人類乙醯膽鹼酯脢 (huAChE) 和Arthrobacter globiformis 組織胺氧化酵素 (AGHO) 之 QSAR 模型研究
70
0
0
全文
(2) 人類乙醯膽鹼酯脢 (huAChE) 和 Arthrobacter globiformis 組織胺氧化酵素 (AGHO) 之 QSAR 模型研究 Integrating GEMDOCK with GEMPLS and GEMkNN for QSAR model of huAChE and AGHO. 研 究 生:張立人. Student:Li-Jen Chang. 指導教授:楊進木. Advisor:Jinn-Moon Yang. 國 立 交 通 大 學 生 物 資 訊 所 碩 士 論 文. A Thesis Submitted to Institute of Bioinformatics National Chiao Tung University in partial Fulfillment of the Requirements for the Degree of Master in Bioinformatics. July 2005 Hsinchu, Taiwan, Republic of China. 中華民國九十四年八月.
(3) 人類乙醯膽鹼酯脢 (huAChE) 和 Arthrobacter globiformis 組織胺氧化酵素 (AGHO) 之 QSAR 模型研究 學生:張立人. 指導教授:楊進木 博士 國立交通大學生物資訊學系﹙研究所﹚碩士班 摘. 要. 在新藥開發的過程中,電腦輔助藥物設計技術的應用,可以大幅度減少新藥開發過程 中所耗費的時間和與金錢,而分子嵌合(Molecular docking)和 QSAR 模型是電腦輔助藥物 設計的關鍵技術。在本研究中我們利用分子嵌合工具— GEMDOCK 產生蛋白質-配體原子 交 互 作 用 描 述 表 (profiles) , 並 將 交 互 作 用 描 述 表 作 為 建 立 QSAR 模 型 的 敘 述 子 (descriptors) ,輔以演化式方法為基之 QSAR 建模工具— GEMPLS 與 GEMkNN 篩選並建 立人類乙醯膽鹼酯脢 (huAChE) 和 Arthrobacter globiformis 組織胺氧化酵素 (AGHO)之 QSAR 模型。我們的 QSAR 建模方法主要是以蛋白質-配體的原子交互作用情況來描述蛋 白質活性區域與分子間的作用特徵。而後我們將會藉由 GEMPLS 與 GEMkNN 建立的初步 模型中,篩選一致性的敘述子群(consensus feature set)以及化合物結構差異部分(specific skeleton)產生敘述子群,建立並增進最終的 QSAR 模型預測能力。目前我們的方法已驗證 於 huAChE 之 QASR 模型建立,其 leave-one-out 交互驗證之 q2 達到 0.818、實驗值與預測 值之相關係數 r2 亦達 0.781。除此之外,本方法也實際應用在 AGHO 之 QSAR 模型建立。 此為目前首次應用在 AGHO 之 QSAR 模型,其實驗值與預測值之相關係數 r2 高達 0.983。 藉由本研究發展的 AGHO 之 QSAR 模型,我們探討了 AGHO 與一系列的受質及其衍生物 之結合親合力和疏水特性的關係,其中包括取代基的長度和環的大小對其催化能力之影 響。此外我們由 AGHO 之 QSAR 模型預測新的受質結構— benzylamine,並藉酵素催化實 驗證實預測結果之正確性。藉由成功的發展具有高度預測準度的 QSAR 模型與新受質發 現,說明了我們的 QSAR 模型之應用性,並證明本研究發展之 QSAR 建模方法是有用且 有效的工具。. i.
(4) Integrating GEMDOCK with GEMPLS and GEMkNN for QSAR model of huAChE and AGHO Student: Li-Jen Chang. Advisor: Jinn-Moon Yang Institute of Bioinformatics National Chiao Tung University ABSTRACT. Molecular docking and quantitative structure activity relationships (QSAR) are the core technologies in computer-aided drug design. These technologies would help to save much time and cost to find out potential leads for the target protein in drug discovery. In this study, we introduced molecular docking tool, GEMDOCK to generate the atom-based protein-ligand interaction profile. We utilized the interaction profile to be descriptor and integrate with GEMPLS and GEMkNN for QSAR model of human acetylcholinesterase (huAChE) and Arthrobacter globiformis histamine oxidase (AGHO). Our method has adopted the atom-based interaction profile of protein-ligand complex to represent the molecular descriptor. The atom-based interaction profile would be used in GEMPLS and GEMkNN to construct the preliminary QSAR models. By collecting the selected feature of preliminary models, we generated the consensus feature set. Finally, the consensus feature set and ligand specific skeleton set were used to generate the final QSAR model and improve the prediction accuracy of model. We have verified our method for QSAR model of human acetylcholinesterase (huAChE). The model shows the leave-one-out cross validation of q2 is 0.818 and the correlation of r2 is 0.781 between the predicted and experimental values. After verifying the utility of our method on huAChE, we applied it to develop a novel QSAR model for Arthrobacter globiformis histamine oxidase (AGHO). This model is the first QSAR model for AGHO, and it shows a correlation of r2 is 0.983 between the predicted values and experimental values. This model has also been employed to a series of substrates and derivatives to probe the relationship between affinities of AGHO and hydrophobicities of ligands (including the length of substitution group and ring size). From QSAR models of AGHO, we discovered a novel substrate, which was called benzylamine and was evaluated by experiments. Experiments show that our QSAR model was capable of predicting with reasonable accuracy even that the activity of novel compounds not included in the original dataset. The successful development of highly predictive QSAR models implies that our method is a robust and useful tool for QSAR models.. ii.
(5) 誌謝 一轉眼,兩年的碩士求學生活即將進入倒數,在寫誌謝的此刻,腦海中浮 起兩年來的點點滴滴,心中百感交集。一路走來,因為有大家的鼓勵、支持, 才促成今日我能夠順利取得學位。 這篇論文得以順利完成,最要感謝的是我的指導教授. 楊進木博士,謝謝. 老師在學業上的敦敦教誨,不厭其煩的賜導,每每當我在研究上偏離軌道時, 適時將我拉回正軌,在遇到瓶頸挫折時,給我鼓勵和關心。亦感謝口試委員中 央研究院生醫所 黃明經老師、台灣大學藥學系 陳基旺老師的不吝指教,並給 予寶貴的意見。此外也感謝所有曾在研究上曾給予協助的夥伴們,俊辰、阿甫 (彥甫)、沈老(再威)、PIKI(凱程)、台大蔡其杭學長以及實驗室所有同學們,謝 謝你們的幫忙和支持。子明、偉晉、岳賢、文鴻謝謝你們的精神鼓勵。 最後,僅以此論文獻給我最親愛的老爸、老媽、老哥立群、大嫂佳慧,謝 謝你們總在我的背後給予最大的支持,給予無限的關懷和包容,一路陪著我渡 過所有低潮與挫折,謝謝你們。. iii.
