For Peer Review
Helicobacter pylori attenuates lipopolysaccharide-induced nitric oxide production by murine macrophages
Journal: Innate Immunity Manuscript ID: INI-11-0004.R1 Manuscript Type: Original Manuscript Date Submitted by the
Author: 24-Apr-2011
Complete List of Authors: Lu, Dah-Yuu; China Medical University, Graduate Institute of Neural and Cognitive Sciences
Tang, Chih-Hsin; China Medical University, Graduate Institute of Basic Medical Science and School of Medicine
Chang, Chia-Hsian; China Medical University, Graduate Institute of Basic Medical Science and School of Medicine
Maa, Ming-Chei; China Medical University, Graduate Institute of Basic Medical Science and School of Medicine
Fang, Shih-Hua; National Taiwan Sport University, Institute of Athletes
Lin, Hwai-Jeng; Taipei Medical University Hospital, Division of Gastroenterology and Hepatology
Lee, Che-Hsin; China Medical University, Graduate Institute of Basic Medical Science and School of Medicine
Lai, Chih-Ho; China Medical University, Graduate Institute of Basic Medical Science and School of Medicine
Keywords: Lipopolysaccharide, Nitric oxide, Helicobacter pylori, Macrophage, Nuclear factor (NF)-kappa B
Abstract:
Intragastric growth of Helicobacter pylori (H. pylori) and non-Helicobacter microorganisms is thought to be associated with elevated levels of proinflammatory cytokines and the production of nitric oxide (NO); these effects can lead to chronic inflammation. Microorganisms can activate the expression of inducible nitric oxide
For Peer Review
synthase (iNOS) and the production of NO by macrophages through stimulation with bacterial lipopolysaccharide (LPS). H. pylori can evade these vigorous immune responses, but the underlying mechanism remains unknown. In this study, we used a murine model of macrophage infection to demonstrate that H. pylori inhibits LPS-induced expression of iNOS and production of NO by macrophages. Suppression of LPS-induced NO production by macrophages led to elevated survival of H. pylori in a trans-well system. This effect was abrogated in macrophages from iNOS-/- mice. Analysis of iNOS mRNA and protein levels revealed that H. pylori inhibits iNOS expression at both transcriptional and post-transcriptional levels, and these effects occurred with live bacteria. Furthermore, the effect of H. pylori involved down-regulation of the mitogen-activated protein kinase pathway and the translocation of active nuclear factor (NF)-kappa B into the nucleus. Taken
together, our results reveal a new mechanism by which H. pylori modulates the innate immune responses of the host and maintains a persistent infection within the stomach.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59
For Peer Review
Helicobacter
pylori
attenuates
lipopolysaccharide-induced
nitric
oxide
production by murine macrophages
Running title: H. pylori modulates LPS-induced NO production
Dah-Yuu Lu1, Chih-Hsin Tang2, Chia-Hsian Chang2†, Ming-Chei Maa2, Shih-Hua Fang3,Yuan-Man Hsu4, Yu-Hsin Lin4, Chun-Jung Lin2, Wan-Chi Lee2, Hwai-Jeng Lin5, Che-Hsin Lee2, and Chih-Ho Lai2,6*
1
Graduate Institute of Neural and Cognitive Sciences, China Medical University, Taichung, Taiwan
2
Graduate Institute of Basic and Clinical Medical Science, School of Medicine, China Medical University,
Taichung, Taiwan
3
Institute of Athletes, National Taiwan Sport University, Taichung, Taiwan
4
Department of Biological Science and Technology, China Medical University, Taichung, Taiwan
5
Division of Gastroenterology and Hepatology, Taipei Medical University Hospital, Taipei, Taiwan
6
Department of Urology, University of Texas Southwestern Medical Center, Dallas, Texas
†
Contributed equally to the first author Correspondence to:
Chih-Ho Lai, Ph.D.
