Synthesis of Hyperbranched Aromatic Poly(amide-imide):
Copolymerization of B
′
B
2Monomer with A
2Monomer
Yao-Te Chang and Ching-Fong Shu*Department of Applied Chemistry, National Chiao Tung University, Hsin-Chu, Taiwan 30035, Republic of China
ABSTRACT: A hyperbranched aromatic poly(amide-imide) was prepared by the copolymerization of 4-(3,5-dicarboxyphenoxy)phthalic anhydride, a B′B2type monomer, and 1,4-phenylenediamine, an A2type monomer. The rapid reaction between the anhydride and amino group led to the formation of the dominant imide intermediate which can be regarded as a new AB2type of monomer. The intermediate, without isolation, was subjected to further polymerization in the presence of TPP/pyridine, as condensing agents, to give the hyperbranched poly(amide-imide), containing carboxylic acid chain ends. In comparison, the AB2monomer was prepared separately, and the conventional self-polymerization of this monomer was also studied. The structures of the obtained polymers were characterized by FTIR and 1H NMR spectroscopy. The spectral data showed that these two polymers, prepared from two different synthetic approaches, have nearly identical structures. The degree of branching of the hyperbranched poly(amide-imide)s was estimated to be 60-61%. The terminal carboxylic acid groups were modified by reaction with a variety of aromatic amines to give the corresponding amide derivatives. The nature of the chain ends was shown to have a significant effect on the solubility and Tgof the hyperbranched poly(amide-imide)s.
Introduction
Hyperbranched macromolecules have received con-siderable attention since their unique, highly branched structure is expected to exhibit some unusual physical and chemical properties.1-4 These polymers can be conveniently synthesized in a single step via a one-pot polymerization,5yet they maintain many of the archi-tectural features and properties found in their well-defined dendrimer counterparts,6-8which are built up by tedious stepwise synthetic sequences.9-12The one-step synthesis allows hyperbranched polymers to be more readily available and prepared on a large scale for potential applications. These attractive features have led to the development of novel synthetic routes for the preparation of such polymers. Most hyperbranched polymers that have been developed were synthesized by the one-step self-polycondensation of ABntype
mono-mers. Only a few studies have concerned the prepara-tion of hyperbranched polymers through the copolym-erization method, even if that route has more flexibility in generating diverse, hyperbranched materials. Fre´chet et al. have reported the synthesis of hyperbranched aliphatic polyethers by the copolymerization of A2and B3 type monomers.13 Using this A2 + B3 strategy, hyperbranched aromatic polyamides and polyimdes have been prepared by Kakimoto’s and Okamoto’s groups.14-16 Recently, Yan et al. have reported a new A2+ BB′2approach for the synthesis of hyperbranched copoly(sulfone-amine)s.17
Linear polyamides and polyimides are well-known as high-performance polymeric materials.18,19Their hyper-branched analogues have been prepared by the self-polymerization of AB2 type monomers20-25 or more recently by copolymerization methods.14-16 Aromatic poly(amide-imide)s are a class of high-performance polymers which may combine the advantages of poly-amides and polyimides; the resulting polymers should possess high thermal stabilities, coupled with good mechanical and chemical properties.26,27To the best of our knowledge, there has been no report regarding the
preparation of hyperbranched aromatic poly(amide-imide)s. In this paper, we report the synthesis of a new hyperbranched poly(amide-imide) by copolymerization of an unsymmetrical B′B2type monomer with an A2type monomer. In the B′B2 monomer, there are one anhy-dride and two carboxylic groups, while in the A2 monomer, there are two amino functionalities. The anhydride (B′) is much more reactive than the carboxylic group (B) toward the amino group in the A2monomer. At the initial stage of the polycondensation, the rapid reaction between B′and A groups results in the forma-tion of the dominant imide intermediate. The interme-diate can be regarded as a new AB2 type of monomer, which contains one aminophenyl and two carboxyl functionalities. Without isolation, the AB2intermediate was subjected to further polymerization in the presence of TPP/pyridine, as condensing agents, to give the hyperbranched poly(amide-imide).28,29For comparison, the AB2 intermediate was prepared separately and characterized; the conventional one-step self-polymer-ization of this AB2monomer was also studied.