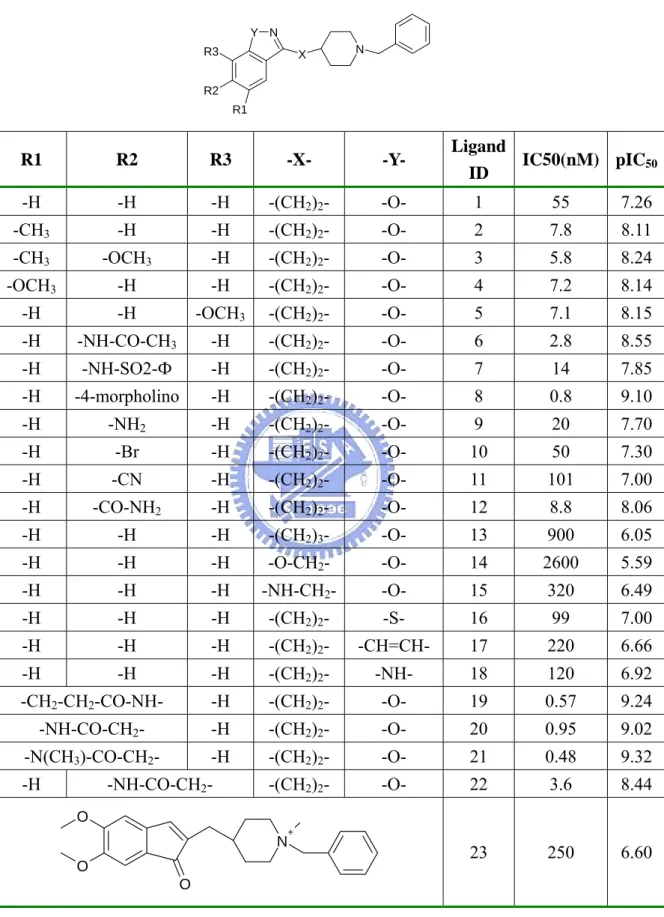
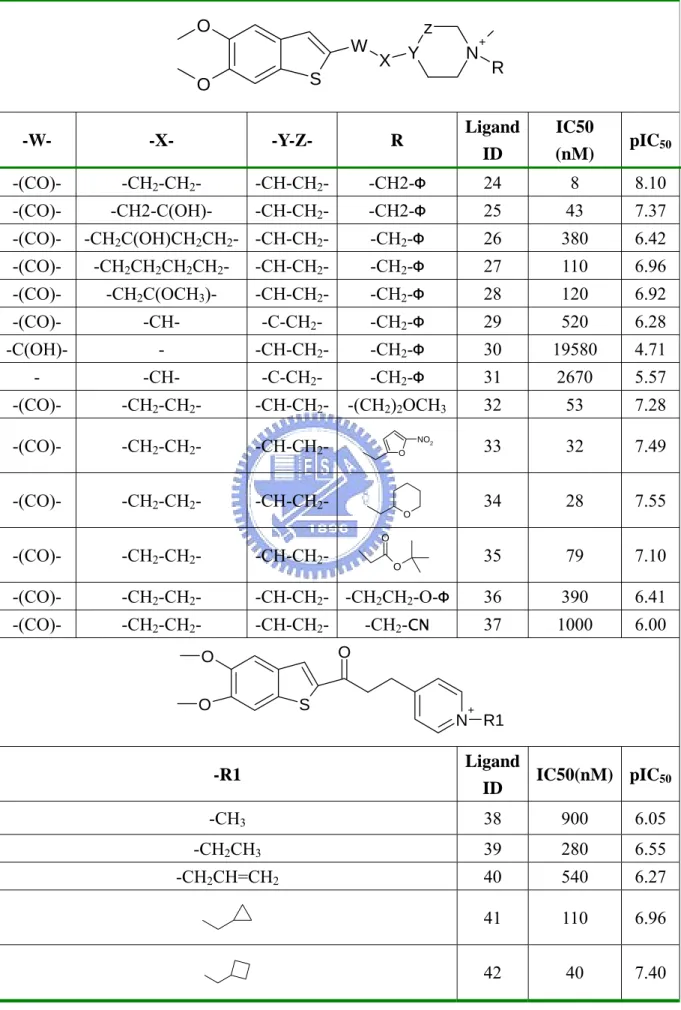
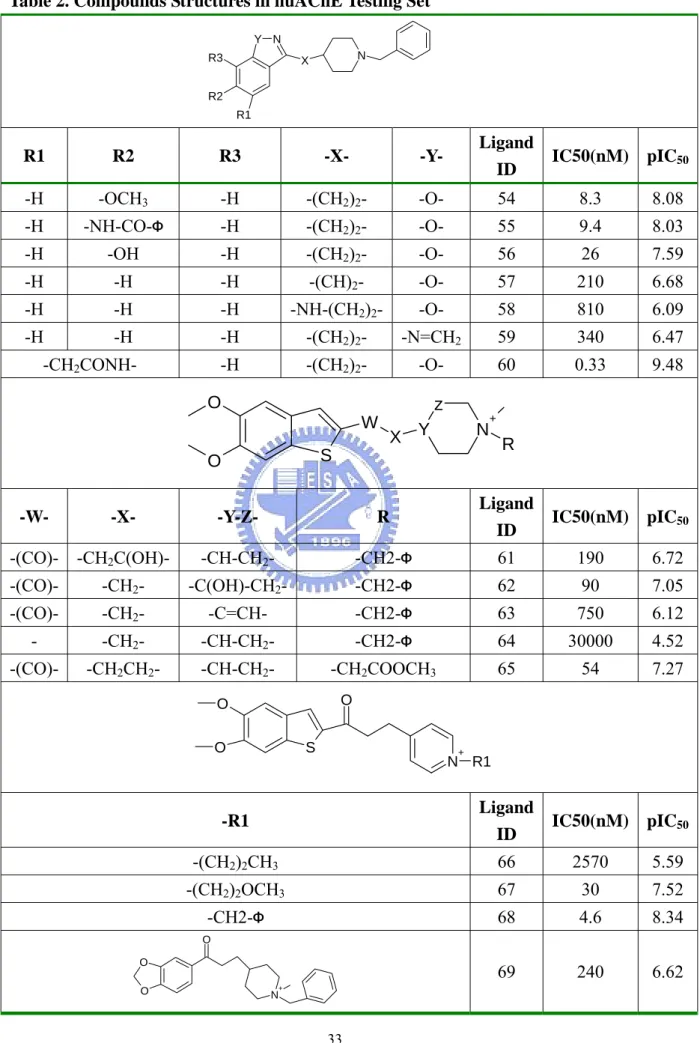
(6) List of Tables Table 1.. Compounds Structures in huAChE Training Set…………………………………30. Table 2.. Compounds Structures in huAChE Testing Set……………………………....…..33. Table 3.. Compound Structures in the AGHO Training Set………………...………....…...34. Table 4.. Structures of AGHO Substrates Derived with Different Lengths of Side Chains..35. Table 5.. Structures of AGHO Substrates Derived with Different Ring Sizes……………..36. Table 6.. Performance of GEMPLS and GEMkNN Relative to Different Interaction Profiles on huAChE Set………………………………...……………………………........37. Table 7.. Performance of GEMPLS with Different Descriptions (features) on huAChE Set………………………………………………………………………………...38. Table 8.. Comparison our Method with the Method by Jianxin et. al………………..…… 39. Table 9.. Selected Residues in the huAChE QSAR Model………………………………...40. Table 10.. Performance of GEMPLS and GEMkNN Relative to Different Protein-Ligand Interactions Profiles on AGHO Set…………………………………………...….41. Table 11.. Performance of GEMPLS with Different Descriptions (features) on AGHO Set……...................................................................................................................42. Table 12.. Selected Residues of the AGHO QSAR Model………………………...………...43. iv.
(7) Table of Figures Figure 1.. The main steps of QSAR model building………………………………...……...44. Figure 2.. The step of compound structures superimposition……………………………….45. Figure 3.. The protein-ligand interaction profile………………..…………………………..46. Figure 4.. The definition of ligand common skeleton………………………………………47. Figure 5.. Proposed mechanism of turnover for amine oxidases…………...……………….48. Figure 6.. The docked pose of phenylethylamine (known substrate) in the active site of AGAO (PDB entry 1IU7)…………………………………………...…………...49. Figure 7.. The structural alignment of 1B41 (yellow), 1EVE (pink) and modeled structure (green)……………………………………………………………………………50. Figure 8.. The docked poses of the inhibitor, E2020, at the active site of the modeled structure…………………………………………………………………………..51. Figure 9.. The structural alignment of homology model (green) and the template, AGAO (1IU7) (yellow)…………………………………………………………………..52. Figure 10.. The docked poses of twelve known substrates at the active site of the modeled AGHO structure………………………………………………………………….53. Figure 11.. (A) The experimental verified inhibitor, spermine………………………………54 (B) The predicted pose of spermine at the active site of AGHO………………...54. Figure 12.. The correlation between the experimental values and predicted value of huAChE QSAR model……………………………………………………………………..55. Figure 13.. The correlation between the experimental values and the predicted values of the AGHO QSAR model……………………………………………………….…….56. v.
(8) Figure 14.. The multiple sequence alignments of CuAOs on the spots of the selected residues in AGHO QSAR model…………………………………….…………………….57. Figure 15.. The relationships between the length of substitution group, -CH2- and the predicted affinity of AGHO QSAR model………………………………….…....58. Figure 16.. The relationship between the ring size and the predicted affinity of AGHO QSAR model……………………………………….…………………………………….59. Figure 17.. The relationship between experimental values and predicted affinity of AGHO QSAR model…………………………….……………………………………….60. vi.
(9) Abstract (in Chinese) .................................................................................... ..........I Abstract ........................................................................................................ ............II Acknowledgements (in Chinese)………………..……………………………….III List of Tables ................................................................................................ ……..IV List of Figures ................................................................................................ …....V. CHAPTER 1 ............................................................................................1 INTRODUCTION ...........................................................................................................................................................1 1.1 MOTIVATIONS AND PURPOSES ...............................................................................................................................1 1.2 THESIS OVERVIEW.................................................................................................................................................1. CHAPTER 2 ............................................................................................3 METHODS AND MATERIALS ........................................................................................................................................3 2-1 MATERIALS ..........................................................................................................................................................3 2-1-1 Compound Set of huAChE..........................................................................................................................3 2-1-2 Compound Set of AGHO ............................................................................................................................3 2-1-3 Preparation of huAChE structure ................................................................................................................4 2-1-4 Preparation of AGHO structure ...................................................................................................................5 2-2 METHODS .............................................................................................................................................................6 2-2-1 Method for QSAR model constructing........................................................................................................6 2-2-2 Tool for QSAR model constructing.............................................................................................................9. CHAPTER 3 ..........................................................................................16 EVALUATION OF GEMDOCK AND MODELING PROTEIN STRUCTURES .....................................................................16 3-1 EVALUATION GEMDOCK ON ACHE AND CUAOS .............................................................................................16 3-1-1 Evaluation GEMDOCK on AChE.............................................................................................................16 3-1-2 Evaluation GEMDOCK on CuAOs...........................................................................................................16 3-2 EVALUATION MODELING PROTEIN STRUCTURES ................................................................................................17 3-2-1 Evaluation Modeling Structure of huAChE ..............................................................................................17 3-2-2 Evaluation Modeling Structure of AGHO .................................................................................................18. CHAPTER 4 ..........................................................................................20 METHOD EVALUATION ON HUACHE.........................................................................................................................20 4-1 VALIDATION OF CONDITIONS AND METHODS......................................................................................................20 4-2 QSAR MODEL FOR HUACHE .............................................................................................................................21. CHAPTER 5 ..........................................................................................23 PRACTICAL APPLICATION .........................................................................................................................................23 5-1 VALIDATION OF CONDITIONS AND METHODS......................................................................................................23 5-2 QSAR MODEL FOR AGHO.................................................................................................................................24 5-3 PREDICTION ON DERIVATIVES .............................................................................................................................25. CHAPTER 6 ..........................................................................................27 CONCLUSIONS ..........................................................................................................................................................27 6-1 SUMMARY ..........................................................................................................................................................27 6-2 FUTURE WORKS .................................................................................................................................................27. REFERENCE........................................................................................61 vii.