Graduate Institute of Basic and Clinical Medical Science, School of Medicine
China Medical University
No. 91, Hsueh-Shih Road, Taichung, 40402 Taiwan
Telephone: 886-4-22052121 ext. 7729; Fax: 886-4-22333641
E-mail: [email protected] 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
ABSTRACT
Intragastric growth of Helicobacter pylori (H. pylori) and non-Helicobacter microorganisms is
thought to be associated with elevated levels of proinflammatory cytokines and the production of
nitric oxide (NO); these effects can lead to chronic inflammation. Microorganisms can activate the
expression of inducible nitric oxide synthase (iNOS) and the production of NO by macrophages
through stimulation with bacterial lipopolysaccharide (LPS). H. pylori can evade these vigorous
immune responses, but the underlying mechanism remains unknown. In this study, we used a
murine model of macrophage infection to demonstrate that H. pylori inhibits LPS-induced
expression of iNOS and production of NO by macrophages. Suppression of LPS-induced NO
production by macrophages led to elevated survival of H. pylori in a trans-well system. This effect
was abrogated in macrophages from iNOS–/– mice. Analysis of iNOS mRNA and protein levels
revealed that H. pylori inhibits iNOS expression at both transcriptional and post-transcriptional
levels, and these effects occurred with live bacteria. Furthermore, the effect of H. pylori involved
down-regulation of the mitogen-activated protein kinase pathway and the translocation of active
nuclear factor (NF)-kappa B into the nucleus. Taken together, our results reveal a new mechanism
by which H. pylori modulates the innate immune responses of the host and maintains a persistent
infection within the stomach.
Keywords: Lipopolysaccharide, nitric oxide, Helicobacter pylori, macrophage, nuclear factor
(NF)-kappa B 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
INTRODUCTION
Helicobacter pylori is the most common causative agent of gastrointestinal disease in humans.
Infection with this pathogen usually occurs in childhood, and the bacteria can persist in the stomach
for an individual’s lifetime.1, 2 Persistent infection with H. pylori in the gastric mucosa induces the
expression of nuclear factor (NF)-κB and the secretion of proinflammatory cytokines, including
interleukin (IL)-1β, IL-6, IL-8, and tumor necrosis factor-α.3, 4 Other inflammatory mediators, such
as nitric oxide (NO), a bactericidal agent generated by inducible nitric oxide synthase (iNOS)
during the conversion of L-arginine to L-citrulline, are activated by H. pylori infection in both
macrophages 5 and the gastric epithelium.6 These findings indicate that H. pylori is an important
factor for the induction of proinflammatory cytokines and NO in the host stomach.
Nitric oxide is derived from iNOS in lipopolysaccharide (LPS)-activated macrophages during
inflammatory responses. Following treatment of macrophages with LPS, the NF-κB heterodimer
rapidly translocates to the nucleus where it activates the transcription of target genes, including
iNOS and several proinflammatory cytokines.7 In addition, p38 mitogen-activated protein kinase
(MAPK), protein kinase C, and extracellular signal–regulated kinase (ERK) are also involved in the
activation of NF-κB and the expression of iNOS in response to LPS.8, 9 Several microorganisms
disrupt the activation of MAPKs or the NF-κB signaling pathway in macrophages to evade immune
attack.10-14 The effect of H. pylori on the modulation of LPS-activated molecules in macrophages
remains unknown.
In addition to H. pylori, non-Helicobacter microorganisms are found in the gastric
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
environment.15, 16 One study reported that H. pylori–associated gastritis was associated with the
presence of several other microbes in the stomach, including Enterococcus, Pseudomonas,
Streptococcus, Staphylococcus, and Stomacoccus.17 A more recent study identified 128 phylotypes
in 23 gastric biopsy samples; however, the presence of H. pylori did not affect the composition of
microbiota in the gastric microbial community.18 These findings indicate that H. pylori and
non-Helicobacter microorganisms are present in the microbiota of the human stomach, and these
microbes can elicit proinflammatory mediators and induce vigorous immune responses.19 These
findings also raise the question of how H. pylori persists in the microbial ecosystem under the harsh
environment of the stomach.