Experimental Section
General Directions. N-Methyl-2-pyrrolidinone (NMP) and
N,N-dimethylformamide (DMF) were distilled over CaH2 under reduced pressure. Pyridine was dried by distillation after being refluxed with KOH. 1,4-Phenylenediamine was recrystallized from MeOH. Triphenyl phosphite (TPP) was purified by distillation under reduced pressure. (2,3-Dihydro-2-thioxo-3-benzoxazolyl)phosphonic acid diphenyl ester (DBOP) was used as received. All other reagents and solvents were used as received from commercial sources unless otherwise stated.1H and13C NMR spectra were recorded on a Varian Unity 300 MHz or a Bruker-DRX 300 MHz spectrometer. IR spectra were obtained on a Nicolet 360 FT-IR spectrometer. Mass spectra were obtained on a JEOL JMS-SX 102A mass spectrometer. Size exclusion chromatography (SEC) was car-ried out on a Waters chromatography unit, interfaced with a Waters 410 differential refractometer. Three 5 µm Waters styragel columns (300 × 7.8 mm), connected in series of decreasing order of pore size (105, 104, and 103Å), were used with DMF as the eluent. Standard samples of PMMA were 10.1021/ma021499f CCC: $25.00 © 2003 American Chemical Society
used for calibration. Differential scanning calorimetry (DSC) was performed using a Du Pont TA 2000 instrument, with a heating/cooling rate of 20 °C min-1. Samples were scanned from 30 to 400 °C and then cooled to 30 °C and scanned a second time from 30 to 400 °C. The glass transition temper-ature (Tg) was determined from the second heating scan. Thermogravimetric analyses (TGA) were made on a Du Pont TGA 2950 instrument. The thermal stability of the samples was determined in nitrogen by measuring weight loss while heating at a rate of 20 °C min-1.
Dimethyl 5-(3,4-Dicyanophenoxy)isophthalate (1). A mixture of dimethyl 5-hydroxyisophthalate (5.00 g, 26.9 mmol), 4-nitrophthalonitrile (4.65 g, 26.9 mmol), K2CO3(5.00 g, 36.2 mmol), and DMF (20 mL) was heated at 130 °C for 4 h. After this period, the reaction mixture was cooled and poured into 200 mL of water. The precipitated crude product was collected by filtration, dried under vacuum, and purified by recrystal-lization from ethyl acetate to afford 1 (8.20 g, 91.4%).1H NMR (DMSO-d6): δ 3.89 (s, 6H), 7.53 (dd, 1H, J ) 8.7, 2.7 Hz), 7.89 (d, 1H, J ) 2.7 Hz), 7.93 (d, 2H, J ) 1.5 Hz), 8.14 (d, 1H, J ) 8.7 Hz), 8.36 (t, 1H, J ) 1.5 Hz).13C NMR (DMSO-d6): δ 52.7, 109.2, 115.2, 115.7, 116.9, 122.8, 123.1, 125.2, 126.4, 132.5, 136.3, 154.4, 160.2, 164.5. HRMS [M+]: 336.0745. Calcd 336.0746 for C18H12N2O5.
4-(3,5-Dicarboxyphenoxy)phthalic Acid (2). To a solu-tion of KOH (14.0 g, 0.25 mol) in a mixture of water-ethanol (100 mL/80 mL) was added compound 1 (7.00 g, 28.3 mmol). The mixture was then refluxed for 24 h. The resulting solution was diluted with water (250 mL) and acidified with 6 N HCl-(aq) to pH∼1. The precipitated product was filtered, washed thoroughly with water, and dried to give 2 (7.00 g, 97.1%).1H NMR (DMSO-d6): δ 7.24 (dd, 1H, J ) 8.7, 2.7 Hz), 7.71 (d, 1H, J ) 2.7 Hz), 7.73 (d, 2H, J ) 1.5 Hz), 8.23 (d, 1H, J ) 8.7 Hz), 8.28 (t, 1H, J ) 1.5 Hz).13C NMR (DMSO-d6): δ 120.6, 121.5, 123.4, 125.6, 130.1, 133.9, 135.4, 137.8, 156.4, 157.8, 166.2, 167.4. HRMS [M+- H]: 345.0236. Calcd 345.0247 for C16H9O9. 4-(3,5-Dicarboxyphenoxy)phthalic Anhydride (3). A mixture of compound 2 (6.00 g, 17.3 mmol), acetic anhydride (6.0 mL), and acetic acid (6.0 mL) was stirred under reflux for 12 h. After cooling, the precipitate was collected by filtration, washed with a small amount of glacial acetic acid, and then dried under vacuum at 140 °C for 12 h to yield 3 (4.85 g, 85. %).1H NMR (DMSO-d6): δ 7.63 (dd, 1H, J ) 8.1, 2.1 Hz), 7.68 (d, 1H, J ) 2.1 Hz), 7.84 (d, 2H, J ) 1.2 Hz), 8.10 (d, 1H, J ) 8.1 Hz), 8.34 (t, 1H, J ) 1.5 Hz), 13.55 (br, 2H). 13C NMR (DMSO-d6): δ 114.2, 124.2, 125.7, 126.0, 126.6, 127.9, 134.1, 134.3, 155.2, 162.5, 162.6, 162.9, 165.9. HRMS [M+]: 328.0209. Calcd 328.0219 for C16H8O8.
