Distant HNF1 Site as a Master Control for the Human Class I
Alcohol Dehydrogenase Gene Expression
*
□SReceived for publication, April 14, 2006 Published, JBC Papers in Press, May 4, 2006, DOI 10.1074/jbc.M603638200
Jih-Shyun Su‡1, Ting-Fen Tsai‡§1, Hua-Mei Chang¶, Kun-Mao Chao储, Tsung-Sheng Su‡**, and Shih-Feng Tsai‡§2
From the‡Faculty of Life Sciences and Institute of Genetics, National Yang-Ming University, Taipei 112,§Division of Molecular
and Genomic Medicine, National Health Research Institutes, Zhunan, Miaoli County 350,¶Vita Genomics, Inc., Taipei County 248,
储Department of Computer Science and Information Engineering, National Taiwan University, Taipei 106, and the **Department
of Medical Research and Education, Taipei Veterans General Hospital, Taipei 112, Taiwan Gene duplication and divergence have contributed to the
biochemical diversity of the alcohol dehydrogenase (ADH) family. Class I ADH is the major enzyme that catalyzes alco-hol to acetaldehyde in the liver. To investigate the mecha-nism(s) controlling tissue-specific and temporal regulation of the three human class I ADH genes (ADH1A, ADH1B, and ADH1C), we compared genomic sequences for the human and mouse ADH loci and analyzed human ADH gene expression in BAC transgenic mice carrying different lengths of the upstream sequences of the class I ADH. A conserved noncod-ing sequence, located between the class I and class IV ADH (ADH7) genes, was found to be essential for directing class I ADH gene expression in fetal and adult livers. Within this region, a 275-bp fragment displaying liver-specific DNase I hypersensitivity was bound by HNF1. The HNF1-containing upstream sequence enhanced all three class I ADH promoters in an orientation-dependent manner, and the transcriptional activation depended on binding to the HNF1 site. Deletion of the conserved HNF1 site in the BAC led to the shutdown of human class I ADH gene expression in the transgenic livers, leaving ADH1C gene expression in the stomach unchanged. Moreover, interaction between the upstream element and the class I ADH gene promoters was demonstrated by chromo-some conformation capture, suggesting a DNA looping mechanism is involved in gene activation. Taken together, our data indicate that HNF1 binding, at⬃51 kb upstream, plays a master role in controlling human class I ADH gene expression and may govern alcohol metabolism in the liver.
Alcohol dehydrogenases (ADHs3; EC 1.1.1.1) are
zinc-con-taining dimeric enzymes, which can reversibly oxidize the
cyto-solic alcohol to acetaldehyde or retinol to retinal (1). Through gene duplications, mammals have evolved unique combina-tions of genes encoding ADHs with different biochemical prop-erties. In humans, there are seven alcohol dehydrogenase (ADH) genes and they can be grouped into five classes as fol-lows: ADH1A, ADH1B, and ADH1C of class I and ADH4,
ADH5, ADH7, and ADH6 of classes II, III, IV, and V, respec-tively. Similarly, mice have seven ADH genes belonging to the five biochemical classes. However, only one class I Adh (Adh1) has been found in the mouse genome, and there are three class V Adh genes (Adh5a, Adh5b, and one pseudogene of Adh5ps) (2, 3).
The human class I ADH genes are clustered head-to-tail
within an ⬃80-kb region on chromosome 4 (4). Proteins
encoded by the class I ADH genes form both homo- and het-erodimeric alcohol dehydrogenase isozymes (5– 8), and they represent the predominant ADHs in the liver. Class I ADH gene expression varies at developmental stages and in extrahepatic tissues (5). The proteins encoded by ADH1A, ADH1B, and
ADH1Cwere designated as␣-, -, and ␥-subunit, respectively. The␣-subunit is first detected during early fetal liver develop-ment and later in adult kidneys.⌻he-subunit can be detected at late stages of fetal liver development and also in adult liver, lung, and kidneys.⌻he ␥-subunit is expressed mainly in the postnatal liver but also in fetal stomach, kidneys, and intestine and in adult stomach (6).
In humans, the genomic location of class IV ADH (ADH7) is next to ADH1C, and they are separated by a distance of⬃60 kb. Compared with class I ADH, the ADH7 gene encodes retinol dehydrogenase with higher activity than proteins encoded by class I ADH (9, 10). The enzyme was first identified from
stom-ach mucosa and designated as or ADH (11, 12).
Subse-quently, a major physiological function of ADH7 was shown to act through retinol dehydrogenase, which was involved in a rate-limiting step of retinol metabolism (13).
Many studies have shown that genetic variation in the human class I ADH genes is associated with the risk of alcoholism, and the physiological basis of the link between ADH1B variants and alcohol tolerance is well understood (14, 15). The control of the three class I ADH genes, however, remains unclear. The ques-tions of how the entire class I gene locus is specifically expressed in the liver and the differential use of the class I ADH
*This work was supported by National Science Council Grant 93-3112-B-400-006- (to S. F. T.) and National Research Program for Genomic Medicine Grant 92GM064 and National Science Council Grant 93-2752-B-010-004-PAE (to T. F. T.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
□S The on-line version of this article (available at http://www.jbc.org) contains
a supplemental table and figure.
1Both authors contributed equally to this work.
2To whom correspondence should be addressed: Division of Molecular and
Genomic Medicine, National Health Research Institutes, Zhunan, Miaoli County 350, Taiwan. Tel.: 886-2-28267043; Fax: 886-2-28200552; E-mail: [email protected].
3The abbreviations used are: ADH, alcohol dehydrogenase; EMSA,
electro-phoretic mobility shift assay; CNS, conserved noncoding sequence; dpc,
days post-coitum; HS, hypersensitive; RT, reverse transcription; SNP, single nucleotide polymorphism.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 29, pp. 19809 –19821, July 21, 2006 © 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
at National Taiwan University on March 11, 2009
www.jbc.org
genes during development are of interest, a situation analogous to the subject of “globin switching” in hematology research. Several studies have investigated the regulation of class I ADH gene expression, and most of them focused on promoter activ-ity and cis-elements in proximal promoter regions (1, 16). The three human class I ADH genes share with each other 80 – 84% sequence identity for about 270 bp upstream of the transcrip-tion start site, yet the promoter sequences showed variable
in vitrobinding affinities with nuclear extracts, as assayed by DNase I footprinting (16). Transfection studies in cell culture systems also revealed differential transcriptional activity in liver and non-liver cancer cell lines, when using different human class I ADH promoters (16). These results indicated that differ-ent regulatory mechanisms might have evolved for each of the
ADHgenes to account for the distinct expression patterns. DNA sequence comparison has proven to be a powerful approach to investigate the evolution of tissue-specific gene expression and to identify regulatory elements in the noncod-ing regions (17). Across the long evolution distance between human and mouse, several conserved regions can be identified in the ADH genes and in the intergenic regions, including an ⬃60-kb interval between ADH1C and ADH7. We speculated that conserved noncoding sequence (CNS) in the intergenic region might play a critical role in regulating ADH gene expres-sion. To test this possibility, we identified CNS between
ADH1Cand ADH7 and examined their regulatory function using a BAC transgenic mouse approach. In parallel, we con-ducted DNase I hypersensitive site mapping to identify regula-tory elements that are involved in controlling class I ADH gene
expression. We found that HNF1 binding at⬃51 kb upstream
of the human class I ADH gene cluster plays an important role in controlling human class I ADH gene expression in vivo.