The aim of the present study was to address the question of how H. pylori evades the vigorous
antimicrobial activities of macrophages. We established an in vitro murine model system and an ex
vivo murine model system to examine whether this bacterium could suppress LPS-induced NO
production through the MAPK or the NF-κB signaling pathway. We showed that H. pylori inhibits
iNOS expression and NO production by murine macrophages stimulated with a high dose of LPS.
We further demonstrated that H. pylori down-regulates the LPS-induced activation of
phosphorylated p38, ERK1/2, and NF-κB, and it subsequently suppresses LPS-induced macrophage
responses. Thus, our study reveals that H. pylori attenuates LPS-induced NO production in
macrophages and consequently evades early host immune responses.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
MATERIALS AND METHODS
Antibodies and reagents
Polyclonal rabbit anti-iNOS, anti-phosphorylated c-Jun-N-terminal kinase (p-JNK), and
anti-α-tubulin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Mouse monoclonal antibodies specific for p38 MAPK, stress-activated protein kinase (SAPK)/JNK,
and p44/42 (ERK 1/2) were purchased from Cell Signaling (Beverly, MA, USA). Mouse
monoclonal anti-phosphorylated p38 MAPK, and the anti-phosphorylated MAPK 1/2 (ERK1/2)
(Thr185/Tyr187) antibodies were purchased from Upstate (Billerica, MA, USA). LPS (Escherichia
coli O55: B5) and aminoguanidine hemisulfate (AG) were purchased from Sigma-Aldrich (St.
Louis, MO, USA). SB203580 (p38 inhibitor), PD98059 (ERK inhibitor), and SP600125 (JNK
inhibitor) were purchased from Calbiochem (San Diego, CA, USA). The AP-1-Luc and NF-κB-Luc
plasmids were purchased from Stratagene (San Diego, CA, USA). The iNOS promoter construct
(piNOSLuc) was a kind gift from Dr. E. A. Ratovitski (Johns Hopkins University, Baltimore, MD,
USA). The pSV-β-galactosidase vector and the luciferase assay kit were purchased from Promega
(Madison, WI, USA). All other reagents were obtained from Sigma-Aldrich.
Bacterial strains, cell culture and mice
H. pylori 26695 (ATCC 700392) was used as a reference strain. The cagA or vacA isogenic mutants
derived from H. pylori 26695 were constructed as described.20 H. pylori strains were recovered
from frozen stocks on Brucella agar plates (Becton Dickinson, Franklin Lakes, NJ, USA) containing
10% sheep blood. H. pylori strains were stored and cultivated as described,21 and H. pylori extracts
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
were prepared as described.22 Heated-killed H. pylori was obtained by boiling 1 × 109/ml of bacteria
suspended in PBS for 30 min. Crude H. pylori extracts were prepared by sonicating 1 × 109/ml of
bacteria suspended in PBS for 5 min on ice. Crude extracts were then centrifuged at 16,000 × g for
5 min at 4° C. The supernatant was filtered through a 0.22-µm filter and used for further analysis.
RAW 264.7 cells (ATCC TIB-71) were cultured in RPMI 1640 medium (Invitrogen, Carlsbad,
CA, USA). De-complement fetal bovine serum (10%; HyClone, Logan, UT, USA) was added to the
culture medium. For bacterial infection experiments, the cell culture medium was not supplemented
with antibiotics.
Male wild-type C3H/HeN and TLR4-deficient C3H/HeJ mice at ages 6 to 8 weeks were kindly
provided by Dr. Ai-Li Shiau (Departments of Microbiology and Immunology, National Cheng
Kung University Medical College). C57BL/6 iNOS knockout (C57BL/6-Nostm1Lau) (iNOS–/–)and
wild-type mice at ages 6 to 8 weeks were kindly provided by Dr. Ming-Chei Maa (Graduate
Institute of Basic Medical Science, China Medical University). Mice were maintained in the animal
center of China Medical University (Taichung, Taiwan). All procedures were performed according
to the “Guide for the Care and Use of Laboratory Animals” (NRC, USA) and were approved by the
animal experiment committee of China Medical University.
