Introduction
Ellis-van Creveld (EvC) syndrome (OMIM 225500), or chondroectodermal dysplasia, is an autosomal reces-sive ciliary disorder associated with a wide spectrum of developing abnormalities involving the ectoderm, skeleton and heart. EvC syndrome is a relatively rare
E
LLIS
-
VAN
C
REVELD
S
YNDROME
: P
RENATAL
D
IAGNOSIS
, M
OLECULAR
A
NALYSIS AND
G
ENETIC
C
OUNSELING
Chih-Ping Chen1,2,3,4,5,6*, Yi-Ning Su7, Chin-Yuan Hsu1, Schu-Rern Chern2, Fuu-Jen Tsai4,8, Pei-Chen Wu1, Po-Tsang Chen2, Wayseen Wang2,9
Departments of 1Obstetrics and Gynecology and 2Medical Research, Mackay Memorial Hospital, 5Institute of Clinical and
Community Health Nursing, 6Department of Obstetrics and Gynecology, National Yang-Ming University, 7Department of Medical Genetics, National Taiwan University Hospital, and 9Department of Bioengineering, Tatung University, Taipei; 3Department of Biotechnology, Asia University, 4School of Chinese Medicine, College of Chinese Medicine, China Medical
University, and 8Departments of Medical Genetics and Medical Research, China Medical University Hospital, Taichung, Taiwan.
SUMMARY
Objective:To present the perinatal findings and molecular genetic analysis of two siblings with Ellis-van Creveld (EvC) syndrome.
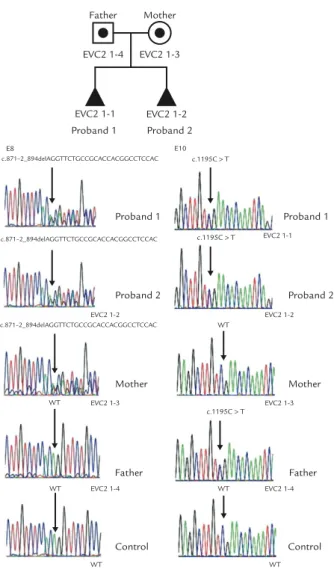
Materials, Methods and Results:A 33-year-old woman, gravida 3, para 1, was referred for genetic counseling at 18 gestational weeks because of recurrent fetal skeletal dysplasia. Two years previously, she had delivered a 1,316-g dead male baby at 28 gestational weeks with a karyotype of 46,XY, postaxial polydactyly of the hands, thoracic narrowness, endocardial cushion defects, transposition of the great arteries, shortening of the long bones, malposition of the toes, and hypoplastic nails. During this pregnancy, prenatal ultrasound at 18 gesta-tional weeks revealed shortening of the long bones (equivalent to 15 weeks), postaxial polydactyly of both hands, thoracic narrowness, and endocardial cushion defects. The pregnancy was subsequently terminated, and a 236-g female fetus was delivered with a karyotype of 46,XX, postaxial polydactyly of the hands, thoracic dysplasia, endocardial cushion defects, shortening of the long bones, and malposition of the toes and hypoplastic nails. The phenotype of each of the two siblings was consistent with EVC syndrome. Molecular analysis of the EVC and EVC2 genes revealed heterozygous mutations in the EVC2 gene. A heterozygous deletion mutation of a 26-bp deletion of c.871-2_894del26 encompassing the junction between intron 7 and exon 8 of the EVC2 gene was found in the mother and two siblings, and a heterozygous nonsense mutation of c.1195C> T, p.R399X in exon 10 of the EVC2 gene was found in the father and two siblings.
Conclusion:Prenatal sonographic identification of endocardial cushion defects in association with shortening of the long bones should alert clinicians to the possibility of EvC syndrome and prompt a careful search of hexa-dactyly of the hands. Molecular analysis of the EVC and EVC2 genes is helpful in genetic counseling in cases with prenatally detected postaxial polydactyly, thoracic narrowness, short limbs and endocardial cushion defects. [Taiwan J Obstet Gynecol 2010;49(4):481–486]
Key Words:Ellis-van Creveld syndrome, EVC, EVC2, prenatal diagnosis, ultrasound
*Correspondence to: Dr Chih-Ping Chen, Department
of Obstetrics and Gynecology, Mackay Memorial Hospital, 92, Section 2, Chung-Shan North Road, Taipei, Taiwan.