EXPERIMENTAL PROCEDURES
BACs Selection—Human BAC clones used are CA(496L13;
182,676 bp; GenBankTM accession number AP002027) and
BI(2138E16; 138,307 bp, including 130,962 bp overlapping sequence with CA and 7345-bp unique sequence). The two BAC sequences cover the entire class I ADH genes but with different upstream conserved noncoding regions (Fig. 1).
Transgenic Mice Generation—We used SrfI to linearize the BAC clone CA and CAscarDNA and NotI to excise the insert of
BAC BI from the pBeloBAC11 vector. BAC transgenic mice were generated by pronucleus microinjection of FVB/N fertil-ized eggs (18). The transgenes in mice were checked by PCR using primers of ADH1A-F3 and ADH1A-R3 (supplemental table) with cycling conditions of 94 °C for 5 min at the first step, followed by 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. To determine copy numbers of the BAC transgenes, Southern blot hybridization was performed. A 983-bp probe (CA112117–113099) was generated by PCR using CA112117F and CA113099R as primers (supplemental table) and labeled
with [␣-32P]dCTP by the Rediprime II kit (Amersham
Bio-sciences). The filter was stripped and re-hybridized with an inter-nal control probe of the mouse endogenous gene MB20 (19).
RT-PCR Analysis of Class I ADH Gene Expression in Trans-genic Tissues—Total RNA was isolated from mouse tissues using the TriReagent (Molecular Research Center). Human
fetal liver RNA was commercially available (Clontech). For RT-PCR, 2g of total RNA was reverse-transcribed with Super-script II reverse tranSuper-scriptase (Invitrogen) using oligo(dT) primer in a total volume of 20l at 42 °C for 1 h. Human ADH expression in transgenic mice was detected by RT-PCR with conditions of initial heating at 94 °C for 5 min followed by 10 cycles of 94 °C for 15 s, 55 °C for 30 s, 68 °C for 1 min and 25 cycles of 94 °C for 15 s, 55 °C for 30 s, 68 °C for 1 min⫹ 15 s/cycle. Human ADH RT-PCR primers were ADH1A (cADH1A-F1 and cADH1A-R9), ADH1B (cADH1B-F1 and cADH1B-R9), and ADH1C (cADH1C-F1 and cADH1C-R9) (supplemental table). RT-PCR of mouse endogenous genes was used for internal control. Amplification conditions for mouse
Adh1 were identical to those for human ADHs mentioned
above. RT-PCR primers for internal controls of Adh1 were cAdh1-F2 and cAdh1-R8 (supplemental table).
DNase I Hypersensitive Site Mapping—The experimental procedure was modified from published methods (20, 21). CA transgenic mice at 2 months old were sacrificed, and⬃1 g of adult mouse liver, brain, or stomach was dissected. Genomic DNA isolated from control or DNase I-treated nuclei was exhaustively digested with EcoRV and analyzed by Southern blot analysis. Probes were generated by PCR using primers of CA130136F and CA131010R for detecting HS sites in CNS-C, -D, and -E regions and primers of CA111558F and CA113099R for detecting HS sites in CNS-A and -B regions. (supplemental table).
DNase I Footprinting Assay—Brain and liver nuclear extracts were prepared as described previously (22). Footprinting assay was modified from the method described previously (23). Two probes (CA133499 –133773 and 138372–138609) were gener-ated by PCR using one32P-end-labeled primer and one
unla-beled primer. The primers of CA133499F and CA133773R were used for amplifying the fragment of CA133499 –133773, and CA138372F and CA138609R for the fragment of CA138372– 138609 (supplemental table). After purification by the G-50
mini column (GeneAid), a total of 2⫻ 104cpm probes was
incubated with 2–16g of mouse brain or liver nuclear extracts at room temperature for 20 min, followed by DNase I digestion. After electrophoresis, the gels were dried, and the signals were scanned with the Molecular Dynamics Typhoon 9410 Phos-phorImager (Amersham Biosciences).
Electrophoretic Mobility Shift Assay (EMSA)—DNA probes (CA133499 –133773 and CA138372–138609) were labeled and purified as mentioned for DNase I footprinting. Competitor oligonucleotides were synthesized according to the footprint sequences as follows: 29 bp (CA133549 –133577), 32 bp (CA138479 –138510), and 50 bp (CA138481–138530), and the consensus or mutant oligonucleotides for transcription factor
binding sequences AP1, HNF3, HNF1, HLF, NF-1, NF-1mut,
C/EBP, and C/EBPmut (supplemental table). For analyzing the HNF1-binding sites, six oligonucleotides containing HNF1 wild type and mutant sequences were synthesized, and the sequences are shown in Fig. 6A. The probe was end-labeled with [␥-32P]ATP using T4 nucleotide kinase in one strand and
then annealed with the complementary strand. 32P-Labeled
probes were incubated with 1g of mouse liver nuclear extract with 1⫻ binding buffer (10 mMHepes, pH 7.9, 10% glycerol, 50
at National Taiwan University on March 11, 2009
www.jbc.org
mMKCl, 5 mMMgCl2, 5 mMdithiothreitol, 2g of poly(dI䡠dC),
and 0.1% Nonidet P-40) at room temperature for 20 min. Com-petitor oligonucleotides were incubated with 100-fold molar excess in the reaction mixtures for 10 min before adding the probe. For supershift, 2g of antibodies (Santa Cruz Biotech-nology) were added after the incubation of probe and nuclear extract and stood for 10 min at room temperature. DNA-pro-tein preparations were loaded onto native 5% polyacrylamide gels, and electrophoresis was carried out in 0.25⫻ TBE at 200 V for 2.5 h.