(10) Chapter 1 Introduction 1.1 Motivations and Purposes. As the development in computer science, molecular biology and pharmaceutical chemistry, computer-aided drug design become more and more important in drug discovery. Computer-aided drug design is promising directions for shortening the time and reducing the cost for new drug discovery. Molecular docking and quantitative structure activity relationships (QSAR) are the important technologies in computer-aided drug design. However the two methods suffer several challenges: Molecular docking is powerful in characterizing protein-ligand binding but the scoring function of docking still obtains few or no relationship between predicted energy and truly biological activity (e.g., binding affinity or IC50). 3D QSAR analysis such as CoMFA and COMBINE also suffer some problems, such as superposition of steric structures or selection of molecular descriptors1-3. By applying the excellent performance of the molecular docking tool, GEMDOCK4-7, in protein-ligand docking, this thesis makes an attempt to combine the great achievements of GEMPLS8 and GEMkNN in optimization and statistic for QSAR modeling. In order to describe the characteristic of feature more specifically, we have focused the feature on the atom basis. In the process of QSAR model constructing, the generation of consensus feature set and ligand ignored common skeleton set have been evaluated the effect for QSAR modeling. To evaluate our method for QSAR model, we have verified the method for QSAR model of human acetylcholinesterase (huAChE). In addition, we have practically applied the method for the first QSAR model of Arthrobacter globiformis histamine oxidase (AGHO).. 1.2 Thesis overview. We have integrated GEMDOCK with GEMPLS and GEMkNN for QSAR models. In chapter 2, we have prepared the huAChE compound set and AGHO compound set for 1.
(11) verification and application. In addition, we have prepared numbers of AGHO substrate derivatives derived from CHEMSK and CORINA3.09. The target proteins were modified from the template respectively. After preparing compound set and target protein, we integrated GEMDOCK with GEMPLS and GEMkNN for the QSAR models of huAChE and AGHO. In chapter 3, we have evaluated GEMDOCK performance and evaluated the structure of modeling protein. In order to verify the performance of GEMDOCK on acetylcholinesterase (AChE) and Cu2+ amine oxidases (CuAOs), we have applied GEMDOCK on the molecular simulation. of. Torpedocalifornica. AChE. (tcAChE). and. Arthrobacter. globiformis. phenylethylamine oxidase (AGAO). To evaluate the adaptability of the modeling structure, GEMDOCK has been employed to simulate the protein-ligand complex of huAChE and AGHO. In chapter 4, we have verified our method for QSAR modeling of huAChE. In the process of QSAR modeling, the generations of the consensus feature set and the ligand ignored common skeleton set have been used to evaluated the effect for QSAR modeling. There are sixty-nine compounds with IC50 values in the huAChE compound set, and the compounds were divided into the training set (fifty-three compounds) and the testing set (sixteen compounds). The evaluated result of QSAR model shows that our method is useful in QSAR modeling. In chapter 5, we have practically applied our method on QSAR modeling of AGHO. In the process of QSAR modeling, the generations of the consensus feature set and the ligand ignored common skeleton set have been used to evaluated the effect for QSAR modeling too. There are twelve known substrates with Km in the AGHO compound set, and the twelve compounds have been employed in the training set. This is the first specific QSAR model for AGHO. And the evaluated result of the QSAR model shows the predictability. Chapter 6 presents some conclusions and future perspectives. Integrating GEMDOCK with GEMPLS and GEMkNN shows that it is adaptable to QSAR modeling. And the generation of consensus feature and ligand ignored common skeleton set effect the quality of QSAR modeling. And we could make our method an automatically predictive system in the future.. 2.
(12) Chapter 2 Methods and Materials 2-1 Materials. 2-1-1 Compound Set of huAChE Acetylcholinesterase (AChE) is a protein that catalyzes the hydrolysis of acetylcholine (ACh) in cholinergic synapses (Acetylcholine + H2O → Choline + Acetate), and it has one of the fastest reaction rates of any of our enzymes10. In pharmacy industry, AChE is the target of nerve agents, insecticides and therapeutic drugs, in particular the generation of anti-Alzheimer drugs11. To evaluate the method we used, we got the human AChE (huAChE) compound set from reference12. There are sixty-nine compounds with IC50 values measured with huAChE assay in the set, and the compounds are divided into four groups mainly. Within the set, fifty-three compounds are selected for training set (Table 1) and sixteen compounds for testing set (Table 2) to validate the result of our method.. 2-1-2 Compound Set of AGHO CuAOs (EC 1.4.3.6) are ubiquitous in the nature, and the enzymes have a variety of function in the metabolism of biogenic primary amines (RCH2NH2+H2O+O2 → RCHO + NH3 + H2O2)13. In prokaryotic organisms, these enzymes are utilized for growth on amine. In human, these enzymes have been found to be correlated with heart failure14 and chronic medical condition in diabetic patients15,16. To probe into CuAOs, we have applied our method on Arthrobacter globiformis histamine oxidase (AGHO), it is one member of CuAOs family. The twelve compounds with Km values measured with AGHO assay (cooperation with Dr. Chiun-Jye Yuan) (Table 3) were selected for the training model. According to the known twelve substrates of AGHO, we have constructed the first QSAR model for AGHO and found out some significant residues in AGHO by our method. 3.
(13) In addition, in order to study the correlation between binding affinity and structural characteristic of ligand, we have set up a ligand set of AGHO derivatives (Table 4) (Table 5). The 3-D structures of the derivatives are prepared by CHEMSK and CORINA3.0. By this derivatives set, we would probe into the tendency of ligand affinity related to the hydrophobicity in AGHO.. 2-1-3 Preparation of huAChE structure In prior AChE inhibitor studies, many researches were base on using ligand-based design methods such as CoMFA17-21. In our study, in order to simulate the protein-ligand interactions by GEMDOCK, we have modified a proper structure of huAChE to be the target protein. And the docking simulation of protein-ligand complex would be considered for the QSAR model constructing. The AChE X-ray crystallized structures we used in the study were huAChE (PDB entry 1B4122) and tcAChE (PDB entry 1EVE23). The crystallized structure of huAChE (1B41) has no ligand complex with the protein and the structure of tcAChE (1EVE) has one co-crystallized E2020 inhibitor complex with the protein. For the docking simulation, the docking target was the huAChE (1B41) structure but rely on the co-crystallized E2020 inhibitor binding conformation from the tcAChE (1EVE). To ascertain the binding conformation of the co-crystallized E2020 inhibitor relative to the huAChE structure, we have aligned the huAChE structure to the tcAChE structure by a maximal overlap of Cα atoms for the huAChE/tcAChE residues within the proteins. The sequence identity between the two proteins is 57% and the root mean square deviation (RMSD) between huAChE structure and tcAChE structure is 0.88 Å for the set of all Cα atoms in the whole protein, indicating the good overall alignment and substantial structural homology. Because the absence of a solid understanding of the roles of solvent molecules in the huAChE active site, we did not include all waters in considering. After ascertaining the binding conformation of E2020 inhibitor relative to the huAChE structure, the hydrogen atoms were added to the huAChE-E2020 complex via SYBYL7.0 modeling software package from Tripos, Inc., St. Louis, MO. The huAChE-E2020 complex was then energy optimized by Tripos force field, the termination gradient is 0.05 kcal/mol*Å via SYBYL7.0. The resulting structure of protein was extracted for the docking simulation. And the docking simulation of protein-ligand complex would be considered for the QSAR model constructing.. 4.