E-mail: [email protected] Accepted: September 29, 2010
disorder, but is most prevalent in the Amish popula-tion [1,2] and in some Arab populapopula-tions [3] because of consanguinity. The birth prevalence is estimated to be 0.7 per 100,000 of live births in the non-Amish population [4], 5.2 per 100,000 of live births in the United Arab Emirates [3], and 5 per 1,000 of live births in the Amish of Lancaster County, Pennsylvania, USA [5]. EvC syndrome is characterized by short ribs, short limbs, postaxial polydactyly of the hands, poly-dactyly of the feet (in 10% of cases), ectodermal dys-plasia such as dysplastic nails and teeth, sparse hair and an absent gingival sulcus, and congenital heart defects (in 60% of cases) such as a common atrium, atrioventricular septal defects (AVSDs) and patent ductus arteriosus [6,7]. EvC syndrome is caused by mutations in the EVC gene (OMIM 604831) [8] or
EVC2 gene (OMIM 607261) [9] that encodes
cilia-related proteins Evc or Evc2, respectively. Mutations in the EVC gene or EVC2 gene may also cause Weyers acrodental dysostosis (OMIM 193530), an autosomal dominant disorder characterized by postaxial poly-dactyly and abnormalities of the lower jaw, dentition
Figure 1. Whole body X-ray of proband 1 at 28 gestational weeks.
A B
Figure 2.Postaxial polydactyly of the hands in proband 1.
and oral vestibule. The Evc and Evc2 proteins are localized in the basal bodies of primary cilia. Evc is a basal body component of hedgehog signaling indis-pensable for normal endochondral growth and normal transcriptional activation of Indian hedgehog-regulated genes [10]. Both EvC syndrome and Weyers acroden-tal dysostosis are caused by hedgehog signaling de-fects in the primary cilia due to mutations in the cilia-related proteins resulting in an aberrant response to the hedgehog ligands [7]. We previously reported perinatal findings of hexadactyly-associated ciliary dis-orders of Meckel syndrome [11], Joubert syndrome [12], and short rib-polydactyly syndrome (SRPS) [13–16]. We present the perinatal findings and molecular genetic analysis of two siblings affected by hexadactyly and EvC syndrome.
Materials, Methods and Results
A 33-year-old woman, gravida 3, para 1, was referred for genetic counseling at 18 gestational weeks because of recurrent fetal skeletal dysplasia. She and her hus-band were non-consanguineous. She had experienced one spontaneous abortion and delivered a baby with skeletal dysplasia. Two years previously, she had deliv-ered a 1,316-g dead male baby (proband 1) at 28 ges-tational weeks with a karyotype of 46,XY, postaxial polydactyly of the hands, thoracic narrowness, endo-cardial cushion defects, transposition of the great arteries (TGA), shortening of the long bones, malposi-tion of the toes and hypoplastic nails (Figures 1–3). During this pregnancy, prenatal ultrasound at 18 ges-tational weeks revealed shortening of the long bones (equivalent to 15 weeks), postaxial polydactyly of both hands, thoracic narrowness, and endocardial cushion defects (Figures 4–6). The pregnancy was subsequently terminated, and a 236-g female fetus (proband 2) was delivered with a karyotype of 46,XX, postaxial poly-dactyly of the hands, thoracic dysplasia, endocardial
A B
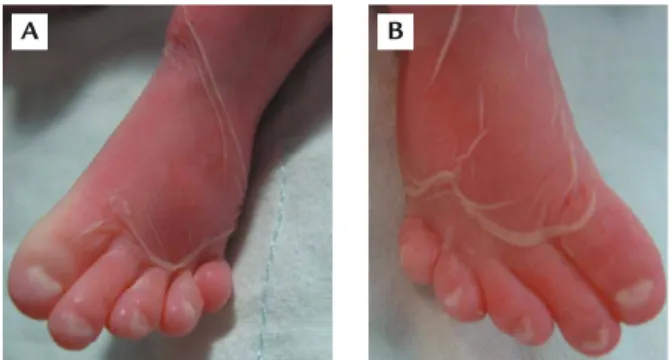
Figure 3.Malposition of the toes with hypoplastic nails in proband 1.