Reporter Gene Assay—Transcription activation of three class I ADH promoters and the SV40 promoter was analyzed using the luciferase reporter gene system. The SV40 promoter of the pGL3 luciferase reporter plasmid (Promega) was removed by BglII and HindIII digestion and exchanged for the ADH1A,
ADH1B, and ADH1C promoters. Upstream sequences of each of the class I ADH genes were generated by PCR. HindIII and BglII compatible ends were created at 5⬘ and 3⬘ for cloning. The primers with restriction sites used in PCR were ADH1A (1A-PR-HindIII and 1A-PF-BglII), ADH1B (1B-(1A-PR-HindIII and 1B-PF-BglII), and ADH1C (1C-PR-HindIII and 1C-PF-BglII) (supplemental table). A 7133-bp DNA fragment (CA132882– 140015) containing CNS-D and CNS-E was isolated from BAC CA with BamHI digestion and inserted upstream of the SV40 or
ADHpromoters. To delete the CNS-E region, DNA sequence
(CA135616 –139331) was removed by PstI and SpeI digestion. To create CNS-D deletion, DNA sequence (CA133496 – 137882) was removed by EcoRI digestion. For the condensed elements of CNS-D and CNS-E, the fragment of CA133499 – 133773 and CA138266 –138699 was amplified by PCR and inserted into the SmaI site of pGL3 vector. To mutate HNF1-binding sites (CA133552–133564), the QuikChange site-di-rected mutagenesis kit (Stratagene) was used. Test plasmids
and internal control cytomegalovirus--galactosidase DNA
plasmid (CMV-Gal) were co-transfected into human hepato-blastoma cell line HepG2 using the calcium phosphate copre-cipitation method (24) or EZfast transfection reagent (Infinigen Biotech), according to the manufacturer’s instructions. Cells were harvested after transient transfection for 48 h to measure transcriptional activity with the luciferase assay system (Applied Biosystems). To normalize transfection efficiency, -galactosidase activity was assayed as described (24).
Generation of BAC CAscarwith HNF1-binding Site Deletion—
Deletion of the HNF1-binding site (CA133538 –133734) in CA was performed using PCR-targeted method (25). A gene replacement cassette containing apramycin-resistant gene and two FRT sites was amplified by PCR. The 59-bp upstream
primer (CA133499-F59 bp) contains 39 bp of 5⬘-flanking
sequence of the HNF1 site (CA133538 –133734) and 20 bp of the P1 sequence of the PIJ773 plasmid. The 58-bp downstream primer (CA133773-R58 bp) includes the 39-bp 3⬘-flanking sequence and 19 bp of the P2 sequence of the plasmid (25) (supplemental table). The PCR product was purified and elec-troporated into Escherichia coli containing the CA BAC clone, and the colonies were selected with apramycin. The cassette containing the apramycin-resistant gene was subsequently excised from the recombinant BAC clone by introducing the FLP recombinase expressing plasmid. Finally, the HNF1 site
deletion and the 81-bp scar (20-bp⫹19-bp priming sequence ⫹ 42-bp FRT) left behind in the recombinant BAC clone, designed as CAscar, were confirmed by Southern blot analysis
and DNA sequencing.
Chromosome Conformation Capture (3C)—The 3C assays were performed according to the method of Vakoc et al. (26). Primers used for different EcoRI fragments are indicated in Fig. 8A (sequences are given in the supplemental table). Brain and liver tissues from the CA78 transgenic mice were homogenized in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, and the cells were isolated by centrifugation at 2000 rpm for 5 min. Approximately 5⫻ 106cells were used for 3C
anal-ysis. Quantitative PCR was performed by using serial dilutions of samples into a linear range of signal for each primers set in individual experiments. DNA templates were adjusted to the same level of self-ligation efficiency for the brain and liver sam-ples. Cross-linking efficiency was determined using the ubiqui-tously expressed mouse Adh5 gene as a reference. Visualization of the experimental results was performed by ethidium bro-mide staining, and the images were quantified using the Image-Quant 5.2 software. To compare signal intensities between dif-ferent primer pairs, control templates were prepared from the BAC clone of CA, which was digested with EcoRI and ligated. The primers of CA133773R and CA137663F were used to con-trol ligation efficiency, and the primers of 393J8 –171440R and 393J8 –168102F were used as control for cross-linking effi-ciency (supplemental table and figure).
RESULTS
To understand how each of the component genes of the human class I ADH cluster is regulated during development, we have taken a comparative genomics approach to identify
non-coding sequences conserved between humans and mice.4 In
human chromosome 4, the class I and class IV ADH genes are aligned with the mouse Adh genes (Adh1 and Adh4) on the corresponding chromosome 3. The order and arrangement for orthologous ADH genes are similar between the two species. Notably, several segments of conserved noncoding sequences are present in the intergenic region between class I ADH and class IV ADH (Fig. 1). These conserved noncoding sequences were named CNS-A through CNS-E, ordering from centro-meric to telocentro-meric direction of human chromosome 4. CNS-A and CNS-B are shared by the BAC clones BI and CA, whereas CNS-C, CNS-D, and CNS-E are present in the clone CA but not BI.
To investigate the molecular mechanism(s) regulating class I
ADH gene expression, we applied transgenics to examine
whether the conserved regulatory elements identified by com-parative genomics analysis could direct class I ADH gene expression in transgenic mice. Two human BAC clones, BI and CA, whose sequences have been completely determined, were linearized and injected into mouse embryos to generate trans-genic lines for expression analysis.
At least two independent transgenic lines were bred for each of the BAC clones to analyze human class I ADH gene expres-sion. Adult mice were sacrificed, and ADH expression from 4J. S. Su and S. F. Tsai, manuscript in preparation.
at National Taiwan University on March 11, 2009
www.jbc.org
multiple tissues was analyzed by reverse-transcription PCR (RT-PCR). As shown in Fig. 2A, mice bearing the BI clone, lines
BI35 (copy number ⫽ 10) and BI38 (copy number ⫽ 3),
expressed variable levels of human class I genes (ADH1A, ADH1B, and
ADH1C) in the liver and other tis-sues. By contrast, mice bearing the CA clone, lines CA78 (copy
num-ber ⫽ 10) and CA74 (copy
num-ber⫽ 2), displayed copy
number-dependent expression of ADH1A and ADH1B in the liver and ADH1C in the liver and stomach. ADH1C was also detected in the intestine of the CA78 line (Fig. 2B). Although the mouse has only one class I ADH gene (Adh1), which was expressed in all tissues with relatively low lev-els in brain and spleen, the three human class I ADH genes were spe-cifically expressed in the transgenic liver, stomach, and/or intestine in both CA transgenic lines carrying the upstream sequences of the human class I ADH cluster.
During development, the three human class I ADH genes are also specifically expressed in the fetal liver. We investigated what con-trol elements are required for
ADH1A gene expression in the fetal livers of the BI and CA trans-genic mice. At E16.5 dpc stage, the two BI transgenic lines showed virtually no expression of ADH1A in fetal livers. By contrast, CA78
and CA06 expressed mainly
human ADH1A in the fetal livers (Fig. 2C). RT-PCR using human
fetal liver RNA (18 –24-week
stage, which was equivalent to the stage of E16.5 dpc in the mouse) (27) showed that ADH1A was
pre-dominantly expressed in the
human fetal liver. Thus, we con-cluded that human class I ADH genes were expressed in liver with temporally correct manner in the CA transgenic mice and that enhanced expression of human class I ADH genes in fetal and adult livers requires the upstream sequence (beyond the extent of the BI clone ending at CA130962)
from ⬃48 kb upstream of the
ADH1C gene (CA66423– 82672) (Fig. 1).