(14) 2-1-4 Preparation of AGHO structure In our study of CuAOs, we focused on the AGHO. There is no X-ray crystallized structure of AGHO so far. In order to simulate the protein-ligand interactions in the binding site of AGHO, we have constructed a homology modeling of AGHO. GEMDOCK has been employed to simulate the binding conformations between the model and substrates. And the docking simulation would be considered for the QSAR model constructing. Homology modeling was a predictive technique to generate 3D-structure of a protein from its amino acid sequence. The method was based on two major opinions (i) the structure of a protein was determined by its amino acid sequence and (ii) the similar sequences have practically identical structures. To generate a homology structure, we need the amino acid sequence of the target protein and the 3D structures of proteins with homologous amino acid sequence (template protein). And the confidence of the homology modeling is critically dependent on the selection of structural templates and the alignment of the amino acid sequence of the target protein and the templates. There are several programs for structure modeling, and in our study, we constructed the AGHO modeling by SWISSMODEL24. First we obtained the amino acid sequence of AGHO from the SwissProt/TrEMBL. Subsequently the amino acid sequence was be used to search for the template by BLAST25, and we selected the AGAO structure (PDB entry 1IU726) to be the template. The sequence identity between the AGHO and AGAO is 61%, suggesting the high structural homology. In the preparation for structure of template, we selected the structure of AGAO A chain (1IU7A) to be the template, and removed the Cu2+ ion and H2O molecules away. Furthermore there was a special cofactor, 2.4.5-trihydroxyphenylalanyl quinine (TPQ) in the protein, and it was generated from an intrinsic tyrosine in the amino acid sequence by a self-processing that required the Cu2+ ion and molecular oxygen27. We have modified the TPQ to tyrosine by removing the O atoms from the side-chain of TPQ. Subsequently, the homology modeling of AGHO was constructed according to the amino acid sequence of target protein and the structure of template by SWISSMODEL. The root mean square deviation (RMSD) between target protein structure and template structure is 0.15 Å for the set of all Cα atoms in the whole protein, indicating the good overall alignment and substantial structural homology. To ascertain the orientation of Cu2+ ion relative to the modeling structure, we have aligned the modeling structure to the AGAO (PDB entry 5.
(15) 1IU7) structure by a maximal overlap of Cα atoms for the residues within the two proteins. To modify the tyrosine to TPQ, we modified the hydrogen atoms of the side-chain of tyrosine to oxygen in position 2, 4 and 5. Because the absence of a solid understanding of the roles of individual solvent molecules in the AGHO active site, we did not include all waters in considering. In order to mimic the structural character of AGHO, we aligned the modeling structure to the structure of AGAO A chain (1IU7A) and AGAO B chain (1IU7B) respectively, and then we adopted the relative coordinate after alignment of each monomer. After the modification, hydrogen atoms were added and the charge of Cu2+ was assigned to the structure via SYBYL7.0. The structure of model was then energy optimized by Tripos force field, the termination gradient is 0.05 kcal/mol*Å via SYBYL7.0. The resulting structure of protein was extracted for the docking simulation.. 2-2 Methods. 2-2-1 Method for QSAR model constructing In the study, we have integrated GEMDOCK with GEMPLS or GEMkNN for QSAR modeling. To find out the significant hot spots in the binding site, we have focused the feature on atom basis. In addition, we have adopted the concept of consensus feature set and ligand ignored common skeleton set to improve the stability and performance of our method. (Figure 1) shows the main step of our method. The main steps involved in QSAR model building included the following: (a) Prepare an adaptable 3D structure of target protein. (b) Transform the 2D information of ligand into 3D structure by CORINA3.0. (c) Prediction of protein-ligand conformation. (d) According to the protein-ligand complex, we gererated protein-ligand interaction profile and adopted the interaction profile to be the molecular descriptor for QSAR model. (e) Feature selection and preliminary QSAR models generation by GEMPLS and GEMkNN. (f) According to the average q2 value of the leave-one-out cross validation correlation in training, selected the proper tool for QSAR modeling. 6.
(16) (g) Generation of consensus feature set by the selected tool. (h) Feature selection and QSAR model evolution by the selected tool. (f) Select the specific model.. Superimposition of compound structures In the process of QSAR model constructing, we employed GEMDOCK to predict the protein-ligand conformation and adopted the conformation to generate protein-ligand interaction profile for molecular descriptors. To derive the reasonable ligand binding conformation in the active site of protein, we have to superimpose structures of compound and select the one compound superimposition set to generate molecular descriptors. Figure 2 shows the step of compound structures superimposition. In the compound set, there are N kinds of compound, and each compound has been generated M conformations derived from GEMDOCK. In the N kinds of compound, we selected the one that is the most similar one to crystal structure to be the reference ligand. Each compound aligned its conserved region to the reference ligand and calculated the RMSD values. Hence there would be M sets of compound superimpositions (because there are M kinds of reference ligand conformation), and each set is made up of N kinds of compound. RM means the sum total of RMSD value between the conserved regions of the reference ligand and the other compounds in the compound superimposition set, M. Among R1、R2…..RM, we select the minimum one and adopt the ligand conformation in this compound superimposition set.. Protein-Ligand interaction profile After prediction of protein-ligand conformation, we generated protein-ligand interaction profile for molecular descriptor. Figure 3 shows the protein-ligand interaction profile. According to predictive ligand binding conformation, we calculated the protein-ligand interaction. The interaction we considered included electrostatic (Eelec), van der Waals (Evdw) and hydrogen bond (Ehb) interactions. We have described the protein-ligand interaction on the atom basis and focused on active site of the target protein. In the interaction profile, we have taken down the interaction between the active site atom of protein and the ligand. On each atom, Eele, Evdw and Ehb would be calculated respectively.. 7.
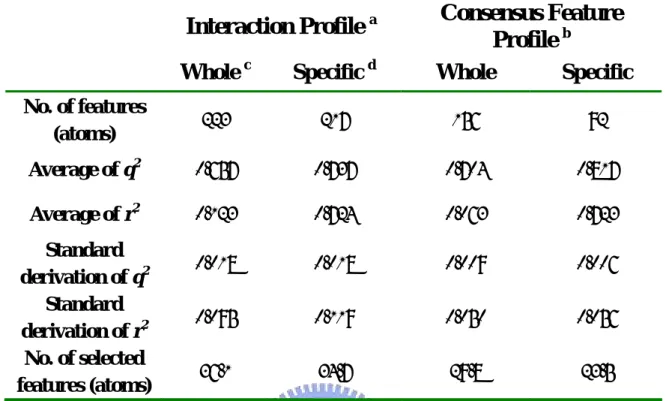
(17) In order to improve the performance of the method for QSAR model constructing, some methods have been introduced into the procedure of QSAR modeling. The methods include the following: (a) Generation of consensus feature set. In the process of model constructing, we have employed the collection of selected feature from GEMPLS or GEMkNN to generate the consensus feature set, and then features were selected again from the consensus feature set by GEMPLS or GEMkNN. And the result caused of this method will be compared. (b) Definition of ligand specific skeleton. Sometimes, ligands in the compound set share high homology, and they contained the same common skeleton. The differences among ligands are in the region of substituent groups and the regions are believed to be distinguishable. In order to manifest the importance of the substituent group, we have ignored the influence from the common regions in feature selection. And the result caused of this method will be compared. Figure 4 shows the definition of common skeleton.. Generation of Consensus Feature set In the process of QSAR modeling, we found it is not very good to the stability of model when GEMPLS or GEMkNN was directly employed to select feature from the molecular feature set. The molecular feature set may include too much feature, so it is difficult to select the significant feature strongly correlating with biological activity. In order to improve this situation, GEMPLS and GEMkNN was employed to carry out ten number of preliminary QSAR models respectively, and ten sets of feature were selected. In the collection of the selected feature, we noted down the number of time that each feature has been selected and then calculated the average (Avg) and the standard deviation (Std) of feature occurrence number. The last, we gathered the consensus feature according to the number of time that each feature has been selected by the following rule, and generated the consensus feature set.. N ≥ ( Avg − Std ) Where N is the number of time that a certain feature has been selected. Finally, GEMPLS or GEMkNN was employed to carry out feature selection from the consensus feature set.. 8.
(18) Definition of Ligand Specific Skeleton Sometimes, the ligands of a certain protein share high homology in the compound set. This is that because the derivatives could form structural complementation with binding site of a protein, and they contained the same common skeleton. On the conformations of those derivatives, the main differences were focused on the region of substituent group and the regions are believed to be distinguishable for biological activity of those derivatives. However, those distinguishable regions accounted for few proportions to make influence power reduce inside the whole ligand. Hence, in order to manifest the importance of the distinguishable groups, we have ignored the influence from the common region and concentrated sight on the substituent group.. 2-2-2 Tool for QSAR model constructing GEMDOCK GEMDOCK is a useful tool for the molecular docking and structure-based virtual screening. The tool was developed by Jinn-Moon Yang, an associate professor of the Institute of Bioinformatics, National Chiao Tung University. GEMDOCK has been evaluated over 300 protein-ligand complexes and applied to identify new substrates or inhibitors for several practical applications, such as sulfotransferase28 and imidase. And the binding-site pharmacophore (hot spots) and ligand preferences are used to substantially enhance GEMDOCK for screening large databases on several target proteins, such as thymidine kinase (TK), estrogen receptor (ER), dihydrofolate reductase (DHFR) and the E protein of dengue virus. The scoring function of GEMDOCK has been developed as the scoring function for both molecular docking and the ranking of screened compounds for post-docking analysis. This function consists of a simple empirical binding score and a pharmacophore-based score to reduce the number of false positives. The energy function can be dissected into the following terms:. E tot = E bind + E pharma + E ligpre where Ebind is the empirical binding energy, Epharma 9. (1) is the energy of binding site.
