行政院國家科學委員會專題研究計畫 期中進度報告
葛瑞夫茲氏病遺傳研究-連鎖及關聯研究(2/3)
期中進度報告(完整版)
計 畫 類 別 : 個別型 計 畫 編 號 : NSC 96-3112-B-002-017- 執 行 期 間 : 96 年 05 月 01 日至 97 年 04 月 30 日 執 行 單 位 : 國立臺灣大學醫學院內科 計 畫 主 持 人 : 張天鈞 共 同 主 持 人 : 楊偉勛、范盛娟 處 理 方 式 : 本計畫可公開查詢中 華 民 國 97 年 03 月 14 日
行政院國家科學委員會補助專題研究計畫 期中進度報告
葛瑞夫茲氏病遺傳研究-連鎖及關聯研究(2/3)
計畫類別:▓個別型計畫 □
整合型計畫
計畫編號:NSC
96-3112-B-002-017
執行期間:96 年 5 月 1 日 至 97 年 4 月 30 日
計畫主持人:張天鈞
共同主持人:
楊偉勛
計畫參與人員:
范盛娟 陳沛隆 張倩青 吳宜凌
成果報告類型(依經費核定清單規定繳交):□精簡報告 ▓完整
報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研
究計畫、列管計畫及下列情形者外,得立即公開查
詢
□涉及專利或其他智慧財產權,□一年□二年後可
公開查詢
執行單位:國立臺灣大學醫學院內科
中 華 民 國 96 年 3 月 14 日
中文摘要
葛瑞夫茲氏病是常見的自體免疫疾病,發病原因一般認為是多重因素,也與遺 傳有關。我們過去的連鎖研究涵蓋122 個葛瑞夫茲氏病家族,共 536 個人。所 有家族皆有兩位以上的兄弟姊妹罹患此病,若有可能,父母之檢體也涵蓋在內。 若父母之一或兩者之血液檢體不能取得,則要包含一位未罹病之兄弟姊妹之檢 體。共計有 270 對罹患葛瑞夫茲氏病之兄弟姊妹(affected sib-pairs)可以做無母數連鎖分析(non-parametric linkage study)。我們從白血球萃取 DNA,並做 short tandem repeat polymorphism(STRP)標記之基因定型。我們選擇涵蓋 HLA 區 13.7 cM 之 8 個 STRP,平均每個標記的密度為 1.9 cM。多點無母數連鎖分析顯示與 HLA 區有相關,尖峰在標記 UniSTS:239159(LOD score 3.46, P = .00003, NPL Z score 4.1, P = .00002) (Clin Endocrinol 2007; 66:646-651)。在 CTLA4 方面,我們
的連鎖研究涵蓋151 個葛瑞夫茲氏病家族,包括 374 個病人和 347 個沒病的家
人。在 CTLA4 基因定型 4 個 single nucleotide polymorphisms (SNP)和一個 STRP 。 我 們 新 發 現 葛 瑞 夫 茲 氏 病 與 5’ 上 游 區 的 SNP CTLA4_-1722_T/C (rs733618)相關(P = 0.0096)。我們也重現 SNP,CTLA4_+49_G/A (rs231775) , 與 葛 瑞 夫 茲 氏 病 相 關 (P = 0.0219) 。 一 個 共 通 的 haplotype , 由 CTLA4_-1722_T/C 和 CTLA4_(AT)n (STRP 標記:UniSTS:48500) 組合而成,顯 示保護的效果 (P = 0.0004)。我們在家族的連鎖研究,和白種人的結果,均顯示 CTLA4 與葛瑞夫茲氏病相關,是跨種族的(Gene and Immunity, 2008; 9:87-92).。
在這次以族群為基礎的研究,我們已完成1026 位檢體收集。其中有 1024 位病 人確診為葛瑞夫茲氏病。包括男性176 位,女性 848 位 (男比女 = 1:4.8) 。平 均年齡為40.9 + 12.8 歲 (範圍 9 – 81 歲)。發病平均年齡為 35.9 + 12.6 歲 (範 圍 6 – 81 歲)。甲狀腺腫程度為 2.1 + 0.9 (範圍 0 – 4)。有 500 位病人(48.8%)有 眼病變(Gr. 1:126, 12.3%; Gr 2:234, 22.9%; Gr. 3:140, 13.7%)。33 位病人(3.2%)有 脛前粘液水腫。19 位病人(1.9%)有週期性癱瘓。13 位病人(1.3%)有重症肌無力。
7 位病人(0.7%) 有白斑。我們已用 Affymetrix 500K chip 在 246 位有眼病變之
女性葛瑞夫茲氏病病人做基因定型,並和224 位女性對照組及 468 位男性及女
性對照組做genomewide association study 。結果顯示 HLA 區之 SNPs 有明顯 之統計意義(-Log10(p)=5.841) 。我們進一步在 721 位家族檢體及 500 位族群病
人做 HLA 基因定型,等取得中研院 500 位對照組做 HLA 基因定型後統計分
析,希望能找到與葛瑞夫茲氏病相關之基因。
英文摘要
Graves’ disease (GD) is an autoimmune disorder. The etiology of GD is accepted to be multifactorial with genetic effect. Our linkage study in the past, included 536 individuals in 122 multiplex families. All the families contained at least two affected siblings. Parents were also enrolled whenever possible; if samples from one or both parents were unavailable, at least one additional unaffected sibling was included. These participants could be analyzed as 270 affected sib-pairs (ASPs) in non-parametric linkage study. Genomic DNA was extracted from peripheral leukocytes. Genotyping of short tandem repeat polymorphism (STRP) markers was performed. Eight STRPs in a 13.7 cM region covering the HLA were chosen, resulting in a 1.9-cM (average) marker density. Multi-point non-parametric linkage analysis yielded evidence of significant linkage to the HLA region, which peaked around the marker UniSTS:239159 (LOD score 3.46, P = .00003; NPL Z score 4.1, P = .00002) (Clin Endocrinol 2007; 66:646-651). For CTLA4 study, we enrolled 374 affected individuals and 347 unaffected family members in 151 GD pedigrees.Four single nucleotide polymorphisms (SNP) and a short tandem repeat polymorphism (STRP) at CTLA4 were genotyped. Association of GD with a novel risk SNP at the 5’ upstream region, CTLA4_-1722_T/C (rs733618), was demonstrated (P = 0.0096). We also replicated the association signal of a coding SNP, CTLA4_+49_G/A (rs231775, P = 0.0219). A common haplotype composed of CTLA4_-1722_T/C and CTLA4_(AT)n (a STRP marker:UniSTS:48500) showed protective effect (P = 0.0004). Our results of family-based association study, taken together with those from the Caucasian population, provide evidence that CTLA4 confers susceptibility to GD across different ethnic backgrounds (Gene and Immunity, 2008; 9:87-92). In the present population-based association study, we have completed the collection of
blood of 1,026 individuals. There are 1024 persons confirmed to be GD, which include 176 men, and 848 women (men to women =1:4.8). The mean age is 40.9 + 12.8 years (range 9 – 81 years). The mean age of onset of hyperthyroidism is 35.9 + 12.6 years (range 6 – 81 years). The degree of goiter is 2.1 + 0.9 (range 0 – 4). There are 500 patients (48.8%) with ophthalmopathy (Gr. 1: 126, 12.3%; Gr 2: 234, 22.9%; Gr. 3: 140, 13.7%), 33 patients (3.2%) with pretibial myxedema, 19 patients (1.9%) with periodic paralysis, 13 patients (1.3%) with myasthenia gravis, 7 patients (0.7%) with vitiligo. Genotyping with Affymetrix 500K chip was performed in 246 female patients of Graves’ disease with ophthalmopathy. We compared with two kinds of control samples :Control I, females only, N = 224; Control II, both males and females, N = 468. The genomewide association study showed the SNPs at HLA region were highly significant (-Log10(p)=5.841). The HLA genotype data of all the individuals (721 familial samples and 500 population-based samples) are completed, but 500 control samples are waiting from Academia Sinica. We will try to find the susceptibility genes at the HLA region.