Preparation of murine peritoneal exudate macrophages (PEMs)
C57BL/6 iNOS knockout (C57BL/6-Nostm1Lau) (iNOS–/–)and wild-type mice of the same age and
gender were used to assess therole of iNOS in H. pylori–induced suppression of LPS-induced NO
production by macrophages. Murine PEMs were obtained after euthanasia by lavaging each mouse
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
with 10 ml of cold PBS 3 days after intraperitoneal injection of 2 ml of 3% thioglycolate in PBS.
Two hours after seeding the cells in culture plates, the non-adherent cells were removed by washing
with PBS, and the adherent cells were used for further experiments.
Mouse inoculations
C3H/HeN (n = 6) and C3H/HeJ (n = 6) mice of 6–8 weeks of age were intragastrically inoculated
with H. pylori. All mice were maintained under fasting for 24 h before inoculation. The protocol of
administration of mouse with LPS was performed as described with slight modifications.23 Each
mouse was administered 1 × 109 CFU/ml of H. pylori and purified LPS (75 µg, phenol extracted
from Escherichia coli O55: B5, Sigma-Aldrich) by intragastric gavage for 3 consecutive days. Six
hours after the final inoculation with H. pylori, the mice were fed with standard food and water and
housed for 1 week. On the 7th day after infection, 6 mice in each group were sacrificed, and the
number of H. pylori in their stomachs was determined by plating on Brucella blood agar plates and
expressed as CFU/g tissue.
Immunoblotting
H. pylori–infected cells were washed three times with PBS and boiled in sodium dodecyl sulfate
(SDS)-polyacrylamide gel electrophoresis (PAGE) sample bufferfor 10 min. The samples were then
resolvedby 10% SDS-PAGE and transferred onto polyvinylidene difluoridemembranes (Millipore,
Billerica, MA, USA). The membranes were incubated with primary antibodies and then with
horseradish peroxidase–conjugated secondary antibodies (Invitrogen). The proteins of interestwere
visualized with ECLTM western blotting reagents (GE Healthcare, Buckinghamshire, UK) and were
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
detected by exposure to X-ray film (Kodak, Boca Raton, FL, USA).
Reverse transcription and quantitative real time-PCR
Total RNA was extracted from PEMs using TRIzol reagent (Invitrogen), and 1 µg of total RNA was
reverse transcribed into cDNA using the oligo(dT) primer. Quantitative real-time PCR using SYBR
Green I Master Mix and a model 7900 Sequence Detector System was conducted according to the
manufacturer's instructions (Applied Biosystems, Foster City, CA, USA). After preincubation at
50ºC for 2 min and 95ºC for 10 min, PCR was performed with 40 cycles of 95ºC for 10 s and 60ºC
for 1 min. The threshold was set above the non-template control background and within the linear
phase of target gene amplification in order to calculate the cycle number at which the transcript was
detected (denoted as CT). The oligonucleotide primers were: iNOS, forward,
5’-CCCAGAGTTCCAGCTTCTGG-3’, and reverse, 5’-CCAAGCCCCTCACCATTATCT-3’; and
GAPDH, forward, 5’-CTCAACTACATGGTCTACATGTTCCA-3’, and reverse,
5’-CTTCCCATTCTCAGCCTTGACT-3’.
Bacterial survival assay
Bacterial survival was assessed in cultures of H. pylori–exposed, LPS-treated macrophages using a
trans-well system, as described22 with slight modification. Briefly, murine PEMs were cultured in
the bottom layer of trans-well plates (Corning, Corning, NY, USA). After 48 h, 1 × 106 H. pylori
were added to the insert membrane (0.1 µm pore size) and co-incubated for an additional 6 h in
culture. The bacteria on the insert membrane were then resuspended and cultured by serial dilution
onto Brucella blood agar plates. Colonies were counted after 4 to 5 days of incubation. Colony
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
forming units (CFU) were used to determine anti-bacterial effects.