cushion defects, shortening of the long bones, malpo-sition of the toes and hypoplastic nails (Figures 7–10). The phenotype of each of the two siblings is consistent with EvC syndrome. Molecular analysis of the EVC and
EVC2 genes showed heterozygous mutations in the
EVC2 gene. A heterozygous deletion mutation of a
26-bp deletion of c.871-2_894del26 encompassing the junction between intron 7 and exon 8 of the EVC2 gene was found in the mother and two siblings, and a het-erozygous nonsense mutation of c.1195C> T, p.R399X
Figure 4.Prenatal ultrasound shows thoracic narrowness in proband 2.
Figure 5. Prenatal ultrasound shows hexadactyly of the hands in proband 2.
Figure 6. Prenatal ultrasound shows endocardial cushion defects in proband 2.
Figure 7.Whole body X-ray of proband 2 at 18 gestational weeks.
Figure 8.Proband 2 at birth.
A B
in exon 10 of the EVC2 gene was found in the father and two siblings (Figure 11).
Discussion
The present case prenatally manifested shortening of the long bones, thoracic dysplasia, hexadactyly of the hands, and AVSD on the second-trimester ultrasound. Prenatal diagnosis of EvC syndrome by direct visualiza-tion of the fetus using fetoscopy has previously been reported [17,18]. First-trimester transabdominal embry-ofetoscopy for the detection of limb or facial abnor-malities has recently been applied to detect skeletal dysplasia such as SRPS [19]. Recurrent EvC syndrome can be diagnosed in the first trimester by the ultra-sound findings of AVSD, polydactyly and short limbs [20] as well as increased fetal nuchal translucency thickness [21]. In the second trimester, the diagnosis of EvC syndrome can be made based on a positive family history and the ultrasound findings of shortness of the long bones, hexadactyly of the hands, a narrow thorax and congenital heart defects, especially abnor-malities of atrial septation and AVSD [22–28].
Our first proband manifested AVSD and TGA, and our second proband manifested AVSD on prenatal ultrasound. Congenital heart defects occur in approxi-mately 60% of patients with EvC syndrome with most patients being variants of AVSD [1]. Atrioventricular septation of the mammalian heart into four chambers requires sonic hedgehog signaling-dependent cellular contributions from the extracardiac tissues of the dor-sal mesocardium as well as contributions from the muscular and mesenchymal atrial septum and the endocardial cushions [29]. Sund et al [30] found that the expression of EVC and EVC2 mRNA and proteins
Figure 10.Malposition of the toes with hypoplastic nails in proband 2. Proband 1 Proband 2 c.871–2_894delAGGTTCTGCCGCACCACGGCCTCCAC c.871–2_894delAGGTTCTGCCGCACCACGGCCTCCAC c.871–2_894delAGGTTCTGCCGCACCACGGCCTCCAC EVC2 1-2 EVC2 1-3 EVC2 1-4 Mother Father Control WT WT WT E8 EVC2 1-1 EVC2 1-2 EVC2 1-3 EVC2 1-4 Proband 1 Proband 2 Mother Father Control c.1195C > T c.1195C > T c.1195C > T E10 WT WT WT EVC2 1-2 EVC2 1-3 EVC2 1-4 EVC2 1-1 Proband 1 Proband 2 Father Mother
Figure 11.A heterozygous deletion mutation of the 26-bp deletion encompassing the junction between intron 7 and exon 8 of the EVC2 gene (c.871-2_894del26) in proband 1, proband 2 and the mother, but not in the father, and a heterozygous nonsense mutation in exon 10 of the EVC2 gene (c.1195C> T, CGA> TGA, Arg399Stop, R399X) in proband 1, proband 2 and the father, but not in the mother. WT= wild type.
was high in the outflow tract and dorsal mesenchymal protrusion and was also present in the mesenchymal structures of the atrial septum and the endocardial cushions. This finding suggested that Evc and Evc2 proteins function coordinately in cardiac development and that loss of this coordinate function results in characteristic EvC syndrome.