Key words: Graves’ disease, Linkage study, Association study, Gene
Linkage of Graves’ disease to the human leucocyte antigen region in the Chinese-Han population in Taiwan (Clin Endocrinol 2007; 66:646-651)
INTRODUCTION
Graves’ disease (GD [MIM 275000], http://www.ncbi.nlm.nih.gov/Omim/) is a common autoimmune disorder characterized by hyperthyroidism, diffuse goiter, thyroid-specific auto-antibodies, with or without ophthalmopathy and dermopathy.1 Its prevalence is estimated to be around 1.0 to 1.6% in the general population.2 The etiology of GD is generally accepted to be multifactorial1,3 with strong evidence of a genetic effect, including family clustering,4 an increased sibling risk (λs) of
approximately 8 to 15,4,5 and a higher concordance rate in monozygotic as compared to dizygotic twins (0.35 vs. 0.03).6 Data from 8,966 Danish twin pairs have suggested that 79% of the predisposition to GD is attributed to genetic factors.6 The lack of a clear inheritance pattern implies that multiple genes are involved in the pathogenesis of GD.7 Previous linkage analysis and association study results have implicated many genomic regions including the HLA region, the cytotoxic T-lymphoctye-associated 4 (CTLA4) gene, and the protein tyrosine phosphatase-22, that might harbor
susceptibility genes for GD.8-10
The HLA region on chromosome 6p21 contains many important immune response genes. A number of population-based genetic association studies supported the association between the HLA region and GD.11-18 Nevertheless the associated genes/alleles have not been consistent across multiple populations and negative studies were also published.8, 9, 19 On the other hand, family-based studies have been more controversial. Only one linkage analysis in Caucasians demonstrated a nominal linkage with an NPL score = 1.95.20 Yet other linkage studies in families from the US, Tunisia, Japan and China have not shown linkage to the HLA region.21-26 The
apparent discrepancy between association studies and linkage analysis, and the discrepancy between different populations, make the HLA region still an intriguing candidate for additional testing.8 However, it should also be noted that differences between HLA allele and haplotype frequencies have been observed in different ethnic backgrounds.27-30 It is therefore not uncommon that the disease-associated HLA haplotypes are not the same across populations.31 Linkage analysis using family samples have suggested more than 20 different loci that might harbor susceptibility genes of GD and/or autoimmune thyroid disease (AITD).8, 9 On the basis of these reports and their biological relevance, we also investigated four other candidate regions in our first attempt for linkage analysis: the CTLA4 region on chromosome 2q33;20 the cytokine gene cluster region on 5q31;24, 25 the pendrin region on 7q22;32
and the GD-1 and the thyroid stimulating hormone receptor regions on 14q31 in addition to the HLA region.25 Here we report
significant linkage of GD to the HLA region on chromosome 6p21 with a
non-parametric LOD score of 3.46 and a NPL Z score of 4.1, but not to the other 6 candidate regions.
MATERIALS AND METHODS
Subjects
Pedigrees were ascertained through a GD proband attending the outpatient clinic of National Taiwan University Hospital or affiliated clinics, Far Eastern Polyclinic. All the individuals enrolled in this study were interviewed and assessed by
board-certified endocrinologists. The diagnosis of GD was made based on thepresence of biochemical hyperthyroidism together with either the presence of thyroid eye disease or a diffuse goiter and a significant titer of
auto-antibodies (including anti-microsomal, anti-thyroglobulin or anti-TSH receptor antibody) as previously reported.19 To enrich phenotypic homogeneity, pedigrees containing any member with
possible Hashimoto’s thyroiditis (HT [MIM603372]), either according to medical records of HT or self-stated history of symptoms or signs of hypothyroidism without previous thyroidectomy or radioactive iodine treatment, were not included in this study. Ethnic background was recorded according to the information from these individuals. Only subjects whose four grandparents were of Chinese Han origin were included, whereas those with ancestors of possible Taiwanese aboriginal (of
Pacific-Polynesian extraction) or other minority Chinese ethnicity were not. This project was approved by the Institutional Review Board of National Taiwan University Hospital. Written informed consent was obtained from each individual.
This study included a total of 536 individuals in 122 multiplex families. All the families contained at least two affected siblings. Parents were also enrolled whenever possible; if samples from one or both parents were unavailable, at least one additional unaffected sibling was included. Among the pedigrees, 73 had two, 32 had three, 9 had four, 3 had five, 4 had six and 1 had eight affected individuals in one family. The pedigrees included a total of 321 affected patients, including 254 females (79.1%) and 67 males (20.9%). Of the 215 unaffected individuals, 113 were females and 102 males. These participants could be analyzed as 270 affected sib-pairs (ASPs) in
non-parametric linkage study.
Short tandem repeat polymorphism (STRP) markers selection and genotyping Genomic DNA was extracted from peripheral leukocytes using the PureGene kit (Gentra Systems, Inc., Minneapolis, MN, USA) according to the manufacturer’s
protocol. Fluorescence-labeled primers were purchased from Applied Biosystems (Foster City, CA, USA). Genotyping of STRP markers was performed on an ABI PRISM 3100 Genetic Analyzer, with allele calling done by Genotyper Software v 3.7 (Applied Biosystems). Each genotype was independently reviewed by two members. Mendelian inconsistency was checked with PedCheck (version 1.1).33 Those
genotypes with initial Mendelian inconsistency were rechecked, were corrected ifobvious mistake was identified, or set as missing. All the genotypes reported here were compatible with Mendelian inheritance. The overall genotype call rate was 97.5%.
Eight STRPs (D6S1660- D6S1691- D6S276- D6S273-
UniSTS:239159-D6S1568- D6S291- D6S1610) in a 13.7 cM region covering the HLA were chosen, resulting in a 1.9-cM (average) marker density. The genetic positions of the markers were determined using the Marshfield (Center for Medical Genetics) genetic maps (http://research.marshfieldclinic.org/genetics/), and the order was verified with the physical map of National Center for Biotechnology Information (NCBI) build 35 (http://www.ncbi.nlm.nih.gov/). One marker (UniSTS:239159) was chosen from UniSTS database in NCBI build 35 without the information in the Marshfield geneticmaps. Its genetic position was approximated based on the physical distances between
flanking markers.
The other 26 markers distributed in the other four candidate regions were as followings: five markers (D2S118- D2S2387- UniSTS:48500- D2S155- D2S2242) in a 15.1-cM region on 2q33, seven markers (D5S2017- D5S436- D5S2090-
D5S434-D5S2014- D5S410- D5S422) in an 18.9-cM region on 5q31, five markers (D7S2446-D7S501- D7S496- D7S2459- D7S486) in a 10.1-cM region on 7q22, and nine markers (D14S276- D14S274- D14S63-
D14S258- D14S74- D14S1044- D14S280-D14S1054- D14S65) in a 61.1-cM region on 14q31. The necessary sequence information for primer design was based on the database in NCBI. The information of primers is available upon request.
Statistical analysis
Non-parametric linkage analyses were performed to locate the position of the susceptibility genes for GD. The allele frequency of markers was estimated on the basis of founders’ genotypes. MERLIN 1.0.1. program was used to carry out two-point and multi-point nonparametric linkage analyses.34 The non-parametric linkage (NPL) Z score and non-parametric LOD score under an exponential model were calculated.35, 36 The information content of the genotypes was estimated with use of entropy information.35 The Sall scoring function was used to capture the
1-LOD support interval was based on our multi-point non-parametric LOD scores.
RESULTS
Our familial collection comprised 270 ASPs (all possible pairs) for the Sall scoring function of non-parametric linkage analysis. For the five promising candidate regions, the maximal multi-point NPL Z scores calculated with MERLIN 1.0.1. program were as followings: 0.97 on chromosome 2q33, 0.82 on chromosome 5q31, 4.1 on chromosome 6p21, -0.89 on chromosome 7q22, and -0.34 on
chromosome14q31 (Table 1).
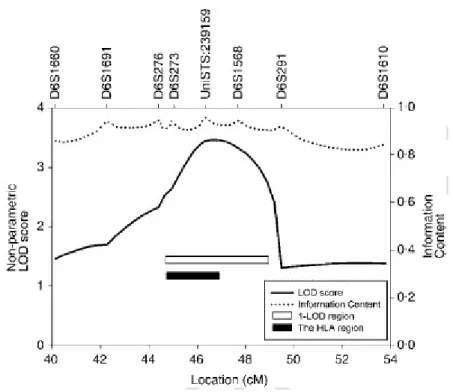
Non-parametric analysis demonstrated linkage of GD to the HLA region onchromosome 6p21. Two-point analysis with MERLIN showed the highest non-parametric LOD score of 2.56 (P = 0.0003) at D6S1568 (Table 2). Multi-point non-parametric LOD score peaked around marker UniSTS:239159, but not exactly at the location of the marker (Figure 1), with the highest score of 3.46 (P =0.00003). The score specifically for this marker is 3.44 (Table 2). Multi-point NPL Z score
correlated very well with the LOD score (Table 2), peaking at the same
UniSTS:239159 marker (maximal NPL Z = 4.1, P = 0.00002). Our 1-LOD support interval was a ~4.5 cM region (44.6 cM – 49.1 cM, sex-average distance) on
theMarshfield genetic map (Figure 1). This 1-LOD support interval corresponds to a ~8.9 Mb region (26.9 Mb – 35.8 Mb) on the NCBI build 35 physical map, which contains the whole ~4.0 Mb HLA region (Figure 1).