(19) pharmacophores (hot spots), and Eligpre is a penalty value if a ligand does not satisfy the ligand preferences. Epharma and Eligpre are especially useful in selecting active compounds from hundreds of thousands of non-active compounds by excluding ligands that violate the characteristics of known active ligands, thereby improving the number of true positives. The values of Epharma and Eligpre are determined according to the pharmacological consensus derived from known active compounds and the target protein. In contrast, the values of Epharma and Eligpre are set to zero if active compounds are not available. The empirical binding energy (Ebind) is given as. Ebind = Einter + Eintra + E penal. where Einter and Eintra. (2) are the intermolecular and intramolecular energy, respectively, and. Epenal is a large penalty value if the ligand is out of range of the search box. For our present work, Epenal is set to 10,000. The intermolecular energy is defined as. ( ). lig pro qq B E inter = ∑ ∑ F rij ij + 332 .0 i 2j 4 rij i =1 j =1 . where. Bij ij. r. (3). is the distance between atoms i and j with interaction type Bij formed by. pair-wise heavy atoms between ligands and proteins, Bij is either a hydrogen bond or a steric state, qi and qj are the formal charges and 332.0 is a factor that converts the electrostatic energy into kilocalories per mole. The terms lig and pro denote the number of heavy atoms in the ligand. ( ) B. and receptor, respectively.. F rij ij. is a simple atomic pair-wise potential function. In this. atomic pair-wise model, the interactive types include only hydrogen bonding and steric potentials having the same function form but different parameters, V1, . . . , V6. The energy value of hydrogen bonding should be larger than that for steric potential. In this model, atoms are divided into four different atom types 9: donor, acceptor, both, and nonpolar. A hydrogen bond can be formed by the following pair-atom types: donor-acceptor (or acceptor-donor), donor-both (or both-donor), acceptor-both (or both-acceptor), and both-both. Other pair-atom combinations are used to form the steric state. We used the atom formal charge to calculate the electrostatic energy, which is set to 5 or -5, respectively, if the electrostatic energy is more than 5 or less than -5. The intramolecular energy of a ligand is lig lig q q dihed B Eintra = ∑ ∑ F rij ij + 332.0 i 2j + ∑ A[1 − cos(mθ k − θ0 )] 4rij k =1 i =1 j = i + 2 . ( ). 10. (4).
(20) ( ) is defined as Equation 3 except the value is set to 1000 when B. where. F rij ij. B. rij ij. < 2.0 Å. and dihed is the number of rotatable bonds of a ligand. We followed the work29 to set the values of A, m, and θ0. For the sp3-sp3 bond A, m, and θ0 are set to 3.0, 3, and π; for the sp3-sp2 bond and. A = 1.5, m = 6, and θ0 = 0. GEMDOCK evolves binding site pharmacological consensuses and ligand preferences. from both known active ligands and the target protein to improve screening accuracy. We used the premise that previously acquired interactions (hot spots) between ligands and the target protein can be used to guide the selection of lead compounds for subsequent investigation and refinement. For each known active ligand, GEMDOCK first yielded ten docked ligand conformations by docking the ligand into the target protein, and only the ligand with the lowest docked conformation energy was retained for pharmacological consensus analysis. The protein-ligand interactions were extracted by overlapping these lowest-energy docked conformations, and the interactions were classified into three different types, including hydrogen bonding, hydrogen-charged interactions, and hydrophobic interactions. After all of the protein-ligand interactions were calculated, the atom interaction-profile weight of the target protein representing the pharmacological consensus of a particular interaction was given as. Q kj =. f jk. (5) 3N where fjk is the number of an atom j (in protein) interacting with ligands with the interaction type k and N is the number of known active ligands. In our present work, an atom j was k considered a hot-spot atom when Q j was more than 0.5.. The pharmacophore-based interaction energy (Epharma) between the ligand and the protein is calculated by summing the binding energies of all hot-spot atoms: lig. ( ). E pharma = ∑∑ CW (Bij )F rij ij hs. B. (6) where CW (Bij) is a pharmacological-weight function of a hot-spot atom j with interaction i =1 j =1. ( ) B. type Bij, F rij ij. is defined as Equation 3, lig is the number of the heavy atoms in a screened. ligand, and hs is the number of hot-spot atoms in the protein. The CW(Bij) is given as 1.0 if Q kj ≤ 0.5 or Bij ≠ k CW ( Bij ) = (7) k if Q kj > 0.5 and Bij = k 1.5 + 5(Q j - 0.5) Qjk is the atomic pharmacological-profile weight (Equation 5) and k is the interact type (e.g., 11.
(21) hydrogen bonding, hydrogen-charged interactions, or hydrophobic interactions) of the hot-spot atom j.. GEMPLS (GEM-Partial Least Squares) GEMPLS is a hybrid approach that combines GA as a robust optimization technique with PLS as a powerful statistical technique for the variable selection and model evolution. GA operates on a population of potential solutions applying the principle of survival of the fittest to produce successively better approximations to optimum solution. PLS deals with strongly collinear input data and make no restriction on the number of variables used. In this thesis, the tool GEMPLS was developed by Cooperating with Prof. Kao Lab. In GEMPLS, the chromosomes consist of some randomly selected features and the latent variables (lv). The squared cross-validated correlation coefficient q2 in the PLS analysis is used as objective function to provide a measure of how the internal predictability with respect to the selected features of the chromosome. And GA will find the fittest features with the highest q2 in the PLS analysis. The main steps involved in GEMPLS included the following: (a) initiation and evaluation of the initial population, (b) selection of the reproductive population, (c) crossover and mutate the reproductive population, (d) evaluation of the child population, (e) reinsertion of the child population to form the population on the next generation. And the cycle of above four steps (from step (b) to (e)) is repeated until the number of generation reaches to the maximum number of generations. In order to improve the performance of GEMPLS for QSAR model building, a number of refinements have been introduced into GEMPLS. The refinements include the following: (a) An extra bit lv, representing the number of latent variables, is appended to the original chromosome of GA and expected to efficiently solve the problem of the optimum number of latent variables though evolutionary process (b) Adopt Mahalanobis distance to discriminate significant features. Mahalanobis distance is a very useful way of determining the deviation of a sample from the mean of the distribution in multivariable calculus. Therefore, the Mahalanobis distance is adopted to identify significant features from all of those. M is the Mahalanobis distance from the feature vector v (column vector of data matrix here) to the mean vector µ, where Σ is the covariance matrix of the features. 12.
(22) (c) Cooperate with biased mutation to lead the evolution. We have recommended that uniform mutation is cooperated with biased mutation to lead the evolution of GA toward significant feature set and to reduce the interference of noise features. N − pi Pi = Pmin + ( Pmax − Pmin ) × s Ns −1 Pi is the probability of setting feature bit i to 1, pi is the position of feature i in the descending order of Mahalanobis distance of all features, Pmin and Pmax are the minimum and maximum values of Pi, and Ns is the number of significant features. Pi is derived from pi only when pi is ahead of Ns, otherwise Pi is set to Pmin. In other words, the significant feature i with higher Mahalanobis distance will obtain the higher Pi. In this study, the corresponding parameters are defined as: Pmax = 0.8, Pmin = 0.2. The predictability of QSAR model was assessed by the conventional correlation coefficient (r2), the cross-validated correlation coefficient (q2), the cross-validated SDEP (SDEPcv), and external SDEP (SDEPex):. q = 1− 2. SDEP =. ∑ ( yi − y pred ,i ) ∑ ( yi − y ). 2. 2. ∑ ( yi − y pred ,i ). 2. N. where yi is the observed biological activity of compound i, ypred, i is the predicted biological activity of compound i in the validation set, y is the average biological activities of the data set, and N is the total number of compounds. After deciding the optimum number of latent variables, the corresponding highest q2, lowest SDEP, together with the conventional squared correlation coefficient r2 can be used to assess the predictability of QSAR model, i.e. the model with more remarkable predictability can provide the higher r2, q2 and the lower SDEP between the observed and predicted biological activities.. 13.
(23) 2-2-3 GEMkNN (GEM- k-Nearest-Neighbor) GEMkNN is a hybrid approach that combines GA as a robust optimization tool with kNN as a pattern recognition method to evaluate the discriminative ability of the subset. GA operates on a population of potential solutions applying the principle of survival of the fittest to produce successively better approximations to optimum solution. kNN is a conceptually simple, nonlinear approach to pattern recognition problems. In this thesis, the tool GEMkNN was developed by In this thesis, the tool GEMPLS was developed by Cooperating with Prof. Kao Lab. In GEMkNN, the chromosomes consist of some randomly selected features and the number of selected similar molecules (k). The similarities between compounds are evaluated by Euclidean distance. The squared cross-validated correlation coefficient q2 in the kNN analysis is used as objective function to provide a measure of how the internal predictability with respect to the selected features of the chromosome. And GA will find the fittest features with the highest q2 in the kNN analysis. The main steps involved in GEMkNN included the following: (a) initiation and evaluation of the initial population, (b) selection of the reproductive population, (c) crossover and mutate the reproductive population, (d) evaluation of the child population, (e) reinsertion of the child population to form the population on the next generation. And the cycle of above four steps (from step (b) to (e)) is repeated until the number of generation reaches to the maximum number of generations. In order to improve the performance of GEMkNN for QSAR model building, a number of refinements have been introduced into GEMkNN. The refinements include the following: (a) Adopt Mahalanobis distance to discriminate significant features. Mahalanobis distance is a very useful way of determining the deviation of a sample from the mean of the distribution in multivariable calculus. Therefore, the Mahalanobis distance is adopted to identify significant features from all of those.. M 2 = (ν − µ )′ ∑ −1 (ν − µ ). M is the Mahalanobis distance from the feature vector v (column vector of data matrix here) to the mean vector µ, where Σ is the covariance matrix of the features. (b) Cooperate with biased mutation to lead the evolution. We have recommended that 14.