To determine how the conserved sequences between ADH1C and ADH7 regulate class I ADH gene expression, we probed the chromatin structure in the CNS segments by DNase I HS site mapping. Nuclei prepared from
FIGURE 1. Comparative analysis of human and mouse ADH genes and genomic coverage of the human
BACs used in this study. The PipMaker program (40) was used to compare the class I and class IV ADH
sequences of human (GenBankTMaccession number AP002027; 182,676 bp) and mouse (GenBankTMaccession
number AC079682; 181,082 bp). Conserved regions are shown in dark gray (highly conserved) or light gray (moderately conserved) in the middle rectangle. Five CNS were shown with the percentage identity plot (pip) and designated CNS-A (CA110797–114170), CNS-B (CA114579 –118379), CNS-C (CA131144 –132782), CNS-D (CA133010 –135543), and CNS-E (CA136015–138640). Human BAC clones BI, CA, and CAscar(CA with a 197-bp
deletion at CA133538 –133734) were used to generate transgenic mice. The genomic position of each BAC and the location of class I ADH (ADH1A, ADH1B, and ADH1C) and class IV ADH (ADH7) in the BACs are indicated by nucleotide number in the CA clone, using AP002027 as a reference. The 5⬘-flanking sequence beyond the CA clone is denoted as⫺ before the number.
FIGURE 2. Expression analysis of human class I ADH genes in BI and CA transgenic mice by RT-PCR. A, mice bearing BI clone sequence, lines 35 and 38, expressed variable levels of human class I genes (ADH1A, ADH1B, and ADH1C) in liver and non-liver tissue. B, lines CA78 and CA74 expressed ADH1A and ADH1B in the liver and ADH1C in the liver and stomach. ADH1C was also detected in the intestine of the CA78 line. Negative controls in A and B showed the PCR results without DNA template. C, at developmental E16.5 dpc stage, the two BI lines showed little expression of ADH1A or ADH1B in fetal livers, whereas lines CA78 and CA06 expressed predomi-nantly human ADH1A in the fetal livers. RNA from human fetal liver at 18 –24 weeks was used as a control.
at National Taiwan University on March 11, 2009
www.jbc.org
the stomach, brain, and liver tissues of the transgenic line CA78 was used to map HS sites in each CNS (A–E). A liver-specific HS site was found at a position near 114 kb (GenBankTM
acces-sion number AP002027) between the CNS-A and CNS-B (des-ignated 4.8kb(A/B)) (Fig. 3A). On the other hand, two HS sites, corresponding to the CNS-D (designated 4.5kb(D)) and CNS-E
FIGURE 3. DNase I hypersensitive sites mapping of conserved noncoding sequences. A, CNS-A and -B 5⬘ upstream of the class I ADH gene cluster. Nuclei from brain and liver of the transgenic line CA78 were treated with increasing concentrations of DNase I. An⬃10-kb parental fragment (CA109326–119376) (arrowhead) was detected using a probe prepared from CA111558 –113099. A liver-specific DNase I hypersensitive site was detected as a 4.8-kb band (A/B) (arrow) between CNS-A and CNS-B. B, CNS-C, -D, and -E 5⬘ upstream of the class I ADH gene cluster. Nuclei from stomach, brain, and liver of the transgenic line CA78 were treated with increasing concentrations of DNase I. An⬃17-kb parental fragment (CA129130–146241) (arrowhead) was detected using a probe prepared from CA130136 –131010. Two liver-specific DNase I hypersensitive sites were detected as bands of 4.5 and 9.5 kb in size (arrows). Asterisks mark the nonspecific bands appearing in all samples.
at National Taiwan University on March 11, 2009
www.jbc.org
(designated 9.5kb(E)) regions, were mapped to position 133.5 and 138.5 kb, respectively (Fig. 3B). We demonstrated that the HS sites were specific to the liver because we did not detect the same sites in the brain or stomach from the CA78 transgenic line.
Because the sequence unique to the CA clone appears to be necessary for developmental stage-specific expression of class I
ADHgenes, and because the CNS-D and CNS-E regions
coin-cided with the HS sites, we further investigated protein-DNA interactions in these regions. A 275-bp D fragment (CA133499 –133773) and a 238-bp E fragment (CA138372– 138609) containing the two HS sites were analyzed by foot-printing assay and EMSA. In the CNS-D region, footprint was detected in nucleotide sequences CA133549 –133577, and we identified consensus binding sequences for HNF1, AP1, and HNF3 (Fig. 4, A and B). A gel shift band was detected and specifically competed by the oligonucleotide sequence of HNF1 but not by HNF3, AP1, C/EBP, or HLF. Furthermore, a super-shift was observed when the HNF1 antibody was included in the reaction (Fig. 4C). In the CNS-E region, NF-1-, CREBP-, and C/EBP-binding sites were identified in the footprint region (Fig. 4, D and E). Gel shift bands were formed with liver nuclear extract, and the bands were competed by the oligonucleotide sequence of NF-1 but not by C/EBP or a 32-bp probe containing the predicted CREBP site (Fig. 4F). Thus, we concluded that the CNS-D and CNS-E regions were bound by HNF1 and NF-1, respectively.
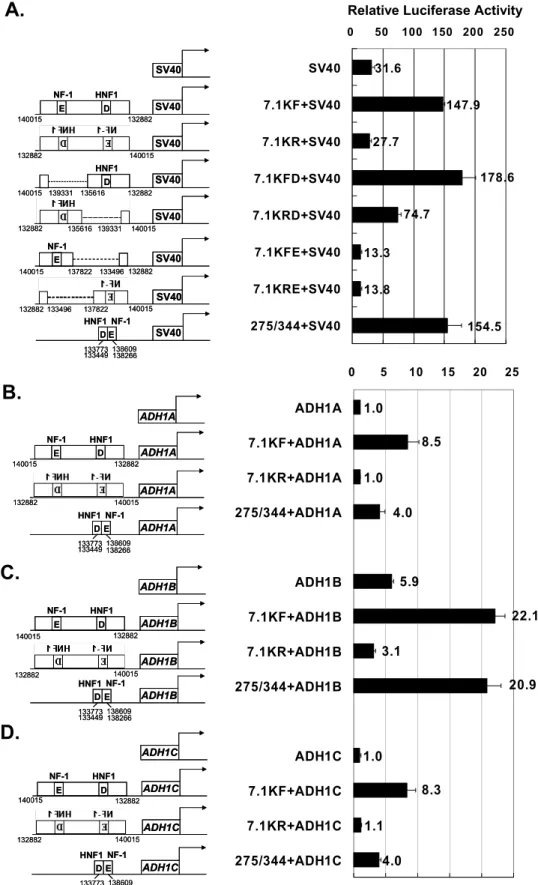
The evolutionarily conserved genomic sequence that dis-played HS and transcription factor binding was examined for the ability to enhance transcription from the ADH1A, ADH1B, and ADH1C promoters. As shown in Fig. 5A, a 7.1-kb fragment with CNS-D/CNS-E sequence (CA132882–140015) could enhance transcriptional activity of the SV40 promoter in the HepG2 hepatoblastoma cell line, in an orientation-dependent manner, because there was little transcriptional activation when the fragment was in the reverse orientation. The CNS-E (NF-1) had little effect on enhancing transcription, when comparing the reporter activities in fragments taining a deletion of CNS-D or CNS-E. Additionally, a con-densed sequence containing HNF1 (CNS-D) and NF-1 (CNS-E) motifs enhanced transcription from the SV40 pro-moter, similar to the effect of the 7.1-kb fragment (Fig. 5A). When ADH promoters of ADH1A (CA21023–21571),
ADH1B(CA51353–51885), and ADH1C (CA82672– 83202) were used, transcriptional activation by the 7.1-kb fragment was 8.5-, 3.7-, and 8.3-fold, respectively (Fig. 5, B–D). Like the SV40 promoter, the activation of the ADH promoters was also dependent on the orientation of the 7.1-kb frag-ment. Finally, the condensed CNS-D/E sequence displayed similar activation effect as that of the 7.1-kb region for the
ADH1B promoter, but much less for the ADH1A and
ADH1Cpromoters (Fig. 5, B–D).