Determination of nitric oxide production and cell viability assay
NO production was estimated from the accumulation of nitrite (NO2−), a stable end product of NO
metabolism, in the culture medium, using the Griess reagent (Sigma-Aldrich).24 The MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to measure the
effects of LPS and H. pylori on macrophage viability.25 RAW 264.7 cells or PEMs were infected
with various multiplicities of infection (MOI) of H. pylori for 24 or 48 h, respectively. Cell viability
was then measured by examining the ability of viable cells to chemically reduce MTT to formazan,
which was quantified by measurement of optical density at 570 nm.
Transfection and reporter gene assay
RAW 264.7 cells were grown to 90% confluency in a 12-well plate and transfected with
NF-κB-Luc, AP-1-Luc, or iNOS-Luc reporter plasmid using Lipofectamine 2000 (Invitrogen).26, 27
After 24 h, cells were incubated without or with LPS and then infected with H. pylori during an
additional 24 h culture. To prepare cell lysates, 100 µl of reporter lysis buffer (Promega) was added
to each well, and cells were scraped from dishes. An equal volume of luciferase substrate was added
to all samples, and luminescence was measured using a microplate luminometer (Biotek, Winooski,
VT, USA). Luciferase activity was normalized to the transfection efficiency as determined by
co-transfection of the β-galactosidase expression vector (Promega).28
Immunofluorescence labeling of phosphorylated p65
To visualize H. pylori–induced inhibition of the translocation of phosphorylated p65 into the
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
nucleus of macrophages, RAW 264.7 cells were seeded onto cover-slips and treated without or with
LPS for 2 h, and then with H. pylori for an additional 1 h incubation at 37ºC. Cells were fixed in
3.7% (w/v) paraformaldehyde and permeabilized with 0.5% (v/v) Triton X-100 in PBS for 2 min.
For labeling of p65, cells were incubated for 30 min with rabbit polyclonal anti-p65 (H-286; Santa
Cruz Biotechnology) and propidium iodide (Calbiochem). Cells were then incubated with a
secondary antibody, fluorescein isothiocyanate–conjugated anti-mouse IgG (Chemicon), and they
were fixed in paraformaldehyde. Fixed cells were mounted and observed with a confocal laser
scanning microscope (Zeiss LSM 510, Carl Zeiss, Göttingen, Germany). The quantification of
fluorescence intensity for p65 was analyzed by ZEN software (Carl Zeiss).
Statistical analysis
The Student's t test was used to calculate statistical significance; a P value of <0.05was considered
significant.
RESULTS
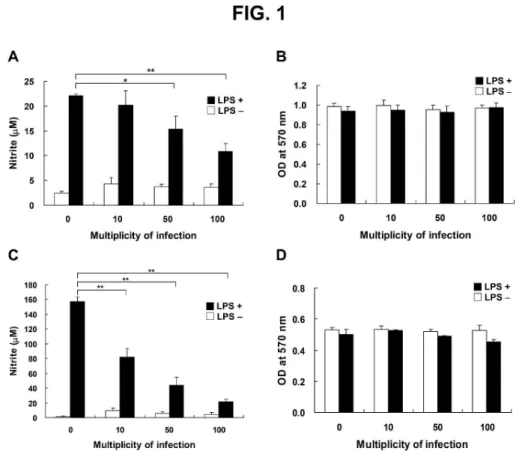
H. pylori inhibits LPS-induced NO production by macrophages
To assess whether H. pylori inhibits LPS-induced NO production by macrophages, mouse
macrophage RAW 264.7 cells were cultured with LPS (2 µg/ml) and infected with H. pylori at MOI
of 0 to 100 for 24 h. Nitric oxide production, measured by nitrite levels, was not suppressed when
LPS-stimulated RAW 264.7 cells were infected with H. pylori at a low MOI of 10 (Fig. 1A). NO
production by LPS-stimulated cells was decreased, however, at MOI of 50 to 100. When cells were
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
infected with H. pylori at MOI of 100, LPS-induced NO production was reduced by approximately
50%. Macrophage viability, as determined by the MTT assay, did not change after 24 h of
incubation with LPS and H. pylori (Fig. 1B).