(24) uniform mutation is cooperated with biased mutation to lead the evolution of GA toward significant feature set and to reduce the interference of noise features.. N − pi Pi = Pmin + ( Pmax − Pmin ) × s Ns −1 Pi is the probability of setting feature bit i to 1, pi is the position of feature i in the descending order of Mahalanobis distance of all features, Pmin and Pmax are the minimum and maximum values of Pi, and Ns is the number of significant features. Pi is derived from pi only when pi is ahead of Ns, otherwise Pi is set to Pmin. In other words, the significant feature i with higher Mahalanobis distance will obtain the higher Pi. In this study, the corresponding parameters are defined as: Pmax = 0.8, Pmin = 0.2. The predictability of QSAR model was assessed by the conventional cross-validated correlation coefficient (q2), the cross-validated SDEP (SDEPcv), and external SDEP (SDEPex):. q2 = 1 −. SDEP =. ∑ ( yi − y pred ,i ) ∑ ( yi − y ). 2. 2. ∑ ( yi − y pred ,i ). 2. N. where yi is the observed biological activity of compound i, ypred, i is the predicted biological activity of compound i in the validation set, y is the average biological activities of the data set, and N is the total number of compounds. After deciding the optimum number of latent variables, the corresponding highest q2, lowest SDEP can be used to assess the predictability of QSAR model, i.e. the model with more remarkable predictability can provide the higher q2 and the lower SDEP between the observed and predicted biological activities.. 15.
(25) Chapter 3 Evaluation of GEMDOCK and Modeling Protein Structures. 3-1 Evaluation GEMDOCK on AChE and CuAOs. In order to evaluation GEMDOCK on AChEs and CuAOs, we have docked the ligands into their target proteins respectively. We based the results on root mean square deviation (RMSD) error in ligand heavy atoms between the docked conformation and the crystal structure or the physical meaning of binding conformation to verify the GEMDOCK performance on the two kinds of protein.. 3-1-1 Evaluation GEMDOCK on AChE To evaluate the performance of docking tool on AChE, we have docked one known E2020 inhibitor back into its reference protein, 1EVE. The structure, 1EVE, is the structure of tcAChE. According to the protein-ligand complex, the ligand forms stable stack force with W84, W279 and F330, and the nearest distance between the atom N of ligand and the water is 2.90 Å. During the molecular docking, because there is no known ligand so far, we set the CW (Bij) (in equation 6) 3.0 for side-chain atoms of F330, 4.0 for side-chain atoms of W84 and 5.0 for side-chain atoms of W279 to simulate the binding state. The active site for the tcAChE docking calculations is the region within a radius of 8 Å relative to E2020. And the RMSD values between the docked conformation and the crystal structure is 1.73 Å. The docked ligand forms stable stack force with W84, W279 and F330, and the nearest distance between the atom N of ligand and the water is 3.69 Å.. 3-1-2 Evaluation GEMDOCK on CuAOs To evaluate the performance of GEMDOCK on CuAOs, we have docked one known substrate, phenylethylamine, into the active site of AGAO and the PDB entry of the protein is 16.
(26) 1IU7. There is no x-ray crystal structure of AGAO complex with ligand so far, according to the pathway for catalytic cycle of CuAOs (Figure 5), the TPQ-O5 of the enzyme reacts with primary amines and releases the product aldehyde, ammonia ions and hydrogen peroxide. And the conserved Asp will be a general base to abstract the proton from the substrate in the reductive half-reaction. Because the crystal structure has no ligand complex with the protein, we defined that the binding site is the collection of amino acids enclosed within a radius of 8 Å relative to the cofactor, 2.4.5-trihydroxyphenylalanyl quinine (TPQ382). Figure 6 shows the binding conformation of docked ligand at the active site of AGAO. By the docking simulation, the function group –NH2 of the ligand forms hydrogen bond with the cofactor TPQ382-O5, D298-OD2 (general base) and I379-O, and the aromatic ring of the ligand stays in the hydrophobic pocket.. 3-2 Evaluation Modeling Protein Structures. In order to verify the modeling structure, we inspected the modeling structure by structural alignments to relative proteins and docked several known ligands (substrates or inhibitors) into the binding site of the modeling structure by GEMDOCK respectively. Before this, we have evaluated GEMDOCK on two kinds of protein: tcAChE and AGHO. And we based the results on the physical meaning of binding conformation to verify the model.. 3-2-1 Evaluation Modeling Structure of huAChE To evaluate the model of huAChE, we inspected the difference among the model and X-ray crystal structure of huAChE (PDB entry 1B41) and tcAChE (PDB entry 1EVE). The root mean square deviation (RMSD) between model and 1B41 is 0.24 Å for the set of all Cα atoms in the whole protein, and 0.89 Å between homology structure and 1EVE. In the active site, the main difference among the three structures is the side-chain conformation of Y337 (residue number of 1B41, relatively to F330 in 1EVE) (Figure 7). In previous study, it has been found that F330 in tcAChE can adopt a wide range of conformations in the complex structure and may play a role as gate19,23,30. It is natural to expect similar behavior of Y337 in the huAChE compared with the analogous F330 in tcAChE. 17.
(27) The tcAChE crystal structure (1EVE) has an inhibitor (E2020) complex with the protein, so the F330 was in the opened form to stabilize the protein-ligand complex. The huAChE crystal structure (1B41) did not have an inhibitor in the active site, thus did not reflect such a conformation shift in the Y337 such as F330 in the tcAChE (1EVE). During the modeling, because we have utilized the ligand binding information from a tcAChE (1EVE) to structure modeling, the Y337 is in the opened form in the model. We have docked one known inhibitor, E2020, into the binding site of the modeling structure. Because there is no X-ray structure of protein-ligand complex, we defined that the binding site is the collection of amino acids enclosed within a radius of 8 Å relative to the E2020. Docking calculations were carried out with the GEMDOCK program. In order to simulate the protein-ligand complex, we set the CW (Bij) (in equation 6 ) was 3.0 for side-chain atoms of Y337, 4.0 for side-chain atoms of W86 and 5.0 for side-chain atoms of W286 just like the parameters in tcAChE. After considering the interaction preference, the inhibitors form stable stack force with W86 and W286. Figure 8 is the docking poses of the inhibitor, E2020, in the active site of the modeling structure. The interaction preference would be considered when we simulated the binding poses of other compounds.. 3-2-2 Evaluation Modeling Structure of AGHO To evaluate the homology structure of AGHO, we inspected the difference between the homology structure and X-ray crystal structure of AGAO (PDB entry 1IU7). Figure 9A shows the whole structural alignments between the homology structure and 1IU7. The root mean square deviation (RMSD) between homology structure and 1IU7 is 0.24 Å for the set of all Cα atoms in the whole protein. In the active site, the residues in the two proteins are identical, besides D326 and V398 in AGHO. D326 in AGHO is relative to Y307 in AGAO, and V398 is relative to I379 in AGAO. Figure 9B shows the structural difference between the two proteins. The O5 of important cofactor 2.4.5-trihydroxyphenylalanyl quinine (TPQ) (TPQ401 in AGHO, TPQ382 in AGAO) slight shift 0.58 Å between homology structure and AGAO structure (1IU7). The OD1 of general base Asp (D317 in AGHO, D298 in AGAO)31 slight shift 0.2 Å between homology structure and AGAO structure (1IU7). And the Tyr (Y296 in AGAO, Y315 in AGHO) that play a role as gate32, are in the opened form in both protein structure. In the AGAO structure (1IU7), the distance between TPQ382-O4 and Y284 is 2.51 Å, and the distance between TPQ401-O5 18.
(28) and Y303 in the AGHO structure is 2.55 Å. We have docked the twelve known substrates of AGHO to the active site of homology structure (Figure 10). Because there is no X-ray structure of AGHO so far, we defined that the binding site is the collection of amino acids enclosed within a radius of 8 Å relative to the TPQ401. Docking calculations were carried out with the GEMDOCK program. In order to simulate the protein-ligand complex, we set the CW (Bij) (in equation 6) was 2.3 for O5 of TPQ401. After considering the interaction preference, the function groups –NH2 of the substrates form hydrogen bonds with the cofactor TPQ401-O5, D317-OD1 (general base), and the –NH on the ring of tryptamine and histamine form hydrogen bonds with P156-O. The aromatic rings of the substrates stay in the hydrophobic pocket. The interaction preference would be considered when we simulated the binding poses of other compounds. By the docking simulation of other compounds from the derivatives set we have setup, we identified a new inhibitor, spermine (Figure 11A). According to the docking conformation in the binding site of the homology structure (Figure 11B), the long flexible spermine passes through the channel and forms hydrogen bonds with TPQ401-O5, D317-OD1, Y321-OH and P156-O. The binding conformation of spermine not only forms hydrogen bonds with TPQ401-O5, D317-OD1 and P156-O that was similar to the substrates, but also forms hydrogen bonds with Y321-OH. By the comparison of protein-ligand interaction between spermine and known substrates in the AGHO binding site, it was natural to expect that spermine could bind stably in the binding site of AGHO protein because it could form more interactions with the protein. And the inhibitive effect of spermine has been verified by experiment.. 19.
