The (PS)2method, taking advantage of a con-sensus strategy for template selection and target-template alignment, compares favorably with most homology modeling methods (15). pKNOT detects knots using Taylor’s algorithm (1), which is basically a smoothing pro-cedure of a 3D curve—it first fixes the protein’s N and C termini, and then repeatedly smoothes and straightens the protein chain. For an unknotted structure, it will reduce the chain into a simple straight line; for a knotted struc-ture, with its structural details being smoothed out, its knot can be easily detected. Although the method usually converges in <50 iterations, it happens that the method may not converge after a given number of iter-ations. This may cause misidentification of protein knots. We have implemented a file, named Convergence File, to provide results at each iteration. The Convergence File, as well as the 3D structure viewer, will help users to ensure the convergence of results. The knot type can be topo-logically detected by computing the corresponding Alexander polynomial (18). However, in the current version, knot types are identified by manual inspection. The simple work flow of pKNOT is schematically shown in Figure 2.
With a modeled structure or a crystal structure avail-able, the pKNOT server can compute the knot core and the knot depth of the knotted region of a knotted protein. The knot core is defined as the smallest region that will remain as a knot (1). The knot depth represents the product of the number of residues that must be deleted from both ends in order to free the knot (1). Both values provide useful information for a further investigation of the structural characteristics of the knotted region.
Input format
The pKNOT web server can accept two types of input (Figure 3): in STRUCTURE QUERY, users can either type PDB ID or upload a structural file in the PDB format and in SEQUENCE QUERY, users can enter protein sequences in FASTA format. In STRUCTURE QUERY, several advanced options are available: user can toggle either IGNORE (default) or PRESERVE option during chain smoothing processes. It sometimes happens that there are missing residues in protein struc-tures. The IGNORE option will close the breaks by using the shortest line segment connecting the breaks, while the PRESERVE option preserves the breaks in the chain, keeping the endpoints of each segment fixed. Users can also set the number of iterations (the default values is 500) and the collision threshold (the default threshold 0.5 A˚). The collision threshold is the minimal distance to determine whether line segments will intersect during smoothing procedures (12). However, users are advised to try the default values first. It usually takes <10 s in run time to detect a protein knot, but it takes longer time to model a homologous structure, around 3–10 min in run time. However, for a very long sequence, say, of 2000 amino acids, it may take >20 min in run time. Output format
Upon structure query, pKNOT will return the results con-cerning each chain of the structure, including its length (CHAIN LENGTH), the type of knot (KNOT TYPE), the length of each knot (KNOT LENGTH) and the visu-alization of each knotted structure (DISPLAY STRUCTURE). If clicked on the KNOT TYPE, the server will return a complete list of knotted structures. The server provides a 3D molecular viewer for users to view 3D structures and manipulate their orientations in space. The original and the smoothed structures can be visualized together or individually in the 3D graphics viewer for easy comparison.
Upon sequence query, if the 3D structure being success-fully built, its modeled structure will be shown in the 3D graphics viewer and its knot type based on sequence homology will be returned (Figure 3).
Figure 1. The yearly growth of knotted proteins in PDB from 1984 to 2011.
Nucleic Acids Research, 2012, Vol. 40, Web Server issue W229
at National Chiao Tung University Library on April 28, 2014
http://nar.oxfordjournals.org/
Database
We have identified 566 knotted structures, which are almost twice as many as those of the last version of pKNOT (12). We have currently identified four types of knots—(i) the proteins with trefoil knots including (a) methyltransferase, (b) transcarbamylase, (c) methio-nine adenosyltransferase, (d) carbonic anhydrase, (e) pre-mRNA-splicing factor RDS3, (f) VirC2-like pro-teins and (g) MJ0366-like protein; (ii) the propro-teins with figure-eight knots including (a) phytochrome, (b) the core proteins of bluetongue virus and (c) ketol-acid reductoisomerase; (iii) ubiquitin hydrolase identified with a 52knot and (iv) a-haloacid dehalogenase (PDB ID: 1jbx)
being only structure identified with currently the most complicated knot, i.e. a Stevedore’s knot. Knotted structures can be classified into 10 SCOP folds, comprising
17 SCOP families (2). However, it should be noted that, as the date of writing, 1bjx has not yet been classified in SCOP. User can download the list of the complete knotted proteins.
DISCUSSION
Herein, we present an updated version of pKNOT web server. The size of the updated database of knotted struc-tures is almost double the size of the previous version. Each knotted structure of the 566 knotted structures is manually validated to reduce false positives usually plagued a fully automated detection system. One of the unique features of pKNOT v.2 is the integration of homology modeling with the existing knot detection and analysis functions such that the updated server can accept
Figure 2. The schematic work flow of pKNOT: accepting a query protein sequence, modeling a 3D structure through homology modeling (PS)2and
smoothing out its backbone for the detection of its knot. The knot shown in this example is a 31knot.