N-(4-Aminophenyl)-4-(3,5-dicarboxyphenoxy)phthal-imide (4). To a solution of compound 3 (400 mg, 1.22 mmol) in NMP (4.0 mL) was added 1,4-phenylenediamine (132 mg, 1.22 mmol). The mixture was stirred at 25 °C under nitrogen for 2 h and then heated at 150 °C for 12 h. The resulting solution was cooled and then poured into dilute HCl(aq) (60 mL). The precipitated product was filtered, washed with water, and dried to give 4 (421 mg, 82.6%).1H NMR (DMSO-d6): δ 6.67 (d, 2H, J ) 8.4 Hz), 7.03 (d, 2H, J ) 8.4 Hz), 7.40-7.54 (m, 2H), 7.81 (d, 2H, J ) 1.2 Hz), 7.96 (d, 1H, J ) 8.9 Hz), 8.31 (s, 1H).13C NMR (DMSO-d6): δ 113.2, 114.3, 120.5, 123.9, 124.2, 125.8, 126.1, 126.8, 128.3, 133.6, 134.5, 147.7, 156.0, 161.4, 165.8, 166.7, 166.9. HRMS [M+]: 418.0785. Calcd 418.0801 for C22H14N2O7. N-(4-Aminophenyl)phthalimide (5). A solution of 1,4-phenylenediamine (3.00 g, 27.8 mmol) and phthalic anhydride (4.11 g, 27.8 mmol) in DMF (6.0 mL) was heated at 150 °C for 4 h. The solution was poured into water (200 mL). The precipitated product was filtered and dried to give 5 (5.95 g, 90.0%).1H NMR (DMSO-d6): δ 5.33 (s, 2H), 6.62 (d, 2H, J ) 8.4 Hz), 7.00 (d, 2H, J ) 8.4 Hz), 7.81-7.95 (m, 4H).13C NMR (DMSO-d6): δ 113.6, 119.6, 123.2, 128.2, 131.6, 134.5, 148.8, 167.6. HRMS [M+]: 238.0737. Calcd 238.0742 for C14H10N2O2. Synthesis of Model Compound 6. A solution of compound 3 (1.00 g, 3.05 mmol) and aniline (284 mg, 3.05 mmol) in NMP (6.0 mL) was heated at 150 °C under nitrogen for 12 h. The
solution was then added dropwise to water (200 mL). The precipitated product was collected by filtration and purified by recrystallization from acetic acid to afford 6 (1.01 g, 82.2%). 1H NMR (DMSO-d6): δ 7.38-7.58 (m, 7H), 7.82 (d, 2H, J ) 1.2 Hz), 8.01 (d, 1H, J ) 8.1 Hz), 8.32 (t, 1H, J ) 1.2 Hz), 13.51 (br, 2H).13C NMR (DMSO-d6): δ 113.3, 124.0, 124.3, 126.0, 126.2, 126.7, 127.4, 128.2, 129.0, 132.0, 133.7, 134.4, 155.9, 161.6, 165.9, 166.2, 166.4. HRMS [M+]: 403.0694. Calcd 403.0692 for C22H13NO7.
Synthesis of Model Compounds 7 and 8. A solution of 5 (531 mg, 2.24 mmol), 6 (600 mg, 1.49 mmol), DBOP (570 mg, 1.49 mmol), and 3 drops of triethylamine in NMP (6.0 mL) was stirred at 25 °C under nitrogen for 12 h. The resulting solution was added dropwise to a mixture of methanol (50 mL) and water (50 mL). The precipitated product was collected by filtration and purified by column chromatography, eluting with CHCl3/ethyl acetate (4:1) to give 8 (0.44 g, 35%). Further elution with CHCl3/CH3OH (3:1) and recrystallization from acetic acid afforded 7 (0.31 g, 33%). Compound 7: 1H NMR (DMSO-d6): δ 7.37-7.61 (m, 9H), 7.80 (br, 1H), 7.88-8.10 (m, 8H), 8.47 (br, 1H), 10.69 (s, 1H), 12.68 (br, 1H).13C NMR (DMSO-d6): δ 113.1, 120.9, 122.8, 123.3, 123.4, 124.1, 124.9, 126.0, 126.6, 127.4, 127.5, 127.7, 128.1, 128.9, 131.6, 131.9, 133.5, 134.5, 134.7, 137.3, 138.5, 155.6, 161.7, 163.8, 166.1, 166.2, 166.4, 167.1. HRMS [M+ + H]: 624.1415. Calcd 624.1407 for C36H22N3O8. Compound 8: 1H NMR (DMSO-d6):
δ 7.40-7.63 (m, 9H), 7.82-8.10 (m, 9H), 8.55 (br, 1H), 10.59 (s, 2H).13C NMR (DMSO-d6): δ 113.4, 121.5, 122.7, 124.1, 124.4, 124.6, 126.7, 127.1, 127.9, 128.3, 128.7, 129.5, 132.3, 132.7, 135.1, 135.4, 138.1, 139.2, 156.0, 162.7, 164.8, 166.8, 166.9, 167.7. HRMS [M++ H]: 844.2046. Calcd 844.2044 for C50H30N5O9.