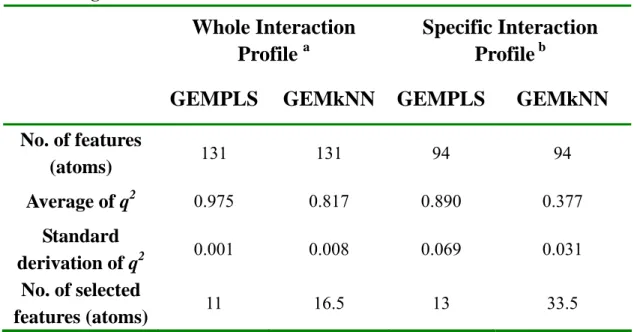
(29) Chapter 4 Method Evaluation on huAChE We have evaluated the method of QSAR model constructing on a public compound set, human acetylcholinesterase (huAChE) compound set from reference. The compound set includes sixty-nine compounds with IC50 values and those ligands belong to four kinds of derivative. Within the set, fifty-three compounds are selected for training set (Table 1) and sixteen compounds for testing set (Table 2). The IC50 values of the ligand rang from 0.48 nM to 19580 nM in the training set and 0.33nM to 30000nM in the testing set.. 4-1 Validation of Conditions and Methods. The common metrics were used to evaluate the QSAR model quality, including the q2 (cross-validated correlation coefficient) in training and r2 (correlation coefficient) in testing. In order to validate the performance and stability of the method for QSAR model building, we have built ten models under each kind of condition and then evaluated the mean and standard deviation values of the q2 and r2. To select the proper tool for huAChE QSAR model building, GEMPLS and GEMkNN both have been employed to construct the QSAR model. In addition, we have made some tests, including the condition that generating ligand ignored common skeleton set and generating consensus feature set. And we would compare the results in each condition. Table 6 shows the performance of GEMPLS and GEMkNN relative to the raw ligand feature set and the ligand ignored common skeleton set. GEMPLS and GEMkNN have been employed in the raw feature sets and the feature set that ignored ligand common skeleton. In the raw feature set, the result of cross-validated correlation coefficient in GEMkNN is a little better than GEMPLS. In ligand ignored common skeleton set, the q2 value of GEMkNN is much better than GEMPLS. The q2 value of GEMPLS would become worse if the ligand common skeleton have been ignored. This phenomenon is different in GEMkNN, and the evaluated performance would be better when ligand common skeleton have been ignored. In the raw feature set of huAChE, the result of cross-validated correlation coefficient in 20.
(30) GEMkNN is better than GEMPLS. Thus GEMkNN has been employed in the QSAR model constructing for huAChE. Table 7 shows the relationship between the result of GEMkNN and generation of consensus feature set. No matter the common skeleton have been ignored or not, the generation of consensus feature set in the process of QSAR model building would improve obviously the quality of the QSAR model. And the result of cross-validated correlation coefficient in GEMkNN would become better if the ligand common skeleton have been ignored no matter the consensus feature set have been generated or not. Table 8 shows the comparison of our method with the method in the reference. In the literature, the docking tool GOLD have been employed in molecular simulation, the feature basis have been focus on residue-based, the leave-one-out cross validation correlation of q2 is 0.72 and the r2 between the predicted values and the experimental values is 0.69. The docking tool, GEMDOCK, have been employed to simulate the protein-ligand complex, we have focused the molecular feature set on the atom basis, the average q2 values of leave-one-out cross validation is 0.81 and the average correlation of r2 = 0.72 between the predicted values and the experimental values. Our QSAR model has been built via GEMkNN over interaction profile within compound in the training set. And the model has been built via PLS regression in the literature.. 4-2 QSAR Model for huAChE. To construct an effective QSAR model for huAChE, we have adopted models by the following step: (a) Predicted the molecular geometry in the binding site of the target protein by GEMEDOCK. (b) According to the protein-ligand complex, we used the interaction profile to be molecular feature set on atom basis. (c) Generation of ligand ignored common skeleton set. (d) Feature selection and model evolution by GEMkNN. (e) Generation of consensus feature set (f) Feature selection and model evolution by GEMkNN (g) Select the specific model. 21.
(31) With such a procedure, the average q2 values of leave-one-out cross validation is 0.817 and the average correlation of r2 = 0.723 between the predicted values and the experimental values. In order to construct a specific QSAR model, we have adopted the one that the q2 value is most close to the average q2 value in 10 times when training. This is because we hope to select a steady model and to avoid over-fitting in QSAR model building. At last we adopted the model with a leave-one-out cross validation of q2 = 0.818 and a correlation of r2 = 0.781 between the predictive values and experimental values. Figure 12A and Figure 12B show the correlation between the experimental values and the predicted values from the QSAR modeling. Table 9 shows the selected feature and their physical meaning. Several residues have been found very important in previous study. Residue Y72 could form a wall to stabilize ligand and W86 forms π-π interaction with choline. N87 and Y337 contribute the electrostatic force in the active site. Residues Y124 and F338 provide hydrophobic contacts with ligand. S203 and H447 are significant in huAChE; they are catalytic triad in the enzyme. In previous study, residue H287 has been found that could affect the binding affinity of AChE inhibitors and W286 might play the same role as H287. Residue Y341 forms the local pocket in the active site. This result reveals that most of the feature we have selected have already been verified their physical meanings and to help confirm the sensibility of our model.. 22.
(32) Chapter 5 Practical Application In this study, our method has been employed to practically apply on AGHO model building. There are twelve known substrates (cooperation with Dr. Chiun-Jye Yuan) with Km values in the compound set. Table 3 shows the structures of the known substrates in the compound set. The Km values of the ligand rang from 0.0025 mM to 0.1221 mM in the set. To construct the QSAR. model for AGHO, twelve known substrates have been employed in the training set.. 5-1 Validation of Conditions and Methods. In order to validate the performance and stability of the method for QSAR model building, we have built ten models under each kind of condition and then evaluated the mean and standard deviation values of the q2. To select the proper tool for huAChE QSAR model building, GEMPLS and GEMkNN both have been employed to construct the QSAR model. And we have done some test and attempted to find out the influence in the conditions, including the condition that generating ligand ignored common skeleton set and generating consensus feature set by the same token. Table 10 shows the performance of GEMPLS and GEMkNN relative to the raw ligand feature set and the ligand ignored common skeleton feature set. GEMPLS and GEMkNN have been employed in the raw feature sets and the ligand ignored common skeleton set. In the raw feature set, the performance of GEMPLS is better than GEMkNN. In ligand ignored common skeleton set, the performance of GEMPLS is much better than GEMkNN. No matter which tool have been employed, the evaluated performance would become worse if the ligand common skeleton have been ignored. In the raw feature set of AGHO, the result of cross-validated correlation coefficient in GEMPLS is better than GEMkNN. Thus GEMPLS has been employed in the QSAR model constructing for AGHO. Table 11 shows the relationship between the performance of GEMPLS and generation of consensus feature set. GEMPLS has been employed in the raw feature set and the ligand ignored common skeleton set. The performance of GEMPLS would be worse if the 23.
(33) ligand common skeleton have been ignored. No matter the common skeleton have been ignored or not, the generation of consensus feature set in the process of QSAR model building would improve obviously the quality of the QSAR model.. 5-2 QSAR Model for AGHO. To construct an effective QSAR model for AGHO, we have adopted models by the following step: (a) Predicted the molecular geometry in the binding site of the target protein by GEMEDOCK. (b) According to the protein-ligand complex, we used the interaction profile to be molecular features set on atom basis. (c) Feature selection and model evolution by GEMPLS. (e) Generation of consensus feature set (f) Feature selection and model evolution by GEMPLS (g) Select the specific model. With such a procedure, the average q2 values of leave-one-out cross validation is 0.977 and standard deviation of the q2 values is 0.001. Such a result means that our method is an effective and stable method for QSAR model building. In order to construct a specific QSAR model, we have adopted the one that the q2 value is most close to the average q2 value in 10 times when training. This is because we hope to select a steady model and to avoid over-fitting in QSAR model building. At last we adopted the model with a leave-one-out cross validation of q2 = 0.979. Figure 13 shows the correlation between predicted values and experimental values. Table 12 shows the selected feature and their physical meaning. In previous research, some functions of residues in AGAO have been studied33. The sequence identity between the AGHO and AGAO is 61%, suggesting the high structural homology. The selected residues of AGHO QSAR modeling are almost conserved in the two proteins, indicting the similar physical function in the active site, so we could understand the AGHO residue functions through the understanding in AGAO. Residue A155 is related to F105 in AGAO, P156 is related to P136 in AGAO, Y315 is related to Y296 in AGAO, Y321 is related to Y302 in AGAO and F426 is related to F407 in AGAO. 24.
