題目: 簡介免疫系統對抗惡性腫瘤之機轉 作者: 李聰亮 林子鈞 黃建仁 機構:中國醫藥大學附設醫院皮膚科;中國醫藥大學皮膚科 通訊作者:李聰亮 醫師 通訊地址:台中市 404 北區育德路 2 號 電話: 04-22120843 傳真:04-22061096 E-mail:sikilover@pchome.com.tw
前言 惡性腫瘤在人體內生成並非單一因素,其牽涉頗為複雜的病理發生。在正常生理狀況下, 各個不同的細胞生長、分化以及細胞之存活皆受到精準的控制,而生理機能要維持在恆定狀 態必須體內基因及其蛋白質產物執行頗複雜細微的工作,當某些基因發生突變時即可能促使 細胞不正常的分裂及增生,終於發生細胞癌變(1-3),我們務必明瞭免疫系統在細胞不正常分 化、異常增生以及惡性細胞生長過程所扮演的角色。 惡性腫瘤分類及癌症標記 癌症是一群疾病必須表現出無法受調控的細胞快速的生長,同時這些細胞具有侵犯和從 原始發生部位擴散到身體其他部位之特性。在癌症分類上已有百種以上不同型態之惡性腫瘤 , 但 80 ~ 85% 之 癌 症 屬 上 皮 細 胞 癌 , 如 : 皮 膚 之 鱗 狀 上 皮 細 胞 癌 ( squamous cell carcinioma)以及肺、乳房、腸胃道、甲狀腺之腺癌(adenocarcinoma)。由中胚層發育形 成的細胞,例如:肌肉、骨骼、軟骨組織所源生之惡性腫瘤即為肉瘤(sarcoma),所佔極 少的比例。除外,另一血球生成細胞來源的癌症包括白血病及淋巴癌等約佔17%。(4)
公元2000 年,Douglas Hanahan 和 Robert Weinberg.提出六項癌症標記(5):自發的生
長訊息、躲避生長抑制訊息、躲避細胞凋亡、無限的細胞複製潛能、血管新生、侵襲性及移轉。 這些皆是惡性腫瘤細胞異於正常細胞之特性。
DNA突變導致細胞重要基因喪失調控功能
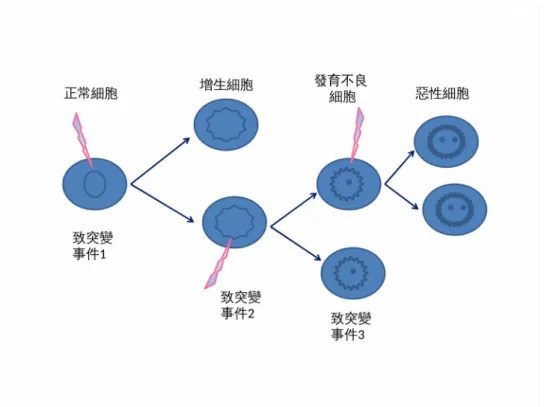
生活周遭存在許多會導致DNA序列改變或突變的物質,稱為致突變物質,例如化學致 癌物、紫外線照射 、輻射、致癌病毒等致使DNA突變為造成癌症極重要的因子。現已知癌細
胞之DNA改變會不斷的累積,此極可能與癌症之惡性度增大有關。而那些導致癌症發生, 明確而言為-多步驟過程(圖 1)。 致癌基因、腫瘤抑制基因與癌症發生之關係 多數的癌細胞起因於某些基因突變之共同結果,致癌基因(oncogenes) (6)促使正常細 胞轉型為癌細胞,而這些致癌基因其實是由正常基因發生突變所產生的,當這些掌控細胞生 長、分化以及細胞凋亡的基因失去正常的機能時,細胞就會不受節制地增生及喪失原有的生 理功能。另一類的腫瘤抑制基因(tumor suppressor genes)(7)也會因突變而失去功能;
此基因必需是其基因位之兩個對偶基因(alleles)皆已發生突變才會表現出來;例如:P53 及RB 基因為最好的例子 (8)。當細胞遭受壓力時,例如輻射、致癌物質導致DNA 受損,致癌 基因活化、低氧和核糖核苷酸存量不足時,此訊息皆會使P53 活化,使得細胞分裂週期停下 來,以爭取時間讓受損的DNA 修復;如無法修復的 DNA 則使細胞邁向凋亡。P53 也會抑制 血管生成之作用,因此使腫瘤缺乏生長所需的養分及氧氣來源(9)。RB 基因其所支配合成 RB 蛋白質可控制細胞週期 G1 期進入 S 期之限制點,RB 基因缺陷將導致視網膜胚細胞瘤 (retinoblastoma)的發生。P53 和 PRB 這些基因產物會形成訊息網路以鑑測細胞基因族 之完整性和確定已收到正確的增生訊息才允許細胞週期順利進行。 吞噬細胞表面模型認知受體與微生物體表病理-相關連分子模型之作用 經長期的演化免疫系統建立一完整的防禦機轉,當遭遇外來入侵的微生物時,天生的免 疫力(innate immunity)為一非特異性,立即的對抗微生物系統,會馬上動員對付之, 其主要成員為上皮黏膜結構形成的屏障、中性白血球、吞噬細胞、自然殺手細胞、補體系統以
及細胞激素等(10)。樹枝狀細胞(dendritic cells)其細胞膜表面具有模型認知受體(pattern
recognition receptors,PRR)(11),其功能是與微生物體表之病理-相關連分子模型