Preparation of Polymer PAI-1. (A) Direct polymerization of monomer 3 with 1,4-phenylenediamine: The monomer 3 (200 mg, 610 µmol) was added in one portion to a stirred solution of 1,4-phenylenediamine (65.9 mg, 610 µmol) in NMP (2.0 mL) under nitrogen at 25 °C. Stirring was continued at 25 °C for 2 h and at 150 °C for 12 h. The solution was cooled, added pyridine (0.50 mL) and TPP (0.32 mL, 1.22 mmol), and then heated at 120 °C for 12 h. After cooling, the resulting polymer was precipitated into methanol. The polymer was collected, washed with hot water, purified by reprecipitation from DMF into methanol twice, and dried under vacuum to give PAI-1 (202 mg, 83.2%). (B) Self-polycondensation of monomer 4: A solution of compound 4 (800 mg, 1.61 mmol), TPP (1.0 mL, 3.2 mmol), and LiCl (160 mg, 3.77 mmol) in NMP/pyridine (10.0 mL, 4:1 v/v) was heated at 120 °C under nitrogen for 12 h. After cooling, the resulting polymer was precipitated into methanol. The polymer was collected, washed with hot water, purified by reprecipitation from DMF into methanol twice, and dried under vacuum to give PAI-1 (732 mg, 95.6%).1H NMR (DMSO-d6): δ 7.30-7.68 (m, 4H), 7.75-8.08 (m, 5H), 8.28-8.56 (m, 1H), 10.69 (s, 1H), 13.51 (br, 1H). IR (KBr): 2500-3500 (broad, O-H and N-H), 1772, 1716 (CdO, imide ring), 1665 (CdO, amide), 1373 cm-1(C-N, imide).
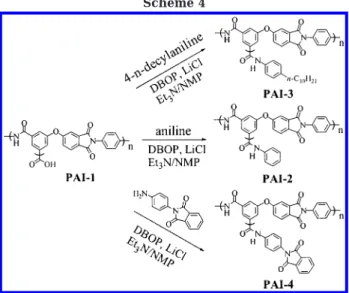
Preparation of Polymer PAI-2. A solution of PAI-1 (100 mg, 250 µmol), aniline (85 mg, 0.91 mmol), DBOP (383 mg, 1.00 mmol), and 3 drops of triethylamine in NMP (2.0 mL) was stirred at 25 °C under nitrogen for 12 h. The resulting solution was added dropwise to methanol (100 mL) with agitation. The polymer was collected by filtration, washed with warm water, and purified by reprecipitation from DMF into methanol twice to give PAI-2 (114 mg, 96.0%). 1H NMR (DMSO-d6): δ 6.80-7.65 (m, 8H), 7.70-8.10 (m, 6H), 8.42-8.58 (m, 1H), 10.48 (br, 1H), 10.68 (br, 1H). IR (KBr): 1772, 1716 (CdO, imide ring), 1665 (CdO, amide), 1373 cm-1 (C-N, imide).
Preparation of Polymer PAI-3. PAI-3 was prepared from PAI-1 (100 mg, 250 µmol) and 4-n-decylaniline (233 mg, 1.00 mmol) following the same procedure as described for the preparation of PAI-2. Yield: 138 mg, 89.8%.1H NMR
(DMSO-d6): δ 0.55-1.62 (m, 21H), 6.80-7.80 (m, 9H), 7.82-8.20 (m, 4H), 8.40-8.56 (m, 1H), 10.39 (br, 1H), 10.69 (br, 1H). IR (KBr): 1772, 1716 (CdO, imide ring), 1665 (CdO, amide), 1373 cm-1(C-N, imide).
Preparation of Polymer PAI-4. PAI-4 was prepared from PAI-1 (100 mg, 250 µmol) and compound 5 (238 mg, 1.00 mmol) following the same procedure as described for the preparation of PAI-2. Yield: 146 mg, 94.2%.1H NMR
(DMSO-d6): δ 7.30-7.68 (m, 6H), 7.80-8.08 (m, 11H), 8.54 (br, 1H), 10.69 (br, 2H). IR (KBr): 1772, 1716 (CdO, imide ring), 1665 (CdO, amide), 1378 cm-1(C-N, imide).