DISCUSSION
Our results strongly support linkage of GD to the HLA region on chromosome 6p21. Among previous five studies of familial linkage analysis, the only positive signal at the HLA region was reported from the United Kingdom, with a nominal level of significance with an NPL score =1.95.20 The other linkage studies, using pedigrees from the United States, United Kingdom, Japan, China, and Tunisia did not detect significant effect at the HLA region.21-26 However, the results from various population-based association studies also offer support for the roles of HLA as a genetic contributor to GD.7-9
In this study, we attempted to decrease genetic heterogeneity, one major problem in genetic studies.38, 39 We enrolled only pedigrees with Chinese-Han ethnic
background. We also try to reduce the genetic heterogeneity by excluding families with a history of hypothyroidism or HT, although it could not be possibly complete. There are both advantages and disadvantages of our
decision to focus on GD. This strategy obviously would negatively influence our sample size, and thus decreased our power to detect susceptibility genes with effect
on both GD and HT. However, although GD and HT may share some common pathophysiological pathways, these two diseases have substantial differences in terms of clinical manifestations, laboratory abnormality and histological findings.40 Previous reports have suggested that there might be different sets of susceptibility genes for GD, HT and AITD.21, 22 In a recent large-scale genome-wide linkage screen with 1,119 AITD relative-pairs, none of the linkages obtained from GD or autoimmune hypothyroidism (AIH) was the same.26 In addition, the diagnostic definition of HT is more controversial,8 and the etiology of HT may be even more heterogeneous than GD.21 In fact, the clinical course is variable in HT, and thyroid function could be normal or abnormal (overt hypothyroidism, subclinical
hypothyroidism, and hyperthyroidism).41 The measurement of anti-thyroglobulin and thyroid peroxidase antibodies can not provide any help in distinguishing various types of AITD.42
Our 1-LOD support interval was a ~4.5 cM region (44.6 cM–49.1 cM,
sex-average distance) on the Marshfield genetic map, which corresponds to an ~8.9 Mb region (26.9 Mb – 35.8 Mb) on the NCBI build 35 physical map. This interval contains the whole ~4 Mb HLA region.43 Class II loci, especially the HLA DRB1*03 and the DRB1*03-DQB1*02-DQA1*0501 haplotype, have repeatedly demonstrated to associate with GD in Caucasian populations.8, 19, 44 However, in studies of
populations of Chinese, Thai, Japanese and Korean ancestry, the DRB1*03 and its haplotype did not show association with GD; instead, there were reports for
association with class I and other class II loci.8, 14, 18, 44, 45 One plausible explanation is that different associated haplotypes reported in different populations actually carry some common polymorphisms driving the same autoimmune response. Due to the extended haplotype spectrum of the HLA genes, it has been extremely difficult to identify the primary etiological variants or to “split” the effect of individual loci from the effect of the whole haplotype.19, 44 Comparison between association studies from populations with different composition of HLA haplotypes may provide very useful clues. Our result provides strong evidence for the involvement of the HLA region in GD in the Chinese-Han population in Taiwan, which advocates for the importance of a larger association study to help clarify the susceptibility HLA loci and/or alleles. Besides, it is still possible that some genes, other than the classical HLA loci, account for our linkage and the previous association signals. The recent linkage-disequilibrium map at the HLA region will facilitate the interpretation of future association studies.27, 43 In addition, Simmonds et al. recently using various statistical techniques were able to dissect the effect of individual loci from the effects
of neighboring variants, all associated with GD because of strong
eventual identification of the primary etiological genetic variants for GD in the future. We also tested the linkage of GD to four other candidate regions, including the
CTLA4 region on chromosome 2q33,20 the cytokine gene cluster region on 5q31,24, 25
the pendrin region on 7q22,32 and the GD-1 and the TSHR regions on 14q31.25 The average marker density at different regions varied from 1 marker per 2.5 cM to 1 marker per 7.6 cM. None of the NPL Z scores reached suggestive level of significance, although we did see some positive signal at 2q33 (maximal NPL Z score = 0.97) and 5q31 (maximal NPL Z score = 0.82). Our results can not exclude any of the four regions, as the lack of signals could
be due to power limitation because of the underlying gene effects and our modest sample size and marker density. Linkage analysis has had great success in genetic mapping of monogenic diseases. Unfortunately, the power for detecting linkage signal of loci with moderate or small effect, the most possible situation in complex traits, is limited.46-48 Risch's calculation demonstrates that it takes more than 3000 affected sib-pairs to have sufficient power of detecting linkage signal of a
susceptibility locus with genotype risk ratio of 2, while less than 1000 cases and controls are needed to find association using case control studies.46 Therefore, further studies are necessary for these regions.
In conclusion, our work provides strong support of linkage of GD to the HLA region. Taken together with the previous linkage report in Caucasians and other association studies, our results suggest that the HLA region harbors one or more susceptibility genes for GD. Further dissection of this region in our population may provide insight into the immunogentics of GD pathogenesis, therefore is warranted.
Acknowledgements
We thank all the GD patients and their families who graciously agreed to participate in the study. We are grateful to Dr. Pei-Jer Chen and Ms. Shu-Fen Lu for equipment and technical support in genotyping. We also thank Drs. David Valle and Dimitrios Avramopoulos for helpful discussion. This work was supported by grants NSC# 91-3112-B-002-003, 92-3112-B-002-009 and 93-3112-B-002-015 from National Science Council, Taiwan. We also apologize to the authors whose work regarding this issue was not included in the references due to word count limitation.
References
1. Weetman, A.P. (2000) Graves' disease. New England Journal of Medicine
343,1236-1248.
2. Hollowell, J.G., Staehling, N.W., Flanders, W.D., Hannon, W.H., Gunter, E.W.,
in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). Journal of Clinical Endocrinology &
Metabolism 87,489-499.
3. DeGroot, L.J. & Quintans, J. (1989) The causes of autoimmune thyroid disease.
Endocrine Review 10,537-562.
4. Brix, T.H., Kyvik, K.O. & Hegedus, L. (1998) What is the evidence of genetic
factors in the etiology of Graves' disease? A brief review. Thyroid 8,727-734.
5. Vyse, T.J. & Todd, J.A. (1996) Genetic analysis of autoimmune disease. Cell
85,311-318.
6. Brix, T.H., Kyvik, K.O., Christensen, K. & Hegedus, L. (2001) Evidence for a
major role of heredity in Graves' disease: a population-based study of two Danish twin cohorts. Journal of Clinical Endocrinology & Metabolism 86,930-934.
7. Vaidya, B., Kendall-Taylor, P. & Pearce, S.H. (2002) The genetics of autoimmune
thyroid disease. Journal of Clinical Endocrinology & Metabolism87,5385-5397.
8. Tomer, Y. & Davies, T.F. (2003) Searching for the autoimmune thyroid disease
susceptibility genes: from gene mapping to gene function. Endocrine
Review24,694-717.
9. Ayadi, H., Hadj Kacem, H., Rebai, A. & Farid, N.R. (2004) The genetics of
autoimmune
thyroid disease. Trends in Endocrinology & Metabolism 15,234-239.
10. Velaga, M.R., Wilson, V., Jennings, C.E., Owen, C.J., Herington, S., Donaldson,
P.T., Ball, S.G., James, R.A., Quinton, R., Perros, P. & Pearce, S.H. (2004) The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves' disease. Journal of Clinical Endocrinology &
Metabolism 89,5862-5865.
11. Yanagawa, T., Mangklabruks, A., Chang, Y.B., Okamoto, Y., Fisfalen, M.E.,
Curran, P.G.. & DeGroot, L.J. (1993) Human histocompatibility leukocyte antigen-DQA1*0501 allele associated with genetic susceptibility to Graves' disease in a Caucasian population. Journal of Clinical Endocrinology
&Metabolism 76,1569-1574.
12. Badenhoop, K., Walfish, P.G., Rau, H., Fischer, S., Nicolay, A., Bogner, U.,
Schleusener, H. & Usadel, K.H. (1995) Susceptibility and resistance alleles ofhuman leukocyte antigen (HLA) DQA1 and HLA DQB1 are shared in
endocrine autoimmune disease. Journal of Clinical Endocrinology & Metabolism 80,2112-2117.
13. Heward, J.M., Allahabadia, A., Daykin, J., Carr-Smith, J., Daly, A., Armitage, M.,
Dodson, P.M., Sheppard, M.C., Barnett, A.H., Franklyn, J.A. & Gough, S.C. (1998) Linkage disequilibrium between the human leukocyte antigen class II
region of the major histocompatibility complex and Graves' disease: replication using a population case control and family-based study. Journal of Clinical
Endocrinology & Metabolism 83,3394-3397.
14. Huang, S.M., Wu, T.J., Lee, T.D., Yang, E.K., Shaw, C.K. & Yeh, C.C. (2003)
The association of HLA -A, -B, and -DRB1 genotypes with Graves' disease in Taiwanese people. Tissue Antigens 61,154-158.
15. Yeo, P.P., Chan, S.H., Thai, A.C., Ng, W.Y., Lui, K.F., Wee, G.B., Tan, S.H., Lee,
B.W., Wong, H.B. & Cheah, J.S. (1989) HLA Bw46 and DR9 associations in Graves' disease of Chinese patients are age- and sex-related. Tissue Antigens 34,179-184.
16. Onuma, H., Ota, M., Sugenoya, A. & Inoko, H. (1994) Association of
HLA-DPB1*0501 with early-onset Graves' disease in Japanese. Human
Immunology 39,195-201.