(pathogen-associated molecular patterns,PAMP)相結合,再將此訊息傳遞到細胞 內使一些轉譯因子(例如:NFκB 和 IRF)活化,再指使合成許多抗微生物之基因產物。 免疫系統如何防衛惡性腫瘤 免疫系統恆常被教育成可分辨〝自己的〞和〝非自己的〞,當免疫系統成員遭遇外來微生 物入侵時必定會啟動一定的步驟以對付入侵者。尤其是駐在於皮膚、口腔、呼吸道、腸胃道、生 殖系統及泌尿道等黏膜下層之單核細胞、吞噬細胞以及中性血球細胞等為第一線的細胞,會 快速動員對抗入侵的微生物。但當免疫系統鑑測到體內癌變細胞時,在某些情況下無法辨識 癌細胞為〝非自己〞的組成,因此針對腫瘤產生的免疫反應大多是不足夠摧毀這些細胞。 由於癌細胞大多為非感染性微生物所導致,惡性腫瘤因而缺乏分子記號也就無法引起免 疫系統去認知體內有無異常發生,而傾向於對於腫瘤產生耐受性之環境。已知免疫系統發動 有效的免疫反應一定是已偵測到PAMPs,吞噬細胞(包括樹枝狀細胞)才會迅速移動過來 利用其PRRs 與 PAMPs 結合,啟動後續一連串反應。事實上,由胚胎組織所生長之腫瘤其 合成的蛋白質以及致癌性病毒所導致腫瘤其表面抗原、免疫系統皆將之認知為外來的物質, 因此樹枝狀細胞會吞噬腫瘤細胞進入細胞質內,再將腫瘤抗原處理成胜肽片斷,此步驟是由 存在於細胞質內之蛋白質體(proteasome)含有的許多蛋白質酵素所負責(12),處理完成 胜 肽 之 大 小 及 核 苷 酸 序 列 恰 好 能 與 由 內 質 網 狀 小 體 所 合 成 的 某 一 特 定 的 major histocompatibility complex (MHC)之溝槽結合,再一起運送到細胞表面,此類 MHC 分子為MHC class I。而攜帶 MHC class I 和 MHC class II 分子的樹枝狀細胞具有呈現抗原
給 其 他 T - 細 胞 的 能 力 , 此 樹 狀 細 胞 即 稱 為 抗 原 - 呈 現 細 胞 ( antigen-presenting cells)。MHC class I 可與細胞毒性 T-細胞(cytotoxic T-cell)之細胞表面受體(T-cell receptor)相結合,而使可認知此特定胜肽之細胞毒性 T-細胞分化及增長出特定細胞株。抗 原-呈現細胞之MHC class II 則提供與幫忙者 T-細胞(helper T-cell)之細胞表面受體相結 合,而使幫忙者T-細胞活化及增生。抗原-呈現細胞吞噬腫瘤細胞或腫瘤抗原後,會將腫 瘤抗原呈現給具有認知此抗原的細胞毒性T-細胞,而在具有認知此腫瘤抗原之幫忙者T- 細胞協助下誘導出其對抗腫瘤之免疫反應。(13,14)
當腫瘤細胞表現出某些不應發生於正常細胞之分子時,由於突變造成新的核苷酸序列, 但適應性的免疫系統(adaptive immune system)仍無法反應(15),這是因為在此免疫系
統背景下必需另一項適當的共同刺激訊息才能使T-細胞活化。而提供此共同刺激訊息者與 駐在於組織中的樹枝狀細胞有關,此細胞由未成熟轉變成成熟狀態必定要與微環境中外來的 PAMP結合,再經由淋巴循環回到局部淋巴結。到達淋巴結時樹枝狀細胞已成熟為具有呈 現抗原的功能,因此能使淋巴結內從未接觸過外來抗原的T-細胞活化成可認知此特定抗原 的T-細胞株,再分化及生長成許多可有效對抗腫瘤的T-細胞。 腫瘤演變成躲避免疫反應之特性 惡性腫瘤在體內也會演變出對於惡性細胞生長及存活有利的條件,因此腫瘤會演變成能 躲避免疫系統攻擊以達成其不斷增生及轉移到其他器官及組織(10)。許多腫瘤抗原對於免疫系 統而言為低致敏性,也許是因腫瘤細胞本是由正常細胞變形而來,僅表現出些微的差異於自 體的抗原。有些腫瘤失去細胞表面抗原,或是細胞表面MCH class I 分子不表現出來(正常 情況有核的細胞皆會具有MHC class I),因而無法讓細胞毒性T-細胞認識其存在,但此
時另一種自然殺手細胞(natural killer cell)可負責摧毀這些不帶有 MCH class I 分子的惡 性細胞,然而自然殺手細胞數量僅占血循環中淋巴細胞的十分之一,是否足夠完全防禦體內 惡性腫瘤生長則是非確定性的。有些腫瘤會釋放細胞激素,例如:TGF-β(transforming growth factor-β)、IL-10 (interleukin-10)以及 VEGF(vascular endothelial growth factor)。TGF-β 會抑制T細胞的分化和增生,以及抑制吞噬細胞活化;IL-10 亦會抑制免疫 反應,而VEGF 已有證據顯示其會抑制樹枝狀細胞成熟,導致免疫系統對腫瘤抗原產生耐受 性而無法有效發動攻擊。 惡性腫瘤趨使腫瘤生長環境進行組織癒合模式(16) 免疫系統除了強健的防禦功能以對抗入侵的微生物和體內浮現的腫瘤細胞外, 實際上免 疫反應於這些危險訊息消除後仍然必須面對後續的任務。 常見於感染所導致發炎反應使組織 間呈現組織水腫壞死等變化, 當感染受控制後,免疫系統仍須負責重建組織完整性工作。 這是由吞噬胞細和其他本有的免疫細胞釋放生長因子包括TGF-β 及 VEGF,前者可刺激纖 維芽細胞增生同時合成組織修復必需的細胞外基質和膠原纖維;後者為新建立的組織提供新 的血管生成。 某些惡性腫瘤已被發現能誘導,腫瘤生長的環境進行如上述 " 組織癒合 " 的 模式, 當進入此模式時免疫系統似已進入休止狀態, 那麼腫瘤也因而逃脫被免疫細胞攻擊 而能持續不斷長大。 慢性感染和慢性炎症與惡性腫瘤發生有關(13,17) 往昔醫學認知大多以為腫瘤組織間發炎細胞可能扮演對抗腫瘤的角色,但近年來證據顯 示慢性炎症反應會促使腫瘤細胞形成及更具侵襲性。 以老鼠所作實驗 : 長了腫瘤的老鼠以