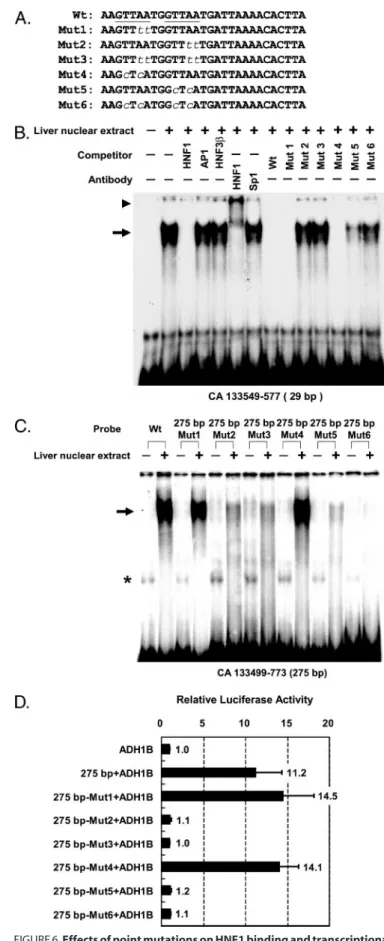
To verify the regulatory function of two predicted HNF1 sites identified from the 29-bp footprint region (CA133549 – 133577), the GTTAA core sequences of the HNF1-binding sites were mutated at one or both sites. We first used the 29-bp double-stranded synthetic oligonucleotides as probes for EMSA to examine DNA binding ability of the wild type and
mutant sequences (Fig. 6, A and B). Additionally, mutants in the putative HNF1 sites were generated by oligonucleotide-di-rected mutagenesis in the 275-bp probe (CA133499 –133773). We found that the mutations at the 3⬘ HNF1 core abolished the DNA complex formed by the nuclear protein with the 29-bp oligonucleotide probe (Fig. 6B) or the 275-bp probe (Fig. 6C). Taken together, these results indicated that the 3⬘ HNF1 con-sensus sequence in the footprint region could bind HNF1.
Consistent with the EMSA results, we found that the mutants of the 5⬘ putative HNF1 site (Mut1 and Mut4) main-tained transcriptional activation function at about 14.5- and 14.1-fold, respectively, using the ADH1B promoter as a refer-ence. The transcription enhancing activity was comparable with that of the wild type sequence (11.2-fold). On the other hand, single mutation at the 3⬘ putative HNF1 site or double mutations at both sites significantly reduced the enhancing activity (Fig. 6D). These data indicate that the 3⬘ HNF1 site is essential for transcriptional activation.
To demonstrate in vivo the role of HNF1 in controlling class I ADH gene expression, we conducted transgenic mouse exper-iments with a mutated version of the CA BAC clone that was devoid of the HNF1-binding sequence. The HNF1 site at position CA133558 of BAC CA was deleted by the method of PCR-targeted gene replacement (25). Southern analysis and DNA sequencing confirmed that the deletion size was 197 bp (CA133538 –133734) and replaced with an 81-bp sequence
(Scar) from the 42-bp FRT and 20-bp ⫹ 19-bp priming
sequence (Fig. 7A and data not shown). The disrupted BAC, designated CAscar, was used to generate transgenic mice. We
first examined whether the HS sites were affected by the HNF1 site alteration. Most interestingly, beside the HS site in CNS-D, the HS site in CNS-A/B was also abolished in the adult liver (Fig. 7B). To determine transgene expression, three offspring from the CAscartransgenic line (copy number⫽ 1) were
ana-lyzed for human class I ADH gene expression in fetal brain and liver at E16.5 dpc stage and also in tissues collected from 2-month-old adult mice. We found that all three human class I
ADHs were undetectable by RT-PCR in either the adult liver or the fetal liver. By contrast, ADH1C expression in the stomach was not affected (Fig. 7C). Furthermore, there was no signifi-cant difference in ADH7 expression in the stomach between CAscarand CA transgenic mice (data not shown). Thus, data
from the mutant BAC construct support that the upstream conserved sequence with the HNF1-binding site is essential for regulating and enhancing class I gene expression in the liver.