17. Cho, B.Y., Rhee, B.D., Lee, D.S., Lee, M.S., Kim, G.Y., Lee, H.K., Koh, C.S.,
Min, H.K. & Lee, M. (1987) HLA and Graves' disease in Koreans. Tissue
Antigens 30,119-121.
18. Wongsurawat, T., Nakkuntod, J., Charoenwongse, P., Snabboon, T., Sridama, V.
& Hirankarn, N. (2006) The association between HLA class II haplotype with Graves' disease in Thai population. Tissue Antigens 67,79-83.
19. Simmonds, M.J., Howson, J.M., Heward, J.M., Cordell, H.J., Foxall, H.,
Carr-Smith, J., Gibson, S.M., Walker, N., Tomer, Y., Franklyn, J.A., Todd, J.A. & Gough, S.C. (2005) Regression mapping of association between the human
leukocyte antigen region and Graves disease. American Journal of Human
Genetics 76,157-163.
20. Vaidya, B., Imrie, H., Perros, P., Young, E.T., Kelly, W.F., Carr, D., Large, D.M.,
Toft, A.D., McCarthy, M.I., Kendall-Taylor, P. & Pearce, S.H. (1999) The cytotoxic T lymphocyte antigen-4 is a major Graves' disease locus. Human
Molecular Genetics 8,1195-1199.
21. Tomer, Y., Barbesino, G., Greenberg, D.A., Concepcion, E. & Davies, T.F.
(1999) Mapping the major susceptibility loci for familial Graves' and Hashimoto's diseases: evidence for genetic heterogeneity and gene interactions. Journal of
Clinical Endocrinology & Metabolism 84,4656-4664.
22. Sakai, K., Shirasawa, S., Ishikawa, N., Ito, K., Tamai, H., Kuma, K., Akamizu, T.,
Tanimura, M., Furugaki, K., Yamamoto, K. & Sasazuki, T. (2001) Identification of susceptibility loci for autoimmune thyroid disease to 5q31-q33 and Hashimoto's thyroiditis to 8q23-q24 by multipoint affected sib-pair linkage analysis in
Japanese. Human Molecular Genetics 10,1379-1386.
Abid, M. & Ayadi, H. (2001) A full genome screening in a large Tunisian family affected with thyroid autoimmune disorders. Genes Immunity 2,71-75.
24. Jin, Y., Teng, W., Ben, S., Xiong, X., Zhang, J., Xu, S., Shugart, Y.Y., Jin, L.,
Chen, J. & Huang, W. (2003) Genome-wide scan of Graves' disease: evidence for linkage on chromosome 5q31 in Chinese Han pedigrees. Journal of Clinical
Endocrinology & Metabolism 88,1798-1803.
25. Tomer, Y., Ban, Y., Concepcion, E., Barbesino, G., Villanueva, R., Greenberg,
D.A. & Davies, T.F. (2003) Common and unique susceptibility loci in Graves and Hashimoto diseases: results of whole-genome screening in a data set of102
multiplex families. American Journal of Human Genetics 73,736-747.
26. Taylor, J.C., Gough, S.C., Hunt, P.J., Brix, T.H., Chatterjee, K., Connell, J.M.,
Franklyn, J.A., Hegedus, L., Robinson, B.G., Wiersinga, W.M., Wass, J.A., Zabaneh, D., Mackay, I. & Weetman, A.P. (2006) A genome-wide screen in 1119 relative pairs with autoimmune thyroid disease. Journal of Clinical Endocrinology
& Metabolism 91,646-653.
27. Yang, H.C., Lin, C.H., Hsu, C.L., Hung, S.I., Wu, J.Y., Pan, W.H., Chen, Y.T. &
Fann, C.S. (2006) A comparison of major histocompatibility complex SNPs in Han Chinese residing in Taiwan and Caucasians. Journal of Biomedical Sciences 13,489-498.
28. Cao, K., Hollenbach, J., Shi, X., Shi, W., Chopek, M. & Fernandez-Vina, M.A.
(2001) Analysis of the frequencies of HLA-A, B, and C alleles and haplotypes in the five major ethnic groups of the United States reveals high levels of diversity in these loci and contrasting distribution patterns in these populations. Human
Immunology 62,1009-1030.
29. Dean, M., Stephens, J.C., Winkler, C., Lomb, D.A., Ramsburg, M., Boaze, R.,
Stewart, C., Charbonneau, L., Goldman, D. & Albaugh, B.J. (1994) Polymorphic admixture typing in human ethnic populations. American Journal of Human
Genetics 55,788-808.
30. Cao, K., Moormann, A.M., Lyke, K.E., Masaberg, C., Sumba, O.P., Doumbo,
O.K., Koech, D., Lancaster, A., Nelson, M., Meyer, D., Single, R., Hartzman, R.J., Plowe, C.V., Kazura, J., Mann, D.L., Sztein, M.B., Thomson, G. &
Fernandez-Vina, M.A. (2004) Differentiation between African populations is evidenced by the diversity of alleles and haplotypes of HLA class I loci. Tissue
Antigens 63,293-325.
31. Ghodke, Y., Joshi, K., Chopra, A. & Patwardhan, B. (2005) HLA and disease.
European Journal of Epidemiology 20,475-488.
32. Hadj Kacem, H., Rebai, A., Kaffel, N., Masmoudi, S., Abid, M. & Ayadi, H.
association and linkage study. Journal of Clinical Endocrinology & Metabolism 88,2274-2280.
33. O'Connell, J.R. & Weeks, D.E. (1998) PedCheck: a program for identification of
genotype incompatibilities in linkage analysis. American Journal of Human
Genetics 63,259-266.
34. Abecasis, G.R., Cherny, S.S., Cookson, W.O. & Cardon, L.R. (2002)
Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nature
Genetics 30,97-101.
35. Kruglyak, L., Daly, M.J., Reeve-Daly, M.P. & Lander, E.S. (1996) Parametric and
nonparametric linkage analysis: a unified multipoint approach. AmericanJournal
of Human Genetics 58,1347-1363.
36. Kong, A. & Cox, N.J. (1997) Allele-sharing models: LOD scores and accurate
linkage tests. American Journal of Human Genetics 61,1179-1188.
37. Whittemore, A.S. & Halpern, J. (1994) A class of tests for linkage using affected
pedigree members. Biometrics 50,118-127.
38. Cardon, L.R. & Bell, J.I. (2001) Association study designs for complex diseases.
Nature Review Genetics 2,91-99.
39. Glazier, A.M., Nadeau, J.H. & Aitman, T.J. (2002) Finding genes that underlie
complex traits. Science 298,2345-2349.
40. Armengol, M.P., Juan, M., Lucas-Martin, A., Fernandez-Figueras, M.T.,
Jaraquemada, D., Gallart, T. & Pujol-Borrell, R. (2001) Thyroid autoimmune disease: demonstration of thyroid antigen-specific B cells and
recombination-activating gene expression in chemokine-containing active intrathyroidal germinal centers. American Journal of Pathology 159,861-873.
41. Lorini, R., Gastaldi, R., Traggiai, C. & Perucchin, P.P. (2003) Hashimoto's
Thyroiditis. Pediatric Endocrinology Review 1 Suppl 2,205-211.
42. Weetman, A.P. (2004) Autoimmune thyroid disease. Autoimmunity 37,337-340. 43. Miretti, M.M., Walsh, E.C., Ke, X., Delgado, M., Griffiths, M., Hunt, S.,
Morrison, J., Whittaker, P., Lander, E.S., Cardon, L.R., Bentley, D.R., Rioux, J.D., Beck, S. & Deloukas, P. (2005) A high-resolution linkage-disequilibrium map of the human major histocompatibility complex and first generation of tag
single-nucleotide polymorphisms. American Journal of Human Genetics 76,634-646.
44. Simmonds, M.J. & Gough, S.C. (2004) Unravelling the genetic complexity of
autoimmune thyroid disease: HLA, CTLA-4 and beyond. Clinical Experiemtal
Immunology 136,1-10.
45. Park, M.H., Park, Y.J., Song, E.Y., Park, H., Kim, T.Y., Park, D.J., Park, K.S. &
Koreans. Human Immunology 66,741-747.
46. Risch, N.J. (2000) Searching for genetic determinants in the new millennium.
Nature 405,847-856.
47. Wang, W.Y., Barratt, B.J., Clayton, D.G. & Todd, J.A. (2005) Genome-wide
association studies: theoretical and practical concerns. Nature Review Genetics 6,109-118.
48. Altmuller, J., Palmer, L.J., Fischer, G., Scherb, H. & Wjst, M. (2001)
Genomewide scans of complex human diseases: true linkage is hard to find.