To further delineate the suppressive effects of H. pylori on LPS-induced NO production,
peritoneal exudate macrophages (PEMs) were prepared and co-incubated with LPS and H. pylori
for 48 h. This ex vivo approach showed that LPS treatment induced NO production in uninfected
PEMs (MOI of 0; Fig. 1C), and LPS-induced NO production was reduced in an MOI-dependent
manner when PEMs were infected with H. pylori (Fig. 1C). The viability of PEMs, as determined
using the MTT assay, was hardly influenced by treatment with LPS and H. pylori at different MOI
(Fig. 1D). Thus, H. pylori inhibited, in an MOI-dependent manner, LPS-induced NO production not
only in RAW 264.7 cells but also in murine primary PEMs, and PEMs were more sensitive than the
macrophage cell line to the suppressive effects of H. pylori.
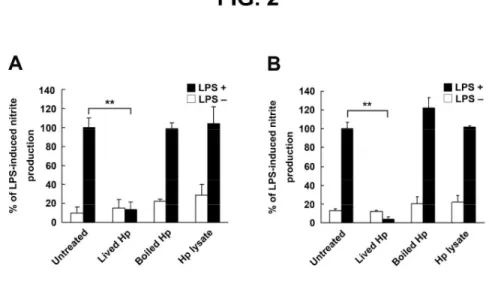
Live H. pylori is essential for the inhibition of LPS-induced NO production
To determine the functional role of H. pylori in inhibiting LPS-induced NO production by RAW
264.7 cells, live bacteria, heat-killed bacteria, and crude bacterial extracts were tested for their
ability to inhibit LPS-induced NO production. As shown in Fig. 2A, live H. pylori attenuated
LPS-induced NO production by RAW 264.7 cells. In contrast to the effects observed with live
bacteria, neither heat-killed bacteria nor crude bacterial extracts inhibited LPS-induced NO
production. We also used PEMs to study the effects of H. pylori on the inhibition of LPS-induced
NO production. Consistent with the results for RAW 264.7 cells, LPS-induced NO production by
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
PEMs was inhibited by live H. pylori but not by heat-killed bacteria or crude bacterial extracts (Fig.
2B). We further investigated the H. pylori–derived components that are responsible for these effects.
Virulence factor isogenic mutants of H. pylori, ∆cagA and ∆vacA, were tested for their ability to inhibit LPS-induced NO production by RAW 264.7 cells. Both isogenic mutants suppressed
LPS-induced NO production, similar to that observed with wild-type H. pylori (Fig. 3).
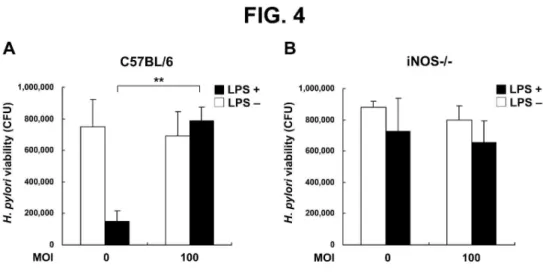
To mimic the environment of the bacterial infection, we further analyzed the ability of H.
pylori to inhibit LPS-induced NO production and to enhance the survival of bacteria adjacent to
infected macrophages in vitro. Using a trans-well culture system to assay bacterial survival of
LPS-stimulated murine PEMs, we found that H. pylori at MOI of 100 increased bacterial viability
(Fig. 4A). To test the hypothesis that H. pylori triggers iNOS activity, which is necessary for NO
production, we tested the effects of H. pylori in C57BL/6 iNOS–/– mice. H. pylori viability was
similar without or with LPS and with H. pylori at MOI of 0 or 100 in PEMs from iNOS–/– mice,
unlike the effects observed in wild-type mice (Fig. 4B). These results revealed that the ability of H.
pylori to inhibit LPS-induced NO production resulted from a reduction in the antimicrobial activity
of macrophages with subsequent enhancement of H. pylori survival.