In the present case, we identified heterozygous
EVC2 mutations in the affected fetuses. The nonsense
mutation of C1195T, R399X, has been described pre-viously [31]. However, the deletion mutation of c.871-2_894del26 is novel. Sequencing the EVC and EVC2 genes has been reported to identify mutations in only two-thirds of EvC patients, indicating the possibility of genetic heterogeneity in EvC syndrome [32]. In a study of 65 individuals with EvC syndrome, Tompson et al
[32] identified EVC mutations in 20 cases (31%), all of whom had mutations on each allele, found EVC2 muta-tions in 25 cases (38%), 22 of whom had mutamuta-tions on each allele, and three had only one mutation, and there was no mutation in either gene in 20 cases (31%). The three patients in their study with an EVC2 mutation on only one allele had a frameshift or a nonsense codon and a more severe phenotype than Weyers acrodental dysostosis.
There is a risk of 25% recurrence in subsequent pregnancies in fetal EvC syndrome. Genetic counseling of fetal EvC syndrome should include differential diag-noses of SRPS, Jeune asphyxiating thoracic dystrophy (JATD), and McKusick-Kaufman syndrome (MKKS). SRPSs are a heterogeneous group of lethal autosomal recessive skeletal dysplasias. Four types of SRPS have been recognized [33]. Type I SRPS (Saldino-Noonan) (OMIM 263530) is characterized by flipper-like extrem-ities, polydactyly, polycystic kidneys and pointed meta-physes. Type II SRPS (Majewski) (OMIM 263520) is characterized by polydactyly, micromelia, cleft lip/palate, polycystic kidneys, a disproportionately short ovoid tibia and occasionally hypoplastic epiglottis and larynx. Type III SRPS (Verma-Naumoff) (OMIM 263510) is characterized by polydactyly, micromelia, metaphyseal spurs and occasionally situs inversus totalis. Type IV SRPS (Beemer-Langer) (OMIM 269860) clinically resembles type II SRPS other than polydactyly. Overlapping clinical and radiological manifestations have led to the hypoth-esis that the different subtypes may be a single genetic disorder with variable expressivity [34–37]. Type III SRPS is caused by mutations of the DYNC2H1 gene (OMIM 603297) [38,39]. JATD (OMIM 208500) is an autoso-mal recessive disorder characterized by thoracic dys-trophy, chondrodysplasia, short ribs, short long bones, inconstant polydactyly and a trident acetabular roof with occasional involvement of the liver, retinal degen-eration, and cystic renal disease. JATD is caused by muta-tions of the IFT80 gene (OMIM 611177) and DYNC2H1 gene [38–40]. JATD and type III SRPS have been sug-gested to be variants of a single ciliary disorder [38]. MKKS (OMIM 236700) is an autosomal recessive dis-order characterized by polydactyly, congenital heart defects, hydrometrocolpos, and it clinically overlaps with Bardet-Biedl syndrome (OMIM 209900) comprising obesity, retinitis pigmentosa, polydactyly, mental retardation, renal malformation and genital hypoplasia. MKKS and Bardet-Biedl syndrome can be caused by mutations of the MKKS gene (OMIM 604896) [41,42]. In summary, we have presented prenatal diagnosis, molecular analysis and genetic counseling of recurrent EvC syndrome. Prenatal sonographic identification of en-docardial cushion defects in association with shortening
of the long bones should alert clinicians to the possibil-ity of EvC syndrome and prompt a careful search of hexa-dactyly of the hands. Molecular analysis of the EVC and
EVC2 genes is helpful in genetic counseling in cases with
prenatally detected postaxial polydactyly, thoracic nar-rowness, short limbs, and endocardial cushion defects.
Acknowledgments
This work was supported by research grants NSC-96-2314-B-195-008-MY3 and NSC-97-2314-B-195-006-MY3 from the National Science Council, and MMH-E-99004 from Mackay Memorial Hospital, Taipei, Taiwan.
References
1. McKusick VA, Egeland JA, Eldridge R, Krusen DE. Dwarfism in the Amish. I. The Ellis-van Creveld syndrome. Bull Johns
Hopkins Hosp 1964;115:306–36.