Figure 1 Multi-point non-parametric LOD scores on 6p21-p22 from linkage analysis
of 270
ASPs in 122 GD families. The scores were calculated with MERLIN v 1.0.1. Multi-point
non-parametric LOD scores (solid line), information content (dotted line), the 1-LOD region
(empty bar) and the HLA region (filled bar) are plotted. The X-axis values are distances from
Family-based association study of cytotoxic T-lymphocyte antigen-4 with susceptibility to Graves’ disease in the Chinese-Han population in Taiwan (Gene
and Immunity, 2008; 9:87-92)
Introduction
Graves’ disease (GD) is a common organ-specific autoimmune disorder manifested with hyperthyroidism, diffuse goiter, thyroid-specific antibodies, with/without ophthalmopathy and/or dermopathy (1). The prevalence of GD in the general population was estimated to be around 1.0 to 1.6% (2-4). The cause of GD is multifactorial, with considerable genetic influence (1, 5). The evidence of the genetic contribution to its pathogenesis came from the observation of family clustering (6), an increased risk ratio between 8 and 15 of patients’ female siblings (6, 7), and a higher concordance rate in monozygotic twins (0.35) than in dizygotic twins (0.03) (8). A statistic model based on the data from 8,966 pairs of Danish twins demonstrated that nearly 80% of the predisposition to GD could be attributed to genetic factors (8). However, being short of a common Mendelian inheritance pattern suggests a polygenic nature in its pathogenesis (9, 10).
Cytotoxic T lymphocyte antigen 4 (CTLA4) is an immunoregulatory molecule expressed on the surface of T lymphocytes and serves as a key negative regulator (11). CTLA4 gene located on chromosome 2q33 is among the most plausible candidate genes for GD (10, 12, 13). Many population-based association studies in different ethnic backgrounds have shown positive results (10), although negative results have also been reported (14, 15). The most frequently reported association signals are CTLA4_+49_G/A (rs231775, at exon 1), CTLA4_-319_C/T (rs5742909, at the promoter region) and CTLA4_(AT)n (UniSTS:48500, a STRP marker at the 3’ untranslated region of CTLA4) (10, 12, 13). Ueda et al. reported a comprehensive SNP fine mapping and proposed that the SNPs at the 3’ uncoding region, such as CTLA4_CT60_G/A (rs3087243, at the 3’ downstream region), are the primary disease determinants (16). Nevertheless, more studies are probably needed to reveal the genuine causative genetic variants.
In contrast to the population-based studies, there have been few family-based genetic studies providing evidence of the involvement of CTLA4. Up to now, however, only three family-based studies in the United Kingdom observed linkage or association signal of GD at the CTLA4 gene (16-18). Population-based genetic association studies in outbred populations may be prone to false positives because of population stratification (19, 20), which can be avoided by family-based design (21). In this study we enrolled 721 individuals in 151 pedigrees with one or more affected persons in the Chinese-Han population in Taiwan. Family-based association study
was performed with 5 markers previously reported to be associated with GD or other autoimmune diseases. We found that a novel risk SNP of GD at the 5’ upstream region of CTLA4 (rs733618, named as CTLA4_-1722_T/C in this report) and haplotypes containing this SNP are significantly associated with GD.
Results
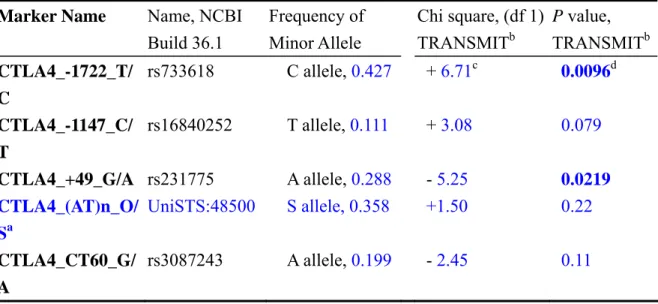
Genotypes of four SNPs (CTLA4_-1722_T/C, CTLA4_-1147_C/T, CTLA4_+49_G/A and CTLA4_CT60_G/A) and a dichotomized STRP marker (CTLA4_(AT)n_O/S) were analyzed with TRANSMIT v2.54 (22) for family-based association study. The basic information of these markers and allele frequency in our samples were summarized in Table 1. The allele frequencies of all these SNP markers were compatible with Hardy-Weinberg equilibrium. The relative positions of these markers at CTLA4 gene were shown in Figure 1. For single marker analysis, our strongest association signal was observed at a 5’ upstream SNP, CTLA4_-1722_T/C, with its minor allele (C allele) over-transmitted to affected individuals (χ2 = 6.714, df = 1, nominal P = 0.0096, Table 1). Our study is the first one to demonstrate association of CTLA4_-1722_T/C with GD. We also replicated the association signal at another SNP, CTLA4_+49_G/A (χ2 = 5.252, df = 1, nominal P = 0.0219, G allele over-transmitted).
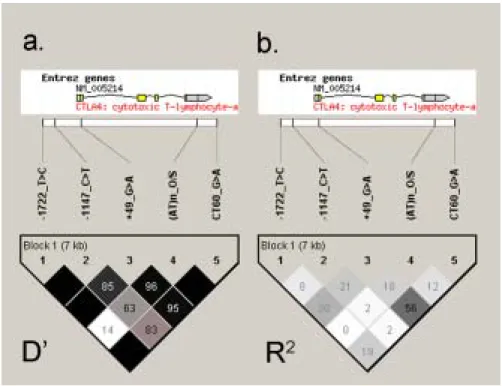
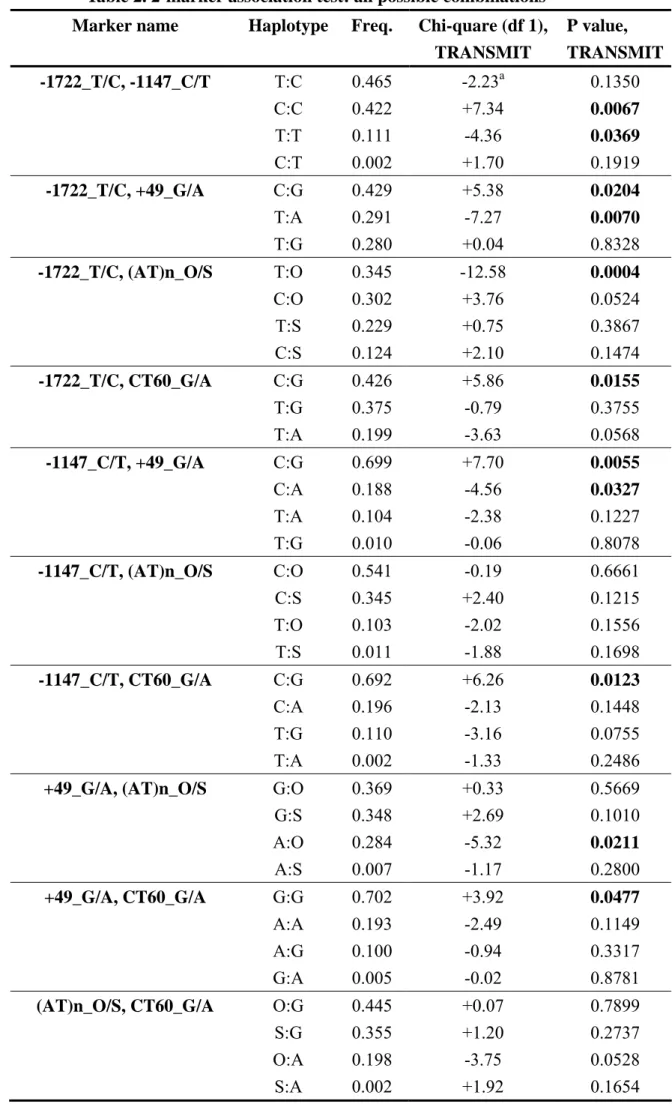
Linkage disequilibrium (LD) between these 5 markers was analyzed with Haploview 3.32 (23). We applied the “solid spine” option to define LD blocks. All the five markers were shown to be grouped into a single LD block (Figure 1), which is compatible with previous reports (16, 24). We applied TRANSMIT to perform family-based association analysis using haplotypes from all the possible 2-marker combinations. The result is presented in Table 2. Eight of the ten combinations had at least one haplotype with a nominal P value less than 0.05. The most significant cluster was related to CTLA4_-1722_T/C; all of its four 2-marker combinations had at least one haplotype with nominal P value less than 0.05, and three of its four 2-marker combinations had one haplotype with nominal P value less than 0.01. Our strongest 2-marker association signal was from a common haplotype (34.5% of the population) composed of the CTLA4_-1722_T allele and the CTLA4_(AT)n_O allele (Chi square = 12.576, df = 1, P = 0.0004). This haplotype was under-transmitted to the affected individuals, which indicated a protective role.
Discussion
We report an association of CTLA4 with GD in a family-based study in the Chinese-Han population in Taiwan. Previously, the only three family-based researches on the association between CTLA4 and GD were all performed in
Caucasians (16-18). Family-based study design has the advantage of overcoming possible spurious positives caused by population stratification (19-21). Our result provides robust evidence of the association between CTLA4 and GD in the Chinese-Han population.