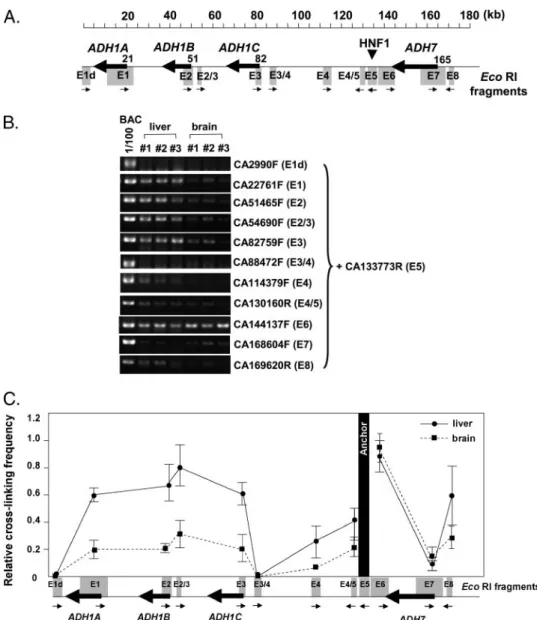
Finally, we used chromosome conformation capture (3C) to investigate the mechanism through which the distant HNF1-binding site activated class I ADH genes. We speculated that the upstream HNF1 site might act upon the ADH promoters through DNA looping. To test this possibility, we used cells from adult liver and brain of CA78 transgenic mice for the 3C analysis. To reliably detect the chromosome conformation, adequate controls were included in the experimental proce-dures, including those for the efficiencies of digestion, ligation, and cross-linking (supplemental figure). As shown in Fig. 8A, the primer in fragment E5 was used as an anchor to perform PCRs with test primers in other fragments, including promoter regions of three class I ADH genes. Consistent with previous
at National Taiwan University on March 11, 2009
www.jbc.org
FIGURE 4. DNase I footprinting and EMSA analysis in HS sites of the CNS-D and CNS-E regions. A, footprint analysis with a probe generated from CA133499 –133773. The protected regions were mapped to the sequence of CA133549 –133577. NE, nuclear extract. B, alignment of human (CA133489 – 133633) and mouse (73455–73600) DNA sequences. The nucleotide positions were based on GenBankTMaccession number AP002027 for human and
AC079682 for mouse. Transcription factor-binding sites in DNase I-protected sequences (shaded) were predicted by MATCH in the public domain and marked above the sequences. C, bands of DNA-protein complexes and supershift detected by EMSA analysis were indicated by arrow and arrowhead, respectively. D, footprint analysis with a probe generated from CA138372–138609. The protected regions were mapped to the sequence of CA138481–138530. Dotted line and solid line indicate the 50-bp (CA138481–138530) and 32-bp (CA138479 –138510) sequence, respectively, corresponding to the footprints generated by CREBP, C/EBP, and NF-1 binding and by CREBP binding alone. E, alignment of human (CA138426 –138569) and mouse (79599 –79735) DNA sequences. F, bands of DNA protein complexes were detected by EMSA and indicated by arrows.
at National Taiwan University on March 11, 2009
www.jbc.org
FIGURE 5. Transcriptional activation function of CNS-D- and CNS-E-containing sequences. Fragment of CA132882–140015 was examined for enhancing transcription from the SV40 promoter and three class I ADH promoters. NF-1- and HNF1-binding sites are marked above the E (CNS-E) and D (CNS-D) boxes. Luciferase reporter activity was calibrated with co-transfected-galactosidase for transfection efficiency and normalized with that of the ADH1A promoter. Average and S.D. from three experiments are shown to the right of each construct. A, the 7.1-kb BamHI fragment (CA132882–140015), bearing CNS-D/CNS-E, enhanced the SV40 promoter activity in the forward direction (7.1KF⫹SV40) but much less in the reverse direction (7.1KR⫹SV40). Derivatives without CNS-E (del CA135616 –139331) resulted in a similar effect as the full-length 7.1-kb fragment in the forward direction (7.1KFD⫹SV40), but the enhancing effect reduced significantly in the reverse direction (7.1KRD⫹SV40). Derivatives without CNS-D (del CA133496–137822) lost enhancing effect in both directions (7.1KFE⫹SV40 and 7.1KRE⫹SV40). A fused sequence of fragments D and E containing the HNF1 and NF-1 sites gave comparable level of transcription activation as the full-length 7.1-kb fragment. B–D, the SV40 promoter was changed to class I ADH promoters.
at National Taiwan University on March 11, 2009
www.jbc.org
studies (28 –30), the cross-linking efficiency in boundaries of the anchor fragment was higher than that of fragments far away from the anchor. We found that the relative cross-linking effi-ciency of fragments E6 was higher than that for E7, and E4/5 was higher than E4 (Fig. 8, B and C). Notably, the promoter regions of class I ADHs (fragments E1, E2, E2/3, and E3) had consistently higher cross-linking efficiencies in liver than in brain. From the data we concluded that there was closer prox-imity between promoter and HNF1 site in the liver than in the brain. Hence, looping interaction between HNF1 site and class I ADH promoters may have contributed to the long range acti-vation of class I ADH gene in the liver.
DISCUSSION
Tandem gene duplication underlines the complexity and diver-sity of the ADH gene families in the vertebrates. Although class I ADH enzymes are highly homologous to each other in their amino acid sequences, the expression pattern and physiological function of the enzymes appear to be distinct. It is apparent that the dupli-cation events generating the three class I ADH genes also involved their proximal promoters. It is fascinating to learn how each of them is regulated and how liver ADH activities are adjusted under physiological conditions. Thus, the ADH gene family provides not only a good model for studying the evolution of gene function but also a fine target to investigate mechanism(s) of differential gene regulation.
Although control elements governing ADH gene expression have been studied in the past, most analysis was done at the sequence level or by transfection experiments in cultured cells (1). Moreover, long distance regulation mechanism has not been addressed for the entire human class I ADH gene cluster in its genomic or evolutionary context. We have taken a different approach, combining genomic sequencing, evolutionary analysis, mouse genetics, and biochemistry, to characterize the human
ADHgene complex. In this study, we focus on class I ADH genes that expressed primarily in the liver at both fetal and adult stages. We discovered that the HNF1 site in the 51-kb upstream region of class I ADH is required for proper gene expression.
Data from independent transgenic mouse studies support our contention that the conserved mechanism acting through the upstream element is crucial for regulating class I ADH gene expression (31, 32). Only the transgenic mice with a BAC containing 110 kb of 5⬘- and 104 kb of 3⬘-flanking sequences of Adh1 in Adh1 knock-out mice showed expres-sion similar to that from endogenous Adh1 of the wild type mice (32). From their findings, we infer that a similar long range regulatory mechanism exists in the mouse, and that human and mouse class I ADH expression requires the distal
cis-linked sequence for proper tissue-specific expression, analogous to the locus control region located upstream of the-globin gene cluster.
FIGURE 6. Effects of point mutations on HNF1 binding and transcriptional
activation. A, alignment of the 29-bp (CA133549 –133577) wild type (Wt) and
six mutant oligonucleotide sequences (Mut1 to Mut6). The core sequence of two predicted HNF1 sites are underlined, and nucleotide substitutions are indicated by lowercase italic. B, 29-bp (CA133549 –133577) probe detected band shifts as indicated by an arrow. The oligonucleotides of wild type, Mut1, and Mut4 competed for the DNA-protein complex, but Mut2, Mut3, Mut5,
and Mut6 did not. HNF1 polyclonal antibodies generated supershift (arrow-head), but the control Sp1 polyclonal antibodies did not. C, PCR probes of CA133499 –133773 wild type sequence and six mutants, designated 275 bp-Mut1 to 275 bp-Mut6, were tested for gel shift (indicated by an arrow) with mouse liver nuclear extract. An asterisk marked the nonspecific bands, which appeared even without nuclear extract. D, wild type and six mutants (Mut1– Mut6) were tested for transcriptional activation of the ADH1B promoter. Lucif-erase reporter activity was adjusted to the construct carrying only the ADH1B promoter. Average and S.D. from three independent experiments are shown.
at National Taiwan University on March 11, 2009
www.jbc.org
at National Taiwan University on March 11, 2009
www.jbc.org
By using DNase I hypersensitive site mapping, DNA binding assays, and transient transfection, we have identified, from within the conserved sequences in the intergenic region between the class I and class IV ADH genes, transcription fac-tor-binding sites that function in liver cell lines. Although the gene numbers for each class of ADH in primates and rodents are different, the liver-enriched transcription factor, HNF1, mediates regulation of human class I gene expression in the
mouse livers with tissue-specific and temporal patterns similar to those observed in humans. We conclude that the transcriptional regulatory mechanism is conserved between the two mammalian species, although the structure of class I ADH gene locus has diverged significantly because
human and mouse separated ⬃92
million years ago.