Results and Discussion
Monomer Synthesis. Two kinds of monomers, 3 and 4, were prepared as shown in Scheme 1. Nucleophilic substitution of the nitro function of 4-nitropthalonitrile with the phenoxide of dimethyl 5-hydroxyisophthalate in a K2CO3/DMF medium yielded dimethyl 5-(3,4-dicyanophenoxy)isophthalate (1). Alkaline hydrolysis of compound 1 in KOH(aq)/ethanol gave 4-(3,5-dicarboxy-phenoxy)phthalic acid (2), which was subsequently dehydrated with acetic anhydride to form the B′B2type monomer 3. The condensation of 3 with 1,4-phenylene-diamine followed by cyclodehydration afforded the AB2 type monomer 4. Monomer 3 has one phthalic anhydride and two carboxylic acid groups, while monomer 4 contains one aminophenyl and two carboxyl function-alities joined with an ether-imide linkage. The struc-tures of the synthesized compounds were verified by1H NMR and 13C NMR spectroscopy as well as by mass spectroscopy. Figure 1 shows the 1H NMR spectra of monomers 3 and 4, which are consistent with the assigned structures.
Polymer Synthesis. As shown in Scheme 2, the hyperbranched poly(amide-imide) PAI-1 was prepared by the direct polymerization of the B′B2monomer 3 with the A2monomer 1,4-phenylenediamine. An equimolar
mixture of the monomer 3 and 1,4-phenylenediamine was reacted in NMP to form the amic acid, which was then thermally cyclized to give the AB2-type intermedi-ate in situ, with an imide linkage. It should be noted that the amino group in the AB2molecule is deactivated in comparison with the original amino groups in the A2 monomer, due to the formation of the imide (amide) group in the para position of the benzene ring system.14 Therefore, the possibility of reaction of the former with the phthalic anhydride (B′) of a second B′B2molecule is diminished and can be neglected under the reaction conditions; the formation of the AB2intermediate is the predominate reaction at the initial stage of the copo-lymerization. This is supported by the fact that the AB2 intermediate, which is identical to the AB2type mono-mer 4, can be isolated with an almost quantitative yield in the first stage of the polymerization. Without isola-tion, the AB2type intermediate was polymerized at 120 °C for 12 h in the presence of TPP/pyridine, as condens-ing agents, to yield PAI-1. No gelation occurred durcondens-ing this polymerization. This polymer could also be synthe-sized by the self-polycondensation of the AB2 type monomer 4. The one-step polymerization of 4 was carried out in NMP at 120 °C for 12 h using TPP/ pyridine as condensing agents. The structure of PAI-1 was characterized by FTIR and1H NMR spectroscopy. It was found that the two different synthetic approaches led to hyperbranched poly(amide-imide)s with almost identical structures. In the IR spectra, the polymers exhibit characteristic carbonyl absorptions correspond-ing to an imide rcorrespond-ing at 1772 (CdO asymmetric stretch-ing) and 1716 cm-1 (CdO symmetric stretching), in addition to the typical aromatic-imide C-N stretching at 1373 cm-1. The carbonyl absorptions of the cyclic anhydride unit at 1849 and 1783 cm-1 in monomer 3 have disappeared. In addition, the CdO stretching of the amide group appears at 1665 cm-1. The carbonyl stretching of the terminal carboxylic acid group at ∼1700 cm-1is obscured by the absorption of the imide ring. The broad bands between 2500 and 3500 cm-1are attributed to amide N-H and acid O-H stretches. In the1H NMR spectrum of PAI-1s, the resonance of the proton of the amide group appears at 10.69 ppm, and a broad peak at 13.51 ppm from the proton of the terminal carboxylic acid group is also observed.
The hyperbranched poly(amide-imide), PAI-1, which has a high number of terminal carboxyl groups, could not be analyzed directly by gel permeation chromatog-raphy (GPC) because the polymer adsorbed to the column, resulting in incomplete elution.30This problem was overcome by the transformation of the carboxylic acids groups into amide groups. By reacting with excess aniline, PAI-1 was readily converted to the amide-terminated polymer PAI-2 (vide post). GPC analyses showed that the weight-average molecular weights (Mw) and polydispersities (Mw/Mn) were approximately 4.4× Scheme 1
Figure 1. 1H NMR spectra of compounds (a) 3 and (b) 4 in DMSO-d6.