Our strongest association signals were observed at a novel risk SNP, CTLA4_-1722_T/C, and haplotypes containing it. This SNP was previously reported to be associated with systemic lupus erythematosus (SLE) in two contradictory studies; the T allele was reported to increase risk in Koreans (25) while the C allele was reported in a Spanish study (26). In a recent association study of celiac disease in the Irish population (27), CTLA4_-1722_T/C itself did not reach statistically significant association but some haplotypes containing the CTLA4_-1722_T allele were shown to be protective. In our study, the T allele of CTLA4_-1722_T/C SNP and haplotypes containing it were under-transmitted to the affected individuals. We conclude that the T allele of CTLA4_-1722_T/C SNP, or other genetic variant(s) in close linkage disequilibrium with it, has protective effect for GD in the Chinese-Han population.
Our most significant result was found in a 2-marker combination containing CTLA4_-1722_T/C and CTLA4_(AT)n markers (P = 0.0004). CTLA4_(AT)n has been shown associated with GD in a couple of studies (12, 28, 29), with the “allele 106” of Yanagawa et al. study (28) as the risk allele. The length of the dinucleotide expansion was shown to lead to T cell hyper-reactivity in myasthenia gravis (30). Ueda et al. (16) dichotomized this STRP marker for single-locus and two-locus association study in their comprehensive SNP fine mapping. In our current report, we dichotomized this STRP marker into “allele S” (standing for the “Specific” allele which is the same as the “allele 106” of Yanagawa et al. study (28)) and “allele O” (standing for the “Other” alleles). The direction of allele effect was found to be consistent between previous reports (“allele 106” as the risk allele) and our study (“allele O” to be in the protective haplotype).
We also notice that the CTLA4_(AT)n marker is not in strong LD with other markers, judging from the measurement of D’ (Figure 1a) and R2 (Figure 1b). One explanation for the highly significant association signal from the two-marker combination is that a single causal genetic variant was not genotyped in our study and its information could be captured by the haplotypes defined by these two markers. However, another possible explanation is that there are two (or even more) different causal variants at the CTLA4 gene, and CTLA4_-1722_T/C and CTLA4_(AT)n represented independent signals. Ueda et al., with the statistic model attempting to detect a single causal variant at CTLA4, proposed that one SNP at the 3’ uncoding region is the primary disease determinant (16). However, their data
turned out unable to exclude the possibility of multiple causative variant(s). We suggest further exploration of the two-causal-variants hypothesis with more samples and markers in the future.
In addition to being compatible with the two-causal-variants hypothesis, our results further raise an intriguing possibility that those two causative variants have some kind of interaction. The P value from the single marker analysis of our association study for CTLA4_(AT)n was 0.22, which did not provide evidence of significant effect. However, when the combination of CTLA4_-1147_T and CTLA4_(AT)n_O alleles were tested, the P value (0.0004) was 24-fold smaller than that of CTLA4_-1147_C/T SNP (P = 0.0096) alone. The observation is remarkably reminiscent of the Ueda et al. study, in which this STRP marker itself showed only very modest single locus association but was one of the few markers that could further increase the association signal under their best model containing the CTLA4_CT60_G/A SNP. We propose that there are two causative variants working together to exert maximal influence on the gene. Replication from other independent samples and evidence from functional studies are necessary to prove this hypothesis.
Our study should be considered as a hypothesis-testing approach because all of the 5 markers have previously been reported to be associated with GD or other autoimmune diseases. This kind of approach affects our analysis in two major ways. First, this set of markers are not tagging markers, which means that further exhaustive haplotype analyses (3-marker, 4-marker and 5-marker haplotype analysis) may not give us interpretable results. The current single-marker and two-marker analyses can be viewed as tests for individual effect and joint (or interaction) effect of these candidate markers. Secondly, it will be very difficult to adapt an appropriate method to correct for multiple testing. Having been previously reported of association, these markers actually have a higher a priori to be the true signal; being in linkage disequilibrium with each other and inside the same LD block (Figure 1), the penalty for multiple testing should be reduced (31). Therefore in this manuscript we only report the nominal P values and leave the issue of correction for multiple testing untouched. Nonetheless, it is obvious that our best association signals remain significant even under the most stringent Bonferroni correction.
By comparing our genotypes of the unrelated founders and those in public database, we noticed allele frequency difference across populations of a couple of SNPs at the CTLA4 gene region. For example, the frequency of the CTLA4_CT60_A allele in our samples was 20.9%, which is very close to that of the Chinese Han in Beijing (CHB) samples of the HapMap project (21.1%) (32), but very different from that of the Utah residents with ancestry from northern and western Europe (CEU) samples of the HapMap project (45.8%). Similarly, the allele
frequency of CTLA4_-1722_C allele in our samples was 42.6%, which is comparable with the CHB samples (33.3%) but differs significantly from the CEU samples (5.8%). Figure 2 demonstrates the allele frequency of CTLA4_-1722_T/C, CTLA4_+49_G/A and CTLA4_CT60_G/A. It intrigues us to find such big allele frequency difference in some specific SNPs which have been repeatedly tested for association with GD and/or other autoimmune diseases. It is hard to say if this kind of difference indicates any biological meaning. However, the allele frequency difference of these three SNPs between Caucasian samples and our Chinese Han samples was very significant. It might partially explain why the association study of
CTLA4 gave inconsistent results. We suggest that any attempt of applying the risk
(or protective) alleles/haplotypes information at CTLA4 across populations should be done with great caution.
With the same collection of familial samples, we did not detect evidence of linkage at the CTLA4 region (33), which could be explained by the effect size of
CTLA4 and by the sample size of our collection. However, we were able to
demonstrate significant linkage (NPL score = 4.1, P = 0.00002) at the human leukocyte antigen (HLA) region (33). With an intention to reduce heterogeneity of our samples, we purposely enrolled individuals with pure Chinese-Han ethnic background, and collected GD-only pedigrees by excluding families with a history of hypothyroidism or Hashimoto’s thyroiditis. With this strategy of sample collection, we successfully detected linkage signal of HLA and association signal of
CTLA4. It is likely that this collection may contribute our genetic study at other
regions in the future.
In conclusion, our family-based association study demonstrates that the CTLA4 gene is associated with GD in the Chinese-Han population in Taiwan. We identified a novel risk SNP in the 5’ flanking region (CTLA4_-1722_T/C, P = 0.0096). A two-marker combination composed of the CTLA4_-1722_T allele and the CTLA4_(AT)n_O allele showed protective effect (P = 0.0004). Our results of family-based association study, taken together with those from the Caucasian population, provide robust evidence that CTLA4 confers susceptibility to GD across different ethnic backgrounds.
Subjects and Methods
Clinical assessment and family ascertainment:
Pedigrees were ascertained through a GD proband as previously described (33). Briefly, the diagnosis of GD was made based on the manifestations of biochemical hyperthyroidism with either ophthalmopathy or a diffuse goiter and a significant titer of auto-antibodies, including anti-microsomal, anti-thyroglobulin or anti-TSH
receptor antibody as in previous reports (33, 34). To increase phenotypic homogeneity, pedigrees containing any member with possible Hashimoto’s thyroiditis (HT [MIM603372], based either on medical records of HT or on self-stated history of symptoms/signs of hypothyroidism without previous treatment of thyroidectomy or radioactive iodine) were excluded. To reduce heterogeneous ethnic background, only subjects with four grandparents of Chinese-Han origin were included. Those patients with ancestors of possible Taiwanese aboriginal (of Pacific-Polynesian origin) or other minority Chinese ethnicity were not enrolled. This project was approved by the Ethic Committee of Human Research of National Taiwan University Hospital. Written informed consent was obtained from each individual.
This report included 721 individuals in 151 pedigrees. Among the pedigrees, 6 (4.0 %) had one affected individual, 100 (66.3 %) had two, 28 (18.5 %) had three, 8 (5.3 %) had four, 7 (4.6 %) had five, 1 (0.7 %) had seven and 1 (0.7 %) had 10 affected individuals in one family. There were 374 affected patients, including 300 females (80.2 %) and 74 males (19.8 %). Of the 347 unaffected family members, 156 were females and 191 males.
Genotyping:
Genomic DNA was extracted from peripheral leukocytes using the PureGene kit following the protocol from the manufacturer (Gentra Systems, Inc., Minneapolis, MN, USA). Four SNPs at the CTLA4 gene region were genotyped, including CTLA4_-1722_T/C (rs733618), CTLA4_-1147_C/T (rs16840252), CTLA4_+49_G/A (rs231775) and CTLA4_CT60_G/A (rs3087243)(Table 1). Genotyping was performed using LightCycler (Roche Diagnostics, Basel, Switzerland) or a matrix-assisted laser desorption ionization-time of flight (MALDI-TOF)-based method with MassARRAY (Sequenorm, Inc., San Diego, CA, USA) at the genotyping core facility at the Academia Sinica in Taipei. The sequence information for primer and probe design was based on the public genome database (http://www.ncbi.nlm.nih.gov). To genotype the STRP marker CTLA4_(AT)n, fluorescence-labeled primers were purchased from Applied Biosystems. PCR was performed in a 10-µl reaction volume containing 25 ng of genomic DNA and 1 unit of AccuPrime Taq polymerase (Invitrogen Corp., Carlsbad, CA, USA) according to the standard protocol. After PCR, 3 µl of the product was mixed with 0.5 µl of internal size standard and 10 µl of deionized formamide, denatured, and separated using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). Allele calling was performed by using Genotyper Software v 3.7 (Applied Biosystems). Each genotype was independently reviewed by two members of the research team. The sequences of primers and probes are available upon request.