(34) Figure 14 shows the multiple sequence alignments of CuAOs on the spots of the selected feature. The amine oxidases belong to bacteria, plants and animals. In previous study, it has been found that although those kinds of enzyme exhibit common mechanistic features, the substrate specificities of them appear to be different. For instance, the amine oxidases from bacteria show a preference for aromatic amines but the ones from animal do not show this preference. We have observed some interesting appearance in the selected feature. It is entirely conserved in all kinds of CuAOs on the positions of D317and N400. The positions of V398, Y321 and Y315 are not entirely conserved in all CuAOs, but the residues on the positions show a similar chemical and physical quality. On the positions of residues P156 and A155, it shows different residue quality in bacteria, plants and animals indicating the residues are significant to the ligand preference.. 5-3 Prediction on Derivatives. According to the ligand set of AGHO, it seems that the ligand affinity of AGHO related to the hydrophobicity of ligand. To probe into the relationship between ligand affinity and ligand characteristic, we have predicted a serial potential affinity of derivatives by the QSAR model we have built. Table 4 and Table 5 show the set of the AGHO derivatives. Here we have focused on the length of side-chain and the size of aromatic ring related to the ligand affinity. Table 4 shows the structures of derivatives and the predicted values. There are six kinds of group in the ligand set. In each kind of ligands, the number of carbon in side-chain rise from one to four. Figure 15 shows the relationship between predicted values and side-chain length. Generally speaking, the predicted values increase with the increase of side-chain length. This tendency is similar to the phenomenon among phenylethylamine, phenylpropylamine and phenylbutylamine. Figure 16 shows the relationship between predicted values and size of aromatic ring. It shows that the predicted values increase with the increase of ring size obviously. In the process of prediction, we have predicted the affinity of benzylamine successfully. The predicted value from QSAR model is 1.077. After experimenting, the measured value of benzylamine is 1.261. Figure 17 shows the relationship between predicted values and experimental values. Although the predicted value is not identical with experimental value, the 25.
(35) tendency between predicted values and experimental values is similar. The results of the prediction show that the predicted values increase with the increase of side-chain length and size of ring. It suggested that hydrophobicity is one of the essential factors that determined the affinity of AGHO to its substrates. In the application on AGHO QSAR modeling, our method has been employed to build a specific QSAR model for AGHO. This model shows evaluation with a leave-one-out cross validation of q2 = 0.979. Besides good behavior in the evaluation, our method also represents the stability in QSAR model building. It shows the average q2 = 0.977 and the standard deviation of the q2 values is 0.001. In the biological field, the selected features derived from our method are meaningful. Most of them have been verified in previous research.. 26.
(36) Chapter 6 Conclusions 6-1 Summary. In summary, we introduced GEMDOCK to generate the atom-based protein-ligand interactions as descriptions, which are used by GEMPLS and GEMkNN to construct the QSAR models. The method has been verified on huAChE QSAR modeling and applied on AGHO for the first novel QSAR model. And the comprehensive effect shows the good performance for QSAR modeling of huAChE and AGHO. In the process of QSAR model constructing, the generation of consensus feature set and ligand specific skeleton set improve the quality and stability of QSAR model. The verification and application show our method is adaptable to QSAR model constructing.. 6-2 Future Works. We would apply our method for more compound set to verify that it is robust and adaptable to QSAR model constructing. In addition, we would make the method an automatically predictive system for drug discovery in the future.. 27.
(37) Table 1. Compounds Structures in huAChE Training Set12. Y N R3. N. X. R2 R1. Ligand IC50(nM) pIC50 ID. R1. R2. R3. -X-. -Y-. -H. -H. -H. -(CH2)2-. -O-. 1. 55. 7.26. -CH3. -H. -H. -(CH2)2-. -O-. 2. 7.8. 8.11. -CH3. -OCH3. -H. -(CH2)2-. -O-. 3. 5.8. 8.24. -OCH3. -H. -H. -(CH2)2-. -O-. 4. 7.2. 8.14. -H. -H. -OCH3. -(CH2)2-. -O-. 5. 7.1. 8.15. -H. -NH-CO-CH3. -H. -(CH2)2-. -O-. 6. 2.8. 8.55. -H. -NH-SO2-Ф. -H. -(CH2)2-. -O-. 7. 14. 7.85. -H. -4-morpholino. -H. -(CH2)2-. -O-. 8. 0.8. 9.10. -H. -NH2. -H. -(CH2)2-. -O-. 9. 20. 7.70. -H. -Br. -H. -(CH2)2-. -O-. 10. 50. 7.30. -H. -CN. -H. -(CH2)2-. -O-. 11. 101. 7.00. -H. -CO-NH2. -H. -(CH2)2-. -O-. 12. 8.8. 8.06. -H. -H. -H. -(CH2)3-. -O-. 13. 900. 6.05. -H. -H. -H. -O-CH2-. -O-. 14. 2600. 5.59. -H. -H. -H. -NH-CH2-. -O-. 15. 320. 6.49. -H. -H. -H. -(CH2)2-. -S-. 16. 99. 7.00. -H. -H. -H. -(CH2)2-. -CH=CH-. 17. 220. 6.66. -H. -H. -H. -(CH2)2-. -NH-. 18. 120. 6.92. -CH2-CH2-CO-NH-. -H. -(CH2)2-. -O-. 19. 0.57. 9.24. -NH-CO-CH2-. -H. -(CH2)2-. -O-. 20. 0.95. 9.02. -N(CH3)-CO-CH2-. -H. -(CH2)2-. -O-. 21. 0.48. 9.32. -(CH2)2-. -O-. 22. 3.6. 8.44. 23. 250. 6.60. -H. -NH-CO-CH2O. N. +. O O. 30.
(38) Table 1. Continued O. z. W. X Y. S. O. +. R. Ligand. IC50. ID. (nM). -CH2-Φ. 24. 8. 8.10. -CH-CH2-. -CH2-Φ. 25. 43. 7.37. -CH2C(OH)CH2CH2- -CH-CH2-. -CH2-Φ. 26. 380. 6.42. -W-. -X-. -Y-Z-. R. -(CO)-. -CH2-CH2-. -CH-CH2-. -(CO)-. -CH2-C(OH)-. -(CO)-. N. pIC50. -(CO)-. -CH2CH2CH2CH2-. -CH-CH2-. -CH2-Φ. 27. 110. 6.96. -(CO)-. -CH2C(OCH3)-. -CH-CH2-. -CH2-Φ. 28. 120. 6.92. -(CO)-. -CH-. -C-CH2-. -CH2-Φ. 29. 520. 6.28. -C(OH)-. -. -CH-CH2-. -CH2-Φ. 30. 19580. 4.71. -. -CH-. -C-CH2-. -CH2-Φ. 31. 2670. 5.57. -(CO)-. -CH2-CH2-. -CH-CH2-. -(CH2)2OCH3. 32. 53. 7.28. -(CO)-. -CH2-CH2-. -CH-CH2-. 33. 32. 7.49. -(CO)-. -CH2-CH2-. -CH-CH2-. 34. 28. 7.55. 35. 79. 7.10. NO2 O. O. O. -(CO)-. -CH2-CH2-. -CH-CH2-. -(CO)-. -CH2-CH2-. -CH-CH2- -CH2CH2-O-Φ. 36. 390. 6.41. -(CO)-. -CH2-CH2-. -CH-CH2-. 37. 1000. 6.00. -CH2-CN O. O O. O. S. +. N R1. Ligand IC50(nM) pIC50 ID. -R1 -CH3. 38. 900. 6.05. -CH2CH3. 39. 280. 6.55. -CH2CH=CH2. 40. 540. 6.27. 41. 110. 6.96. 42. 40. 7.40. 31.
(39) Table 1. Continued Ligand IC50(nM) pIC50 ID. -R1 -CH2CH2-O-CH2CH3. 43. 7. 8.15. 44. 2.6. 8.59. 45. 1000. 6.00. S. 46. 6. 8.22. NO2. 47. 4.5. 8.35. F. O O. O. O N. R1. +. Ligand IC50(nM) pIC50 ID. -R1 O. 48. 100. 7.00. 49. 41.5. 7.38. 50. 139. 6.86. 51. 50. 7.30. 52. 120. 6.92. 53. 22. 7.66. O. O O. O. O O O. N. N N. 32.
數據

+7



Outline
相關文件
Promote project learning, mathematical modeling, and problem-based learning to strengthen the ability to integrate and apply knowledge and skills, and make. calculated
Now, nearly all of the current flows through wire S since it has a much lower resistance than the light bulb. The light bulb does not glow because the current flowing through it
◦ 金屬介電層 (inter-metal dielectric, IMD) 是介於兩 個金屬層中間,就像兩個導電的金屬或是兩條鄰 近的金屬線之間的絕緣薄膜,並以階梯覆蓋 (step
Wang, Solving pseudomonotone variational inequalities and pseudocon- vex optimization problems using the projection neural network, IEEE Transactions on Neural Networks 17
volume suppressed mass: (TeV) 2 /M P ∼ 10 −4 eV → mm range can be experimentally tested for any number of extra dimensions - Light U(1) gauge bosons: no derivative couplings. =>
Define instead the imaginary.. potential, magnetic field, lattice…) Dirac-BdG Hamiltonian:. with small, and matrix
incapable to extract any quantities from QCD, nor to tackle the most interesting physics, namely, the spontaneously chiral symmetry breaking and the color confinement..
• Formation of massive primordial stars as origin of objects in the early universe. • Supernova explosions might be visible to the most