104g/mol and 2.7, respectively, for the amide-terminated polymer of PAI-1 prepared from the direct polymeriza-tion of the B′B2 with the A2 monomers,. The amide derivative of PAI-1, prepared by the self-polyconden-sation of the AB2 type monomer 4 under the same conditions, has similar Mwand Mw/Mn values: 4.1 × 104 g/mol and 2.5. These values, however, are only indicative, since the highly branched nature of PAI-1 deviates strongly from that of linear, coillike poly-(methyl methacrylate) standards. The solution viscosity of PAI-1s was also evaluated in DMAc at a concentra-tion of 0.5 g/dL at 30 °C. The inherent viscosity (ηinh) was 0.29 and 0.28 g/dL respectively for the hyper-branched polymers PAI-1s prepared by the copolymer-ization method and by the self-polymercopolymer-ization of mono-mer 4. The comparison of the solution viscosity reveals that there is no cross-linking taken place in the direct copolymerization. This is consistent with the observa-tion that no gelaobserva-tion occurred during the copolymeri-zation.
Degree of Branching. Hyperbranched polymers, which were formed by a sequence of condensation of AB2 monomers, have an irregular dendritic structure in which three different types of subunits may be present. In this work, the hyperbranched poly(amide-imide) PAI-1 is composed of three kinds of repeating units: the terminal units, which have two carboxylic acid groups, the linear units, which have one amide group and one carboxylic acid group, and the dendritic units, which have two amide groups and no free carboxylic acid group. The degree of branching (DB), a typical property often used to evaluate the structural irregularity of hyperbranched polymers, was defined as the sum of dendritic and terminal units vs the total number of units.31,32 A combination of NMR spectroscopy and comparative studies on a model compound was used to quantify the different subunits present in the hyper-branched polymer and, subsequently, to determine its DB.31
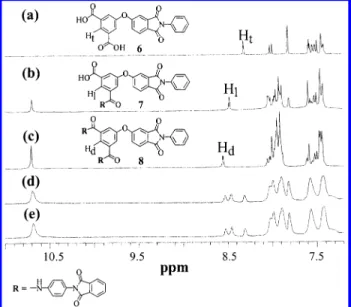
The preparation of the model compounds useful for NMR characterization is illustrated in Scheme 3. Model compounds 6, 7, and 8 resemble the terminal unit, the linear unit, and the dendritic unit, respectively. Their
1H NMR spectra are depicted in Figure 2. A distinct resonance for the terminal model compound, 6, appears at 8.32 ppm (Ht), whereas resonances of the correspond-ing protons for the linear and dendritic model com-pounds, 7 (Hl), and 8 (Hd), are observed at 8.47 and 8.55 ppm, respectively. Figure 2 also shows the 1H NMR spectra of the hyperbranched poly(amide-imide)s. In comparing the1H NMR spectra of PAI-1s with those of the model compounds, one can clearly assign the resonances at 8.32, 8.47, and 8.55 ppm to the proton of the terminal, linear, and dendritic subunits in the hyperbranched polymer, respectively. On the basis of the integration of these well-resolved resonances, the relative percentages of each subunit in PAI-1s can be determined. It was noted that the percentage estimated for the terminal subunit is approximately equal to that for the dendritic subunit. This result is consistent with the theoretical prediction that the number of dendritic units is equal to the number of terminal units for an AB2type hyperbranched polymer possessing high mo-lecular weight.5With these formulas, the DB of the poly-(amide-imide) PAI-1, prepared by the direct polymer-ization of monomer 3 with 1,4-phenylenediamine, is estimated to be 61%. This value is nearly the same as the DB calculated for the PAI-1 synthesized by the self-polycondensation of the AB2type monomer 4, which is 60%.
Chemical Modification. As predicted theoretically by Flory,5the direct polymerization of AB
ntype
mono-mers is expected to produce a highly branched, irregular structure that contains a large number of end groups. In this study, the end groups in the resulting hyper-branched poly(amide-imide) PAI-1 are carboxylic acids, which could be readily functionalized to yield hyper-branched polymers with a variety of functional chain ends. As shown in Scheme 4, the modification of the carboxylic end groups of PAI-1 was carried out with different kinds of aromatic amines, in the presence of DBOP as the condensation agent, to give the corre-sponding amide derivatives. We chose DBOP as the activator33and ran the condensation reaction at room temperature to prevent transamination, which is a side reaction occurring at high temperatures. In the modi-Scheme 3
Figure 2. 1H NMR spectra in DMSO-d
6of model compounds (a) 6, (b) 7, and (c) 8 compared with the hyperbranched poly-(amide-imide)s PAI-1s prepared by either the (d) self-polym-erization of monomer 4 or (e) the copolymself-polym-erization method.
fication reaction, the use of excess amino reagents resulted in almost complete (>95%) functionalization, as confirmed by the1H NMR spectra of the derivative. After end-capping, the broad peak at 13.51 ppm, as-sociated with the proton of the terminal carboxylic acid group, disappeared. For PAI-2-3, an additional reso-nance due to the proton of the terminal amide group appears in the region of 10.4-10.5 ppm. It is interesting to note that for PAI-4 the signals corresponding to the proton of the terminal (Ht), linear (Hl), and dendritic (Hd) subunits shown in Figure 2 coalesce at 8.54 ppm. This is attributed to the similarity of the chemical environments for the three different subunits in PAI-4. This result is consistent with the observation that, in PAI-4, the chemical shift of the proton of the terminal amide group is the same as that of the interior amide group. Thus, there is only one resonance with an integration of 2 H for the protons of the two different kinds of amides. The integration analyses of1H NMR spectra of the derivatives indicate that the modification reactions proceeded almost quantitatively.