酯多醣類( lipopolysaccharide)注射入老鼠體內,結果發現此物質(內毒素)會促使腫瘤成 長及轉移。目前醫界甚為重視慢性感染和慢性炎症反應,為促使腫瘤形成及侵襲性產生極重 要因素。流行病學以及分子病理學研究,皆顯示某些慢性感染與癌症發生有關聯性, 例如人 類乳突病毒(HPV)導致子宮頸癌、Epstein-Barr 病毒會引起鼻咽癌、B-細胞淋巴球瘤、人類 T-細胞白血病病毒(HTLV-1)引發 T-細胞白血病/淋巴瘤、B 型肝炎病毒引起肝細胞癌、幽門螺旋 桿菌與胃癌發生有關。目前醫界也尋獲聯結慢性炎症與惡性腫瘤發生的中介者為核因子κB (一種轉譯因子) (18)。 已知腫瘤細胞表面也可能表現出PRR 如與體內組織中的 PAMP、以及危
險有關分子模型( danger-associated molecular pattern, DAMP)結合,會趨使核因子 κB 活化。此轉譯因子活化時,後續作用機轉為 1.使細胞內之抗凋亡基因表達出來,例如 Bcl-2 家族成員使腫瘤細胞不會凋亡;2.促使細胞製造細胞激素 IL-1 和 IL-6,具有致細胞分 裂而使得細胞增生作用。 至於慢性炎症反應其中極重要成員為吞噬細胞,此細胞釋放腫瘤壞死因子-α,它掌控炎 症反應使組織產生一些作用分子使炎症反應一直持續進行(19)。而由骨髓派遣出來的白血球及 吞噬細胞到達炎症發生部位,尤其是活化的吞噬細胞會釋放活性氧離子(‧O2-)及氮氧化中間 產物(ROS/NO),兩者皆為不穩定性物質易造成細胞 DNA 受損,尤其是活性氧離子使得絲 腺體(mitochondria)和細胞核 DNA 受損因此增加細胞突變發生, 終於使細胞變形伴隨異 常快速增生逐漸使惡性腫瘤進一步形成(圖 2)。(20) 結語 釐清免疫系統在細胞不正常分化、異常增生以及惡性細胞生長過程所扮演的角色,同時 明瞭慢性感染和慢性炎症與惡性腫瘤發生之作用機轉,對於未來腫瘤治療學上的研究及開發
扮演很重要的角色。事實上,惡性腫瘤細胞在寄主體內生長亦需面對某種程度環境壓力,因 此不適於生存之惡性細胞也會經由選擇性淘汰機制而篩選出增大惡性度的細胞。認知惡性細 胞多步驟的演變過程,早期截斷將更有效避免進行最糟的後果。我們知道免疫系統與體內浮 現的惡性細胞之間殘酷的競爭仍然不會終止的。
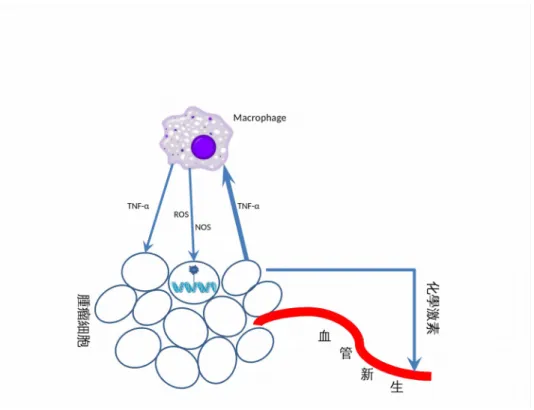
圖2 腫瘤與炎症反應會相互影響:腫瘤細胞釋放化學激素可補充吸引白血球及腫瘤有關吞噬 細胞;吞噬細胞合成細胞激素(TNF-α)及釋放出活性氧離子和氮氧中間產物(ROS/NOS),後 兩者造成DNA 突變。腫瘤細胞釋放的細胞激素會使血管新生。
參考文獻
1. Yu Y, Feng YM: The role of kinesin family proteins in tumorigenesis and progression: potential biomarkers and molecular targets for cancer therapy. Cancer, 2010;116:5150-5160.
2. Holland AJ, Cleveland DW: Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol, 2009;10:478-487.
3. Moynahan ME, Jasin M: Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol, 2010;11:196-207.
4. Sankaranarayanan R, Swaminathan R, Jayant K, et al.: An overview of cancer survival in Africa, Asia, the Caribbean and Central America: the case for investment in cancer health services. IARC Sci Publ, 2011;162:257-291. 5. Hanahan D, Weinberg RA: The hallmarks of cancer. Cell, 2000;100:57-70. 6. Fearon ER, Dang CV: Cancer genetics: tumor suppressor meets oncogene.