HNF1 is not only a liver-enriched transcription factor but is also expressed in other endoderm-de-rived tissues (33). It is notable that HNF1 could activate phenylalanine hydroxylase gene expression in the liver by remodeling the chromatin structure (34). In our study, the
HNF1 site in the regulatory
sequence for class I ADH genes was identified through DNase I HS sites in the chromatin of transgenic mouse livers (Fig. 3) or human HepG2 hepatoblastoma cell line and HuH-7 hepatoma cell line (data not shown). We speculate that the HNF1-binding site could regulate
class I ADH gene expression
through two mechanisms. The first mechanism is the selective interac-tion of the distant HNF1 site with the three class I ADH promoters. It is possible that the HNF1 site and other regulatory elements in the far upstream region could serve as the locus control region for the class I
ADHgene cluster, and the proximity between the HNF1 site and promot-ers of active class I ADH could be dynamically adjusted at different developmental stages. We have dem-onstrated by 3C experiments that the upstream region with a HNF1 site could preferentially interact with the class I ADH gene promoters (Fig. 8C), and we propose that the long range interaction involves a DNA looping mechanism and allows close proximity of the upstream elements to the ADH promoters. The second mechanism is that, besides the looping interac-tion, the HNF1 binding may contribute to the opening of local chromatin and could modulate other ADH gene regulatory sequences. For example, another HS site mapped near the CNS-A and CNS-B regions was diminished when the upstream
FIGURE 7. Class I ADH expression in the CAscartransgenic mice. A, BAC DNA for CA, CA773 (with PIJ 773 cassette integration), and CAscar(with residual PIJ 773
sequence but lacking HNF1-binding site) was analyzed by electrophoreses after EcoRI and SwaI double digestion and stained with ethidium bromide (left). Southern blot analysis (right) with a probe of CA133177–773, shown as a bar below the EcoRI restriction site. Two fragments were detected by the probe as follows: a common band of CA129739 –133495 and the other with variable sizes between CA133495 and 134088. B, loss of DNase I hypersensitive sites in the CNS-D (right) and CNS-A/B regions (left) in the CAscartransgenic mice. Liver nuclei from CA78 transgenic mice were treated with DNase I as controls to reveal the
HS sites in the corresponding A/B, D, and E regions. C, class I ADH expression was analyzed by RT-PCR using RNA from fetal (E16.5 dpc) and adult (2 months old) tissues of the CAscartransgenic mice. Class I ADH gene expression was absent in fetal and adult livers, but ADH1A and ADH1C were expressed in the stomach.
Endogenous mouse Adh1 expression was detected in all tissues.
FIGURE 8. Chromosome conformation capture (3C). A, EcoRI-restricted fragments examined in the 3C anal-ysis are shown as boxes. Arrows under the EcoRI fragments represent primers for the PCR. A primer in E5 fragment, which contained HNF1-binding sites, was used as an anchor. B, nuclear samples prepared from adult liver and brain of CA78 transgenic mice were used for 3C analysis. Cross-linking efficiency between E5 and test fragment was detected by PCR using anchor primer of CA133773R in the presence of another test primer located on various EcoRI fragments. One hundred-fold diluted template of BAC CA, which was digested with EcoRI and ligated, was used for the PCR control for each amplicon. The results from three experiments (#1, #2, and #3) are shown. C, relative cross-linking efficiency, normalized by PCR control, was calculated for the liver and brain samples. Mean and S.E. of relative efficiency were from three experiments.
at National Taiwan University on March 11, 2009
www.jbc.org
HNF1 site was altered (Fig. 7B). It is of interest to compare the
ADHgene expression patterns of the BI lines with that of the CAscarline. In the BI lines, nonspecific but appreciable levels of ADHgene expression can be detected in the liver and other tissues. By contrast, ADH gene expression is not observed in the CAscarline. Thus, the regulatory sequence shared between the
BI and CA constructs (for example CNS-A and CNS-B) might also contain cis-elements that could affect the class I ADH pro-moter activity, but the effect depends on an intact HNF1 site. Therefore, the upstream HNF-1 site could modulate class I
ADH activities through direct and indirect mechanisms. In
extrahepatic tissues, the general chromosome conformation of the class I ADH gene complex might be condensed. HNF1 bind-ing to the upstream regulatory region in liver favors the release of the heterochromatin structure and permits regulatory ele-ments to activate the downstream genes.
Although HNF1 can enhance gene expression in cultured cells, the activation level depends on the orientation of the HNF1-containing fragment, relative to the downstream pro-moter (Fig. 5). One interpretation of the data is that the sequence between the D fragment and the E fragment might include a “boundary element” that functions to separate the class I ADH-expressing domain from the class IV ADH-ex-pressing domain. In the region of CA136371–1363400, we pre-dicted from the sequence CTCF and Ikaros consensus binding sites. We confirmed by EMSA with specific competitors that the 30-bp sequence could be bound by CTCF and Ikaros teins in the HepG2 nuclear extract (data not shown). The pro-tein CTCF has been reported to have enhancer blocking activity in insulators (35). Moreover, insulators can serve as barriers to protect a gene against the encroachment of adjacent inactive condensed chromatin. On the other hand, Ikaros DNA-binding protein has been shown to be essential for lymphocyte develop-ment (36), and it could be co-localized to centromeric foci with inactivated genes and thus have contributed to selective gene silencing (37). It is possible that an insulator can prevent HNF1 from activating on the neighboring class IV ADH gene. Further dissection of the regulatory elements in the CNS-D and CNS-E regions is needed to demonstrate the possible effects of CTCF and Ikaros on HNF1 activation of class I ADH gene expression. The evolution of the class I ADH gene family in humans provides additional mechanism for regulating alcohol metabo-lism. Variant sequences in the human class I and class IV genes have been found to be associated with the risk for alcoholism (14, 15, 38, 39). Enzyme isoforms differing in Vmaxvalues have been found for ADH1B, and the ADH1B R47H variant was shown to have a protective effect against alcoholism in an Asian population (14, 15). It is intriguing to learn that an SNP mapped within the human ADH7 intronic region is epistatic to the class I gene haplotype for alcoholism risk, although it was not clear how the SNP could be associated with the risk for alcoholism (39). One possibility is that the SNP is part of a haplotype block, which might be functionally related to alcohol metabolism. In this regard, we envision that the conserved regulatory sequence identified from our study, which is 15.7 kb apart from the ADH7 intronic SNP (39), might be a region to look for variants that differ in their capacity of enhancing class I ADH gene expres-sion. Identification of regulatory SNPs in the class I to class IV
ADHintergenic region might be instrumental for studying the molecular basis of individual difference in alcohol metabolism and the risk for alcoholism.