The nature of the end groups influences the physical and chemical properties of hyperbranched polymers.34 The glass transition temperatures (Tg’s) of poly(amide-imide)s PAI-1-4 were investigated by differential scan-ning calorimetry (DSC), and the results are presented in Figure 3 and Table 1. As reported, the transition from the polar function to nonpolar end groups results in a decrease in Tg due to the reduction in the extent of
intermolecular interactions in the polymeric mol-ecules.35 The T
g of PAI-1, which has polar carboxylic terminal groups, is 262 °C, while the Tgvalue of PAI-2, which has less polar amide terminal groups, is 248 °C. A further decrease in Tgto 223 °C was observed for PAI-3, due to the introduction of long, flexible n-decyl chain ends. PAI-4, which has rigid, imide chain ends, has a Tg of 283 °C. Because of their highly branched structures, these poly(amide-imide)s have enhanced solubility in organic solvents and are highly soluble in polar solvents such as DMAc, NMP, and pyridine (Table 1). However, the different chain ends led to solubility differences in very polar and in relatively nonpolar solvents. PAI-1-2 and PAI-4 are soluble in DMSO and insoluble in THF, whereas PAI-3, which possesses long
n-decyl chain ends, is soluble in hot DMSO as well as
in THF. Summary
We have synthesized a hyperbranched poly(amide-imide) via the A2+ B′B2approach. The copolymerization of 4-(3,5-dicarboxyphenoxy)phthalic anhydride (3), a B′B2type monomer, and 1,4-phenylenediamine, an A2 type monomer, resulted in a hyperbranched poly-(amide-imide) containing terminal carboxylic acid groups. No gelation occurred during the polymerization. For comparison, we also carried out the synthesis of this hyperbranched polymer via the conventional self-polymerization of N-(4-aminophenyl)-4-(3,5-dicarboxy-phenoxy)phthalimide (4), an AB2 monomer. Both ap-proaches led to hyperbranched poly(amide-imide)s with almost identical structures. The degree of branching of the polymers was approximately 60%, as determined by a combination of model compound studies and1H NMR integration data. End-capping of the terminal carboxylic acid groups was accomplished by reacting with different kinds of aromatic amines. Thermal and solubility prop-erties of the resulting polymers depend on the nature of the chain-end groups.
Acknowledgment. We thank the National Science Council of the Republic of China for financial support. References and Notes
(1) Malmstro¨m, E.; Hult, A. J. Macromol. Sci., Rev. Macromol.
Chem. Phys. 1997, C37, 555.
(2) Kim, Y. H. J. Polym. Sci., Part A: Polym. Chem. 1998, 36, 1685.
(3) Voit, B. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 2505. (4) Jikei, M.; Kakimoto, M. Prog. Polym. Sci. 2001, 26, 1233 and
references cited in the reviews.
(5) Flory, P. J. J. Am. Chem. Soc. 1952, 74, 2718. (6) Fre´chet, J. M. J. Science 1994, 263, 1710.
(7) Newkome, G. R.; Moorefield, C. N.; Vo¨gtle, F. Dendritic
Molecules: Concepts, Syntheses, Perspectives; VCH:
Wein-heim, FRG, 1996. Scheme 4
Figure 3. DSC thermograms of hyperbranched poly(amide-imide)s (a) PAI-1 prepared by self-polymerization of monomer 4, (b) 1 prepared by the copolymeriztion method, (c) PAI-2, (d) PAI-3, and (e) PAI-4.
Table 1. Thermal and Solubility Properties of Hyperbranched Poly(amide-imide)s
solubilitycin
polymer Tg(°C) THF DMF DMAc DMSO pyridine
PAI-1a 262 - + + + +
PAI-1b 263 - + + + +
PAI-2 248 - + + + +
PAI-3 223 + + + + - +
PAI-4 283 - + + + +
aPrepared by the copolymerization approach.bPrepared by
self-polymerization of monomer 4.cSolubility: +, soluble; -, insoluble;
(8) Hecht, S.; Fre´chet, J. M. J. Angew. Chem., Int. Ed. 2001, 40, 74.