2. McKusick VA. Ellis-van Creveld syndrome and the Amish.
Nat Genet 2000;24:203–4.
3. Al-Gazali LI, Bakir M, Hamid Z, et al. Birth prevalence and pattern of osteochondrodysplasias in an inbred high risk pop-ulation. Birth Defects Res A Clin Mol Teratol 2003;67A:125–32. 4. Stoll C, Dott B, Roth MP, Alembik Y. Birth prevalence rates
of skeletal dysplasias. Clin Genet 1989;35:88–92.
5. Zangwill KM, Boal DK, Ladda RL. Dandy-Walker malformation in Ellis-van Creveld syndrome. Am J Med Genet 1988;31:123–9. 6. Baujat G, Le Merrer M. Ellis-van Creveld syndrome. Orphanet
J Rare Dis 2007;2:27.
7. Ruiz-Perez VL, Goodship JA. Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to hedgehog ligands. Am J Med Genet C
Semin Med Genet 2009;151C:341–51.
8. Ruiz-Perez VL, Ide SE, Strom TM, et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet 2000;24:283–6.
9. Galdzicka M, Patnala S, Hirshman MG, Cai JF, Nitowsky H, Egeland JA, Ginns EI. A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Mol Genet Metab 2002;77:291–5. 10. Ruiz-Perez VL, Blair HJ, Rodriguez-Andres ME, et al. Evc is a
positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development 2007;134: 2903–12.
11. Chen CP. Meckel syndrome: genetics, perinatal findings and differential diagnosis. Taiwan J Obstet Gynecol 2007;46:9–14. 12. Chen CP, Su YN, Huang JK, et al. Fetal magnetic resonance
imaging demonstration of central nervous system abnor-malities and polydactyly associated with Joubert syndrome.
Taiwan J Obstet Gynecol 2010;49:243–5.
13. Chen CP, Tzen CY. Short rib-polydactyly syndrome type III (Verma-Naumoff) in a third-trimester fetus with unusual asso-ciations of epiglottic hypoplasia, renal cystic dysplasia, pyelec-tasia and oligohydramnios. Prenat Diagn 2001;21:1101–2.
14. Chen CP, Chang TY, Tzen CY, Lin CJ, Wang W. Sonographic detection of situs inversus totalis, ventricular septal defect, and short rib-polydactyly syndrome type III (Verma-Naumoff) in a second-trimester fetus not known to be at risk.
Ultrasound Obstet Gynecol 2002;19:629–31.
15. Chen CP, Chang TY, Tzen CY, Wang W. Second-trimester sonographic detection of short rib-polydactyly syndrome type II (Majewski) following an abnormal maternal serum biochemical screening result. Prenat Diagn 2003;23:353–5. 16. Chen CP, Shih JC, Tzen CY, Lin YH, Wang W. Recurrent
short-rib polydactyly syndrome: prenatal three-dimensional ultrasound findings and associations with congenital high air-way obstruction and pyelectasia. Prenat Diagn 2005;25:417–8. 17. Mahoney MJ, Hobbins JC. Prenatal diagnosis of chon-droectodermal dysplasia (Ellis-van Creveld syndrome) with fetoscopy and ultrasound. N Engl J Med 1977;297:258–60. 18. Bui TH, Marsk L, Eklöf O, Theorell K. Prenatal diagnosis of
chondroectodermal dysplasia with fetoscopy. Prenat Diagn 1984;4:155–9.
19. Lee K, Lee JW, Chay DB, et al. Transabdominal embryofe-toscopy for the detection of short rib-polydactyly syndrome, type II (Majewski), in the first trimester. J Korean Med Sci 2006; 21:165–8.
20. Dugoff L, Thieme G, Hobbins JC. First trimester prenatal diagnosis of chondroectodermal dysplasia (Ellis-van Creveld syndrome) with ultrasound. Ultrasound Obstet Gynecol 2001; 17:86–8.
21. Venkat-Raman N, Sebire NJ, Murphy KW, Carvalho JS, Hall CM. Increased first-trimester fetal nuchal translucency thickness in association with chondroectodermal dysplasia (Ellis-van Creveld syndrome). Ultrasound Obstet Gynecol 2005; 25:412–4.