Acknowledgments—We thank Drs. Y. Henry Sun and Shih-Jiun Yin for critical reading of the manuscript and the DNA Sequencing Core Facility at the National Yang Ming University Genome Research Cen-ter for sequencing the human and mouse ADH gene clusCen-ters. The Sequencing Core Facility is supported by National Research Program for Genomic Medicine, National Science Council (Taipei, Taiwan).
REFERENCES
1. Edenberg, H. J. (2000) Prog. Nucleic Acids Res. Mol. Biol. 64, 295–341 2. Duester, G., Farres, J., Felder, M. R., Holmes, R. S., Hoog, J. O., Pares, X.,
Plapp, B. V., Yin, S. J., and Jornvall, H. (1999) Biochem. Pharmacol. 58, 389 –395
3. Szalai, G., Duester, G., Friedman, R., Jia, H., Lin, S., Roe, B. A., and Felder, M. R. (2002) Eur. J. Biochem. 269, 224 –232
4. Yasunami, M., Kikuchi, I., Sarapata, D., and Yoshida, A. (1990) Genomics
7,152–158
5. Smith, M., Hopkinson, D. A., and Harris, H. (1971) Ann. Hum. Genet. 34, 251–271
6. Smith, M., Hopkinson, D. A., and Harris, H. (1972) Ann. Hum. Genet. 35, 243–253
7. Smith, M., Hopkinson, D. A., and Harris, H. (1973) Ann. Hum. Genet. 37, 49 – 67
8. Edenberg, H. J., and Brown, C. J. (1992) Pharmacogenetics 2, 185–196 9. Yang, Z. N., Davis, G. J., Hurley, T. D., Stone, C. L., Li, T. K., and Bosron,
W. F. (1994) Alcohol. Clin. Exp. Res. 18, 587–591
10. Yin, S. J., Chou, C. F., Lai, C. L., Lee, S. L., and Han, C. L. (2003) Chem. Biol. Interact. 143–144, 219 –227
11. Yin, S. J., Wang, M. F., Liao, C. S., Chen, C. M., and Wu, C. W. (1990) Biochem. Int. 22,829 – 835
12. Moreno, A., and Pares, X. (1991) J. Biol. Chem. 266, 1128 –1133 13. Duester, G. (1996) Biochemistry 35, 12221–12227
14. Osier, M., Pakstis, A. J., Kidd, J. R., Lee, J. F., Yin, S. J., Ko, H. C., Edenberg, H. J., Lu, R. B., and Kidd, K. K. (1999) Am. J. Hum. Genet. 64, 1147–1157 15. Chen, C. C., Lu, R. B., Chen, Y. C., Wang, M. F., Chang, Y. C., Li, T. K., and
Yin, S. J. (1999) Am. J. Hum. Genet. 65, 795– 807
16. Brown, C. J., Zhang, L., and Edenberg, H. J. (1996) DNA Cell Biol. 15, 187–196
17. Hardison, R. C. (2000) Trends Genet. 16, 369 –372
18. Hogan, B. L., Costantini, F., and Lacy, E. (1986) Manipulating the Mouse Embryo: A Laboratory Manual,Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
19. Shen, H. H., Huang, A. M., Hoheisel, J., and Tsai, S. F. (2001) Genomics 71, 21–33
20. Becker, P., Renkawitz, R., and Schutz, G. (1984) EMBO J. 3, 2015–2020 21. Holloway, M. P., and La Gamma, E. F. (1992) J. Biol. Chem. 267,
19819 –19823
22. Shapiro, D. J., Sharp, P. A., Wahli, W. W., and Keller, M. J. (1988) DNA (N. Y.) 7, 47–55
23. Mai, B., Miles, S., and Breeden, L. L. (2002) Mol. Cell. Biol. 22, 430 – 441 24. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular Cloning: A
Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
25. Gust, B., Challis, G. L., Fowler, K., Kieser, T., and Chater, K. F. (2003) Proc. Natl. Acad. Sci. U. S. A. 100,1541–1546
26. Vakoc, C. R., Letting, D. L., Gheldof, N., Sawado, T., Bender, M. A., Grou-dine, M., Weiss, M. J., Dekker, J., and Blobel, G. A. (2005) Mol. Cell 17, 453– 462
27. Ko, M. S. H. (2001) Trends Biotechnol. 19, 511–518
28. Dekker, J., Rippe, K., Dekker, M., and Kleckner, N. (2002) Science 295, 1306 –1311
29. Tolhuis, B., Palstra, R. J., Splinter, E., Grosveld, F., and de Laat, W. (2002)
at National Taiwan University on March 11, 2009
www.jbc.org
Mol. Cell 10,1453–1456
30. Dekker, J. (2006) Nat. Methods 3, 17–21
31. Xie, D., Narasimhan, P., Zheng, Y. W., Dewey, M. J., and Felder, M. R. (1996) Gene (Amst.) 181, 173–178
32. Szalai, G., Xie, D., Wassenich, M., Veres, M., Ceci, J. D., Dewey, M. J., Molotkov, A., Duester, G., and Felder, M. R. (2002) Gene (Amst.) 291, 259 –270
33. Cereghini, S. (1996) FASEB J. 10, 267–282
34. Pontoglio, M., Faust, D. M., Doyen, A., Yaniv, M., and Weiss, M. C. (1997) Mol. Cell. Biol. 17,4948 – 4956
35. Bell, A. C., West, A. G., and Felsenfeld, G. (1999) Cell 98, 387–396
36. Georgopoulos, K., Moore, D. D., and Derfler, B. (1992) Science 258, 808 – 812
37. Brown, K. E., Guest, S. S., Smale, S. T., Hahm, K., Merkenschlager, M., and Fisher, A. G. (1997) Cell 91, 845– 854
38. Osier, M. V., Pakstis, A. J., Soodyall, H., Comas, D., Goldman, D., Odunsi, A., Okonofua, F., Parnas, J., Schulz, L. O., Bertranpetit, J., Bonne-Tamir, B., Lu, R. B., Kidd, J. R., and Kidd, K. K. (2002) Am. J. Hum. Genet. 71, 84 –99 39. Osier, M. V., Lu, R. B., Pakstis, A. J., Kidd, J. R., Huang, S. Y., and Kidd, K. K.
(2004) Am. J. Med. Genet. B. Neuropsychiatr. Genet. 126, 19 –22 40. Schwartz, S., Zhang, Z., Frazer, K. A., Smit, A., Riemer, C., Bouck, J., Gibbs,
R., Hardison, R., and Miller, W. (2000) Genome Res. 10, 577–586
at National Taiwan University on March 11, 2009
www.jbc.org