(9) Tomalia, D. A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Polym. J. 1985,
17, 117.
(10) Newkome, G. R.; Yao, Z.-Q.; Baker, G. R.; Gupta, V. K. J.
Org. Chem. 1985, 50, 2003.
(11) Hawker, C. J.; Fre´chet, J. M. J. J. Am. Chem. Soc. 1990, 112, 7638.
(12) Xu, Z. F.; Moore, J. S. Angew. Chem., Int. Ed. Engl. 1993,
32, 1354.
(13) Emrick, T.; Chang, H.-T.; Fre´chet, J. M. J. Macromolecules 1999, 32, 6380.
(14) Jikei, M.; Chon, S.-H.; Kakimoto, M.; Kawauchi, S.; Imase, T.; Watanebe, J. Macromolecules 1999, 32, 2061.
(15) Fang, J.; Kita, H.; Okamoto, K. Macromolecules 2000, 33, 4639.
(16) Hao, J.; Jikei, M.; Kakimoto, M. Macromolecules 2002, 35, 5372.
(17) (a) Yan, D.; Gao, C. Macromolecules 2000, 33, 7693. (b) Gao, C.; Yan, D. Macromolecules 2001, 34, 156. (c) Gao, C.; Yan, D. Chem. Commun. 2001, 107. (d) Gao, C.; Yan, D.; Zhu, X.; Huang, W. Polymer 2001, 42, 7603. (e) Gao, C.; Yan, D.; Tang, W. Macromol. Chem. Phys. 2001, 202, 3035.
(18) Cassidy, P. E. Thermally Stable Polymer; Marcel Dekker: New York, 1980.
(19) Polymides; Wilson, D., Stenzenberger, H. D., Hergenrother, P. M., Eds.; Blackie: New York, 1990.
(20) Kim, Y. H. J. Am. Chem. Soc. 1992, 114, 4947.
(21) (a) Yang, G.; Jikei, M.; Kakimoto, M. Macromolecules 1998,
31, 5964. (b) Yang, G.; Jikei, M.; Kakimoto, M. Macromol-ecules 1999, 32, 2215. (c) Jikei, M.; Fujii, K.; Yang, G.;
Kakimoto, M. Macromolecules 2000, 33, 6228. (d) Jikei, M.;
Fujii, K.; Yang, G.; Kakimoto, M. J. Polym. Sci., Part A:
Polym. Chem. 2001, 39, 3304.
(22) Monticilli, O.; Mendichi, R.; Bisbano, S.; Mariani, A.; Russo, S. Macromol. Chem. Phys. 2000, 201, 2123.
(23) Yamanaka, K.; Jikei, M.; Kakimoto, M. Macromolecules 2000,
33, 1111. (b) Yamanaka, K.; Jikei, M.; Kakimoto, M. Macro-molecules 2000, 33, 6937. (c) Yamanaka, K.; Jikei, M.;
Kakimoto, M. Macromolecules 2001, 34, 3910.
(24) Thompson, D. S.; Markoski, L. J.; Moore, J. S. Macromolecules 1999, 32, 4764. (b) Markoski, L. J.; Thompson, J. L.; Moore, J. S. Macromolecules 2000, 33, 5315.
(25) Wu, F.-I.; Shu, C.-F. J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 2536.
(26) Liaw, D.-J.; Hsu, P.-N.; Chen, W.-H.; Liaw, B.-Y. J. Polym.
Sci., Part A: Polym. Chem. 2001, 39, 3498.
(27) Yang, C.-P.; Chen, R.-S.; Wang, M.-J. J. Polym. Sci., Part A:
Polym. Chem. 2002, 40, 1092.
(28) Yamazaki, N.; Matsumoto, M.; Higashi, F. J. Polym. Sci.,
Polym. Chem. Ed. 1975, 13, 1373.
(29) Tullos, G. L.; Mathias, L. J.; Langsam, M. J. Polym. Sci., Part
A: Polym. Chem. 1999, 37, 1183.
(30) Shu, C.-F.; Leu, C.-M. Macromolecules 1999, 32, 100. (31) Hawker, C. J.; Lee, R.; Fre´chet, J. M. J. J. Am. Chem. Soc.
1991, 113, 4583.
(32) Ho¨lter, D.; Burgath, A.; Frey, H. Acta Polym. 1997, 48, 30. (33) Ueda, M.; Maneyama, A.; Hashimoto, K. Macromolecules
1988, 21, 18.
(34) Hawker, C. J.; Chu, F. Macromolecules 1996, 29, 4370. (35) Schmaljohann, D.; Ha¨ussler, L.; Po¨tschke, P.; Voit, B. I.;
Loontjens, T. J. A. Macromol. Chem. Phys. 2000, 201, 49.