22. Horigome H, Hamada H, Sohda S, Oyake Y, Kurosaki Y. Prenatal ultrasonic diagnosis of a case of Ellis-van Creveld syndrome with a single atrium. Pediatr Radiol 1997;27:942–4. 23. Guschmann M, Horn D, Gasiorek-Wiens A, Urban M, Kunze J, Vogel M. Ellis-van Creveld syndrome: estimation at 15 weeks’ gestation. Prenat Diagn 1999;19:879–83. 24. Tongsong T, Chanprapaph P. Prenatal sonographic diagnosis
of Ellis-van Creveld syndrome. J Clin Ultrasound 2000;28:38–41. 25. George E, DeSilva S, Lieber E, Raziuddin K, Gudavalli M. Ellis van Creveld syndrome (chondroectodermal dysplasia, MIM 22550) in three siblings from a non-consanguineous mating. J Perinat Med 2000;28:425–7.
26. Sergi C, Voigtlander T, Zoubaa S, et al. Ellis-van Creveld syndrome: a generalized dysplasia of enchondral ossification.
Pediatr Radiol 2001;31:289–93.
27. Parilla BV, Leeth EA, Kambich MP, Chilis P, MacGregor SN. Antenatal detection of skeletal dysplasias. J Ultrasound Med 2003;22:255–8.
28. Witters I, Moerman P, Fryns JP. Skeletal dysplasias: 38 pre-natal cases. Genet Couns 2008;19:267–75.
29. Goddeeris MM, Rho S, Petiet A, Davenport CL, Johnson GA, Meyers EN, Klingensmith J. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development 2008;135:1887–95. 30. Sund KL, Roelker S, Ramachandran V, Durbin L, Benson
DW. Analysis of Ellis van Creveld syndrome gene products: implications for cardiovascular development and disease.
Hum Mol Genet 2009;18:1813–24.
31. Ruiz-Perez VL, Tompson SWJ, Blair HJ, et al. Mutation in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet 2003;72:728–32. 32. Tompson SWJ, Ruiz-Perez VL, Blair HJ, et al. Sequencing
EVC and EVC2 identifies mutations in two-thirds of Ellis-van
Creveld syndrome patients. Hum Genet 2007;120:663–70. 33. Lachman RS. Skeletal dysplasia. In: Taybi H, Lachman RS,
eds. Radiology of Syndromes, Metabolic Disorders, and Skeletal
Dysplasias, 4thedition. St. Louis: Mosby, 1996:745–951. 34. Martínez-Frías M-L, Bermejo E, Urioste M, Huertas H,
Arroyo I. Lethal short rib polydactyly syndromes: further evidence for their overlapping in a continuous spectrum.
J Med Genet 1993;30:937–41.
35. Sarafoglou K, Funai EF, Fefferman N, et al. Short rib-polydactyly syndrome: more evidence of a continuous spec-trum. Clin Genet 1999;56:145–8.
36. Ho NC, Francomano CA, van Allen M. Jeune asphyxiating thoracic dystrophy and short-rib polydactyly type III (Verma-Naumoff) are variants of the same disorder. Am J
Med Genet 2000;90:310–4.
37. Elçioglu NH, Hall CM. Diagnostic dilemmas in the short rib-polydactyly syndrome group. Am J Med Genet 2002;111: 392–400.
38. Dagoneau N, Goulet M, Geneviève D, et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am J Hum Genet 2009;84: 706–11.
39. Merrill AE, Merriman B, Farrington-Rock C, et al. Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome. Am J
Hum Genet 2009;84:542–9.
40. Beales PL, Bland E, Tobin JL, et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet 2007;39:727–9. 41. Slavotinek AM, Stone EM, Mykytyn K, et al. Mutations in MKKS cause Bardet-Biedl syndrome. Nat Genet 2000;26:15–6. 42. Stone DL, Slavotinek A, Bouffard GG, Banerjee-Basu S, Baxevanis AD, Barr M, Biesecker LG. Mutation of a gene encoding a putative chaperonin causes McKusick-Kaufman syndrome. Nat Genet 2000;26:79–82.