Statistical analyses:
Mendelian inheritance consistency of all markers in individual pedigree was checked with PedCheck version 1.1 (35) to confirm the family structure and genotyping correctness. Those genotypes with initial Mendelian inconsistency were rechecked, and corrected if obvious mistake was identified, or set as missing. All the pedigrees included in this report are compatible with Mendelian inheritance. For all the 4 SNPs, Hardy-Weinberg equilibrium (HWE) was also checked using the genotype frequency of unrelated founders. None of the SNPs reported in this study was against HWE.
We applied TRANSMIT v2.54 software (22) to conduct family-based association study. TRANSMIT adapts a score test based on a partial score function that omits the terms most influenced by hidden population stratification. In a region without evidence of linkage, TRANSMIT can handle uncertainty for multilocus haplotypes, allow parental genotypes to be unknown and deal with more than one affected offspring per family. Therefore it is appropriate for our study. Haploview 3.32 (23) was used to analyze linkage disequilibrium (LD) between markers. The “solid spine” option was applied to define LD blocks.
Acknowledgments: We thank all the GD patients and their families who
graciously agreed to participate in the study. This work was supported by grants NSC# 91-3112-B-002-003, 92-3112-B-002-009, 93-3112-B-002-015 and NSC 95-3112-B-002-026 from National Science Council, Taiwan. We also apologize to the authors whose work regarding this issue was not included in the references due to word count limitation.
References
1. Weetman AP 2000 Graves' disease. N Engl J Med 343:1236-1248
2. Tunbridge WM, Evered DC, Hall R, Appleton D, Brewis M, Clark F, Evans JG, Young E, Bird T, Smith PA 1977 The spectrum of thyroid disease in a community: the Whickham survey. Clin Endocrinol (Oxf) 7:481-493 3. Jacobson DL, Gange SJ, Rose NR, Graham NM 1997 Epidemiology and
estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol 84:223-243
4. Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, Braverman LE 2002 Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and
Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab 87:489-499
Endocr Rev 10:537-562
6. Brix TH, Kyvik KO, Hegedus L 1998 What is the evidence of genetic factors in the etiology of Graves' disease? A brief review. Thyroid 8:727-734
7. Vyse TJ, Todd JA 1996 Genetic analysis of autoimmune disease. Cell 85:311-318
8. Brix TH, Kyvik KO, Christensen K, Hegedus L 2001 Evidence for a major role of heredity in Graves' disease: a population-based study of two Danish twin cohorts. J Clin Endocrinol Metab 86:930-934
9. Farid NR 1992 Understanding the genetics of autoimmune thyroid disease--still an illusive goal! J Clin Endocrinol Metab 74:495A-495B 10. Vaidya B, Kendall-Taylor P, Pearce SH 2002 The genetics of autoimmune
thyroid disease. J Clin Endocrinol Metab 87:5385-5397
11. Rudd CE, Schneider H 2003 Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat Rev Immunol 3:544-556
12. Tomer Y, Davies TF 2003 Searching for the autoimmune thyroid disease susceptibility genes: from gene mapping to gene function. Endocr Rev 24:694-717
13. Simmonds MJ, Gough SC 2004 Unravelling the genetic complexity of autoimmune thyroid disease: HLA, CTLA-4 and beyond. Clin Exp Immunol 136:1-10
14. Heward JM, Allahabadia A, Carr-Smith J, Daykin J, Cockram CS, Gordon C, Barnett AH, Franklyn JA, Gough SC 1998 No evidence for allelic
association of a human CTLA-4 promoter polymorphism with autoimmune thyroid disease in either population-based case-control or family-based studies. Clin Endocrinol (Oxf) 49:331-334
15. Djilali-Saiah I, Larger E, Harfouch-Hammoud E, Timsit J, Clerc J, Bertin E, Assan R, Boitard C, Bach JF, Caillat-Zucman S 1998 No major role for the CTLA-4 gene in the association of autoimmune thyroid disease with IDDM. Diabetes 47:125-127
16. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, Herr MH, Dahlman I, Payne F, Smyth D, Lowe C, Twells RC, Howlett S, Healy B, Nutland S, Rance HE, Everett V, Smink LJ, Lam AC, Cordell HJ, Walker NM, Bordin C, Hulme J, Motzo C, Cucca F, Hess JF, Metzker ML, Rogers J, Gregory S, Allahabadia A, Nithiyananthan R, Tuomilehto-Wolf E, Tuomilehto J, Bingley P, Gillespie KM, Undlien DE, Ronningen KS, Guja C,
Ionescu-Tirgoviste C, Savage DA, Maxwell AP, Carson DJ, Patterson CC, Franklyn JA, Clayton DG, Peterson LB, Wicker LS, Todd JA, Gough SC
2003 Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423:506-511
17. Vaidya B, Imrie H, Perros P, Young ET, Kelly WF, Carr D, Large DM, Toft AD, McCarthy MI, Kendall-Taylor P, Pearce SH 1999 The cytotoxic T lymphocyte antigen-4 is a major Graves' disease locus. Hum Mol Genet 8:1195-1199
18. Heward JM, Allahabadia A, Armitage M, Hattersley A, Dodson PM, Macleod K, Carr-Smith J, Daykin J, Daly A, Sheppard MC, Holder RL, Barnett AH, Franklyn JA, Gough SC 1999 The development of Graves' disease and the CTLA-4 gene on chromosome 2q33. J Clin Endocrinol Metab 84:2398-2401
19. Freedman ML, Reich D, Penney KL, McDonald GJ, Mignault AA, Patterson N, Gabriel SB, Topol EJ, Smoller JW, Pato CN, Pato MT, Petryshen TL, Kolonel LN, Lander ES, Sklar P, Henderson B, Hirschhorn JN, Altshuler D 2004 Assessing the impact of population stratification on genetic association studies. Nat Genet 36:388-393
20. Cardon LR, Palmer LJ 2003 Population stratification and spurious allelic association. Lancet 361:598-604
21. Schulze TG, McMahon FJ 2002 Genetic association mapping at the crossroads: which test and why? Overview and practical guidelines. Am J Med Genet 114:1-11
22. Clayton D 1999 A generalization of the transmission/disequilibrium test for uncertain-haplotype transmission. Am J Hum Genet 65:1170-1177
23. Barrett JC, Fry B, Maller J, Daly MJ 2005 Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263-265
24. Ramirez-Soriano A, Lao O, Soldevila M, Calafell F, Bertranpetit J, Comas D 2005 Haplotype tagging efficiency in worldwide populations in CTLA4 gene. Genes Immun 6:646-657
25. Hudson LL, Rocca K, Song YW, Pandey JP 2002 CTLA-4 gene polymorphisms in systemic lupus erythematosus: a highly significant association with a determinant in the promoter region. Hum Genet 111:452-455
26. Fernandez-Blanco L, Perez-Pampin E, Gomez-Reino JJ, Gonzalez A 2004 A CTLA-4 polymorphism associated with susceptibility to systemic lupus erythematosus. Arthritis Rheum 50:328-329
27. Brophy K, Ryan AW, Thornton JM, Abuzakouk M, Fitzgerald AP, McLoughlin RM, O'Morain C, Kennedy NP, Stevens FM, Feighery C, Kelleher D, McManus R 2006 Haplotypes in the CTLA4 region are
associated with coeliac disease in the Irish population. Genes Immun 7:19-26
28. Yanagawa T, Hidaka Y, Guimaraes V, Soliman M, DeGroot LJ 1995
CTLA-4 gene polymorphism associated with Graves' disease in a Caucasian population. J Clin Endocrinol Metab 80:41-45
29. Kotsa K, Watson PF, Weetman AP 1997 A CTLA-4 gene polymorphism is associated with both Graves disease and autoimmune hypothyroidism. Clin Endocrinol (Oxf) 46:551-554
30. Huang D, Giscombe R, Zhou Y, Pirskanen R, Lefvert AK 2000 Dinucleotide repeat expansion in the CTLA-4 gene leads to T cell hyper-reactivity via the CD28 pathway in myasthenia gravis. J Neuroimmunol 105:69-77
31. Chang YP, Liu X, Kim JD, Ikeda MA, Layton MR, Weder AB, Cooper RS, Kardia SL, Rao DC, Hunt SC, Luke A, Boerwinkle E, Chakravarti A 2007 Multiple genes for essential-hypertension susceptibility on chromosome 1q. Am J Hum Genet 80:253-264
32. Consortium TIH 2003 The International HapMap Project. Nature 426:789-796
33. Chen PL, Fann CS, Chang CC, Wu IL, Chiu WY, Lin CY, Yang WS, Chang TC 2007 Linkage of Graves' disease to the human leucocyte antigen region in the Chinese-Han population in Taiwan. Clin Endocrinol (Oxf) 66:646-651 34. Simmonds MJ, Howson JM, Heward JM, Cordell HJ, Foxall H, Carr-Smith J, Gibson SM, Walker N, Tomer Y, Franklyn JA, Todd JA, Gough SC 2005 Regression mapping of association between the human leukocyte antigen region and Graves disease. Am J Hum Genet 76:157-163
35. O'Connell JR, Weeks DE 1998 PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259-266
Figure legend:
Figure 1. Pairwise LD pattern of markers at CTLA4 measured by (a) D’ and (b) R2.