Figure 3. The features of the pKNOT v.2 web server: (A) STRUCTURE QUERY: users can enter PDB ID or upload PDB file. (B) SEQUENCE QUERY: users can enter or upload protein sequences in FASTA format. (C) Users can view the modeled structure in 3D graphics viewer and inspect its knot region.
W230 Nucleic Acids Research, 2012, Vol. 40, Web Server issue
at National Chiao Tung University Library on April 28, 2014
http://nar.oxfordjournals.org/
protein sequences as well as protein structures. We believe pKNOT v.2 will prove more useful than its pervious version to biologists.
ACKNOWLEDGEMENTS
We are grateful to both the hardware and software supports of the Structural Bioinformatics Core Facility at National Chiao Tung University.
FUNDING
Academic Summit Program of National Science Council [100-2745-B-009-001-ASP, in part]; the Center for Bioinformatics Research of Aiming for the Top University Program of the National Chiao Tung University; Ministry of Education, Taiwan, Republic of China, in part. Funding for open access charge: National Science Council.
Conflict of interest statement. None declared.
REFERENCES
1. Taylor,W.R. (2000) A deeply knotted protein structure and how it might fold. Nature, 406, 916–919.
2. Hwang,J.K., Lai,Y.L. and Yen,S.C. (2010) In: Zhao,Z. (ed.), Sequence and Genome Analysis: Methods and Applications Comprehensive Analysis of Knotted Proteins. iConcept Press, Queensland, pp. 22–39.
3. Prentiss,M.C., Wales,D.J. and Wolynes,P.G. (2010) The energy landscape, folding pathways and the kinetics of a knotted protein. PLoS Comput. Biol., 6, e1000835.
4. Sulkowska,J.I., Sulkowski,P. and Onuchic,J. (2009) Dodging the crisis of folding proteins with knots. Proc. Natl Acad. Sci. USA, 106, 3119–3124.
5. Bolinger,D., Sulkowska,J.I., Hsu,H.P., Mirny,L.A., Kardar,M., Onuchic,J.N. and Virnau,P. (2010) A Stevedore’s protein knot. PLoS Comput. Biol., 6, e1000731.
6. King,N.P., Jacobitz,A.W., Sawaya,M.R., Goldschmidt,L. and Yeates,T.O. (2010) Structure and folding of a designed knotted protein. Proc. Natl Acad. Sci. USA, 107, 20732–20737. 7. Mallam,A.L. and Jackson,S.E. (2005) Folding studies on a
knotted protein. J. Mol. Biol., 346, 1409–1421.
8. Mallam,A.L., Rogers,J.M. and Jackson,S.E. (2010) Experimental detection of knotted conformations in denatured proteins. Proc. Natl Acad. Sci. USA, 107, 8189–8194.
9. Wagner,J.R., Brunzelle,J.S., Forest,K.T. and Vierstra,R.D. (2005) A light-sensing knot revealed by the structure of the
chromophore-binding domain of phytochrome. Nature, 438, 325–331.
10. Shi,D., Morizono,H., Yu,X., Roth,L., Caldovic,L., Allewell,N.M., Malamy,M.H. and Tuchman,M. (2005) Crystal structure of N-acetylornithine transcarbamylase from Xanthomonas campestris: a novel enzyme in a new arginine biosynthetic pathway found in several eubacteria. J. Biol. Chem., 280, 14366–14369.
11. Elkins,P.A., Watts,J.M., Zalacain,M., van Thiel,A., Vitazka,P.R., Redlak,M., Andraos-Selim,C., Rastinejad,F. and Holmes,W.M. (2003) Insights into catalysis by a knotted TrmD tRNA methyltransferase. J. Mol. Biol., 333, 931–949.
12. Lai,Y.L., Yen,S.C., Yu,S.H. and Hwang,J.K. (2007) pKNOT: the protein KNOT web server. Nucleic Acids Res., 35, W420–W424. 13. Kolesov,G., Virnau,P., Kardar,M. and Mirny,L.A. (2007) Protein
knot server: detection of knots in protein structures. Nucleic Acids Res., 35, W425–W428.
14. Chen,C.C., Hwang,J.K. and Yang,J.M. (2006) (PS)2: protein structure prediction server. Nucleic Acids Res., 34, W152–W157. 15. Chen,C.C., Hwang,J.K. and Yang,J.M. (2009) (PS)2-v2:
template-based protein structure prediction server. BMC Bioinformatics, 10, 366.
16. Noguchi,T. and Akiyama,Y. (2003) PDB-REPRDB: a database of representative protein chains from the Protein Data Bank (PDB) in 2003. Nucleic Acids Res., 31, 492–493.
17. Van Petegem,F., Clark,K.A., Chatelain,F.C. and Minor,D.L. Jr (2004) Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature, 429, 671–675.
18. Adams,C.C. (1994) The Knot Book: An Elementary Introduction to the Mathematical Theory of Knots. Freeman, New York.
Nucleic Acids Research, 2012, Vol. 40, Web Server issue W231
at National Chiao Tung University Library on April 28, 2014
http://nar.oxfordjournals.org/