Curr Biol, 1999;9:R62-65.
7. Sherr CJ: Principles of tumor suppression. Cell, 2004;116:235-246.
8. Bouchet BP, de Fromentel CC, Puisieux A, et al.: p53 as a target for anti-cancer drug development. Crit Rev Oncol Hematol, 2006;58:190-207.
9. Assadian S, El-Assaad W, Wang XQ, et al.: p53 inhibits angiogenesis by inducing the production of Arresten. Cancer Res, 2012;72:1270-1279.
inflammation, and cancer. J Clin Invest, 2007;117:1175-1183.
11. Seya T, Shime H, Ebihara T, et al.: Pattern recognition receptors of innate immunity and their application to tumor immunotherapy. Cancer Sci, 2010;101:313-320.
12. Yen HC, Chang EC: INT6--a link between the proteasome and tumorigenesis. Cell Cycle, 2003;2:81-83.
13. Lowe DB, Storkus WJ: Chronic inflammation and immunologic-based constraints in malignant disease. Immunotherapy, 2011;3:1265-1274.
14. Trinchieri G: Inflammation in cancer: a therapeutic target? Oncology (Williston Park), 2011;25:418-420.
15. Krieg C, Boyman O: The role of chemokines in cancer immune surveillance by the adaptive immune system. Semin Cancer Biol, 2009;19:76-83.
16. Delves PJ, Martin SJ, Burton DR, et al.: Roitt's Essential Immunology, 12th ed, San Francisco, Wiley-Blackwell, 2011:446-451.
17. Hu B, Elinav E, Flavell RA: Inflammasome-mediated suppression of inflammation-induced colorectal cancer progression is mediated by direct regulation of epithelial cell proliferation. Cell Cycle, 2011;10:1936-1939. 18. Pikarsky E, Porat RM, Stein I, et la.: NF-kappaB functions as a tumour
promoter in inflammation-associated cancer. Nature, 2004;431:461-466. 19. van Horssen R, Ten Hagen TL, Eggermont AM: TNF-alpha in cancer
Oncologist, 2006;11:397-408.
20. Lauren Pecorino: Molecular Biology of Cancer: Mechanisms, Targets, and Therapeutics, 2nd ed, UK, Oxford University Press, 2008:217-219.