These five markers were grouped as a single block spanning 7 kb, using the “solid spine” block definition of Haploview (23)v3.32. The location of each tested marker relative to CTLA4 is indicated on top. The number in each diamond indicates the magnitude of LD in percent between respective pairs of markers. For example, the pairwise LD for -1722_T/C and (AT)n_O/S is 0.14 in D’ and 0 in R2. Diamonds without values are the ones with D’ equal to 100%.
Figure 2. Allele frequency of three SNPs in different populations. Abbreviation of
the populations: CHT: Chinese Han in Taiwan (our current collection); CEU: CEPH (Utah residents with ancestry from northern and western Europe); CHB: Chinese Han in Beijing; JPT: Japanese in Tokyo; and YRI: Yoruba in Ibadan, Nigeria. The allele frequency for CHT came from our study using the genotypes of unrelated founders. All the other allele frequency was from the HapMap project (32), except for the allele frequency of +49_G/A of CEU which was from dbSNP
(http://www.ncbi.nlm.nih.gov/SNP/index.html) because of the lack of information in HapMap.
Tables:
Table 1. Information and 1-marker association test of the 4 SNPs and 1 STRP marker
aCTLA4_(AT)n_O/S is a dinucleotide (AT) STRP
marker. “S” allele stands for the “Specific” allele which is the same as the “allele 106” of Yanagawa et al. study (28), and “O” allele stands for the collection of all the “Other” alleles.
bTRANSMIT v. 2.54 (22), with the option –ro (to use the robust estimate of the variance of the score vector).
cThe sign in this column is the sign of Chi square statistics of the minor allele. The plus sign means over-transmission of the minor allele, and
the minus sign under-transmission.
dNominal P value less than 0.05 is marked as bold letters
Marker Name Name, NCBI
Build 36.1 Frequency of Minor Allele Chi square, (df 1) TRANSMITb P value, TRANSMITb CTLA4_-1722_T/ C rs733618 C allele, 0.427 + 6.71c 0.0096d CTLA4_-1147_C/ T rs16840252 T allele, 0.111 + 3.08 0.079 CTLA4_+49_G/A rs231775 A allele, 0.288 - 5.25 0.0219 CTLA4_(AT)n_O/ Sa UniSTS:48500 S allele, 0.358 +1.50 0.22 CTLA4_CT60_G/ A rs3087243 A allele, 0.199 - 2.45 0.11
Table 2. 2-marker association test: all possible combinations Marker name Haplotype Freq. Chi-quare (df 1),
TRANSMIT P value, TRANSMIT -1722_T/C, -1147_C/T T:C 0.465 -2.23a 0.1350 C:C 0.422 +7.34 0.0067 T:T 0.111 -4.36 0.0369 C:T 0.002 +1.70 0.1919 -1722_T/C, +49_G/A C:G 0.429 +5.38 0.0204 T:A 0.291 -7.27 0.0070 T:G 0.280 +0.04 0.8328 -1722_T/C, (AT)n_O/S T:O 0.345 -12.58 0.0004 C:O 0.302 +3.76 0.0524 T:S 0.229 +0.75 0.3867 C:S 0.124 +2.10 0.1474 -1722_T/C, CT60_G/A C:G 0.426 +5.86 0.0155 T:G 0.375 -0.79 0.3755 T:A 0.199 -3.63 0.0568 -1147_C/T, +49_G/A C:G 0.699 +7.70 0.0055 C:A 0.188 -4.56 0.0327 T:A 0.104 -2.38 0.1227 T:G 0.010 -0.06 0.8078 -1147_C/T, (AT)n_O/S C:O 0.541 -0.19 0.6661 C:S 0.345 +2.40 0.1215 T:O 0.103 -2.02 0.1556 T:S 0.011 -1.88 0.1698 -1147_C/T, CT60_G/A C:G 0.692 +6.26 0.0123 C:A 0.196 -2.13 0.1448 T:G 0.110 -3.16 0.0755 T:A 0.002 -1.33 0.2486
+49_G/A, (AT)n_O/S G:O 0.369 +0.33 0.5669
G:S 0.348 +2.69 0.1010 A:O 0.284 -5.32 0.0211 A:S 0.007 -1.17 0.2800 +49_G/A, CT60_G/A G:G 0.702 +3.92 0.0477 A:A 0.193 -2.49 0.1149 A:G 0.100 -0.94 0.3317 G:A 0.005 -0.02 0.8781
(AT)n_O/S, CT60_G/A O:G 0.445 +0.07 0.7899
S:G 0.355 +1.20 0.2737
O:A 0.198 -3.75 0.0528
aThe sign in this column is the sign of Chi square statistics of this haplotype. The plus sign means over-transmission of this haplotype, and the minus sign under-transmission.
Genowide Association Study
In the present population-based association study, we have completed the collection of blood of 1,026 individuals. There are 1024 persons confirmed to be GD, which include 176 men, and 848 women (men to women =1:4.8). The mean age is 40.9 + 12.8 years (range 9 – 81 years). The mean age of onset of hyperthyroidism is 35.9 + 12.6 years (range 6 – 81 years). The degree of goiter is 2.1 + 0.9 (range 0 – 4). There are 500 patients (48.8%) with ophthalmopathy (Gr. 1: 126, 12.3%; Gr 2: 234, 22.9%; Gr. 3: 140, 13.7%), 33 patients (3.2%) with pretibial myxedema, 19 patients (1.9%) with periodic paralysis, 13 patients (1.3%) with myasthenia gravis, 7 patients (0.7%) with vitiligo. Genotyping with Affymetrix 500K chip was performed in 246 female patients of Graves’ disease with ophthalmopathy. We compared with two kinds of control samples :Control I, females only, N = 224; Control II, both males and females, N = 468. The genomewide association study showed the SNPs at HLA region were highly significant (-Log10(p)=5.841). The HLA genotype data of all the individuals (721 familial samples and 500 population-based samples) are completed, but 500 control samples are waiting from Academia Sinica. We will try to find the susceptibility genes at the HLA region.
Affymetrix GeneChip® Mapping 500K Array
There are 489,922 SNPs on 22 chromosomes ~ 500,000 SNPs in the 3*109 bp human genome.
“On average” 6 kb per SNP, (the SNPs are NOT evenly distributed.) Exclusion Criteria
1. non-polymorphism in both of case & control
2. HWE test (Hardy-Weinberg equilibrium test, p-value < 5*10-7) in both of case & control 3. MAF(minor allele frequency) < 5% in control
4. CallRate < 95% in combined case and control samples to make sure the quality of genotypes is good
About 317,000 SNPs were used for further analyses SAS/Genetics was applied for single-point association test
Results
In chromosome 6, the HLA region is believed to contain some strong real susceptibility gene(s). The lessons from this HLA signal in our study are (1) The quality of our study has been good, and GWA can work with our samples. (2) The P values from most of other genuine susceptibility genes might NOT as significant as those from the HLA region ~ 10-6. . In this chromosome, the P values using all controls were smaller than using only female controls perhaps due to larger size. There are 5 SNPs in a ~ 120 kb region in chromosome 1, with the best P value ~ 10-5. The P values using all controls are smaller than those using female-only controls.
In chromosome 5, the results are more significant when using female-only controls (than all controls). GD, like several other autoimmune diseases, is predominantly found in females. It is
possible that some risk genes might be gender-specific.
In chromosome 12, the region contains two genes; the function of these genes is poorly
understood at this moment. Interestingly, one gene is highly expressed in B lymphocytes and T lymphocytes, and the other in B lymphocyte, monocyte and dendritic cells.
Association of HLA-A, HLA-B, HLA-C and HLA-DP, DQ, DR, to GD in the Chinese-Han population
Aim, Methods
Find the susceptibility alleles/loci/haplotypes of classical HLA genes to GD Compare the risk alleles/loci/haplotypes between Chinese-Han and Caucasians
In our genotyping results, there were 15 HLA-A alleles, 45 HLA-B alleles, 22 HLA-C alleles, 18 HLA-DP alleles, 16 HLA-DQ alleles and 30 HLA-DR alleles.
The HLA genotype data of all the individuals (721 familial samples and 500 population-based samples are completed, but 500 control samples are waiting from Academia Sinica.
• We can achieve 4-digit of resolution. (For example HLA-DRB1*0301).
• We basically use the sequence-specific oligo (SSO) probe hybridization methods, either LABType SSO system of One Lambda, or Reli SSO method of DYNAL. These two companies/platforms are among the most reliable ones in this field. Sequence-based genotype confirmation was performed if necessary.