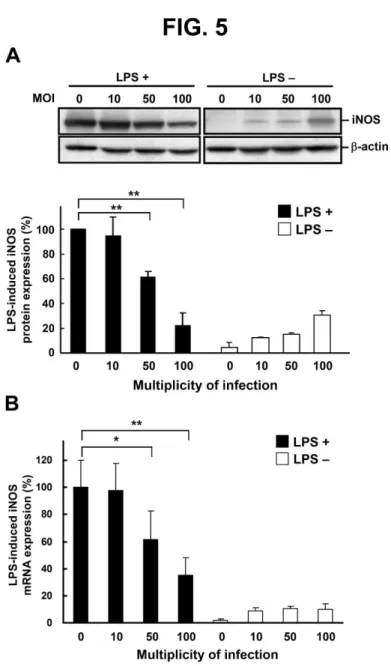
H. pylori inhibits LPS-induced iNOS expression at transcriptional and post-transcriptional levels
To investigate the effects of H. pylori on the regulation of iNOS expression, PEMs were treated (or
left untreated) with LPS and exposed to H. pylori at various MOI for 48 h. Subsequently, iNOS
protein levels were analyzed by western blotting. H. pylori infection led to a significant decrease, in
an MOI-dependent manner, in LPS-induced iNOS level (Fig. 5A). When cells were infected with H.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
pylori at MOI of 100, LPS-induced iNOS expression decreased by approximately 80%. We then
measured expression of iNOS mRNA using reverse transcription and quantitative real-time PCR in
PEMs that were co-cultured without or with LPS and with H. pylori at various MOI for 6 h. The
expression of iNOS mRNA was decreased by H. pylori infection at MOI of 50 to 100 (Fig. 5B).
Taken together, the results indicated that H. pylori inhibited iNOS transcription, which
subsequently influenced the translation of iNOS mRNA and NO production by macrophages.
H. pylori attenuates LPS-induced NO production by macrophages through the p38 and ERK1/2
signaling pathways
LPS-induced NO production by macrophages involves several signaling pathways, including p38,
ERK 1/2 (p42 and p44), and JNK. We used western blotting to analyze the signal transduction
pathways involved in the inhibitory effects of H. pylori on NO production by LPS-stimulated PEMs
for 60 min of culture. The data showed that without treatment with LPS and H. pylori, the
phosphorylated molecules involved in the MAPK signaling pathway were expressed at a basal level
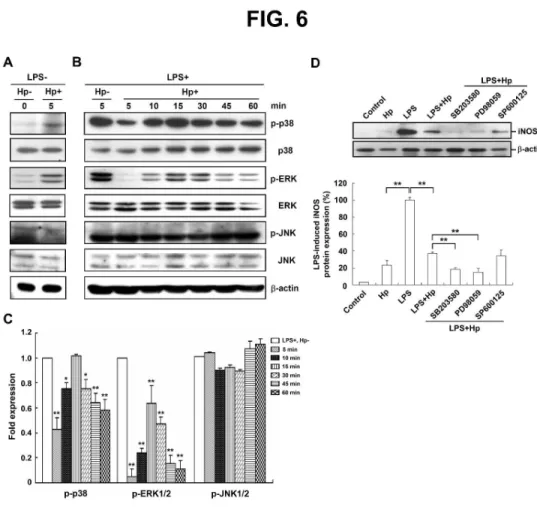
(Fig. 6A). H. pylori infection led to a decrease in phosphorylated p38 in LPS-stimulated
macrophages within 5 to 10 min of culture (Fig. 6B). Phosphorylation of ERK1/2 was also inhibited
by H. pylori infection from 5 to 60 min of culture. In contrast to the results obtained for p38 and
ERK1/2, phosphorylation of JNK1/2 in LPS-stimulated PEMs was not affected by H. pylori
infection. The suppressive effect of H. pylori on LPS-induced iNOS expression was augmented by
SB203580 and PD98059, specific inhibitors of p38 and ERK1/2, respectively, but not by SP600125,
a specific JNK inhibitor (Fig. 6D). Taken together, these results suggested that H. pylori inhibited
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
LPS-stimulated NO production and iNOS expression in macrophages through the p38 and ERK1/2
signaling pathways.
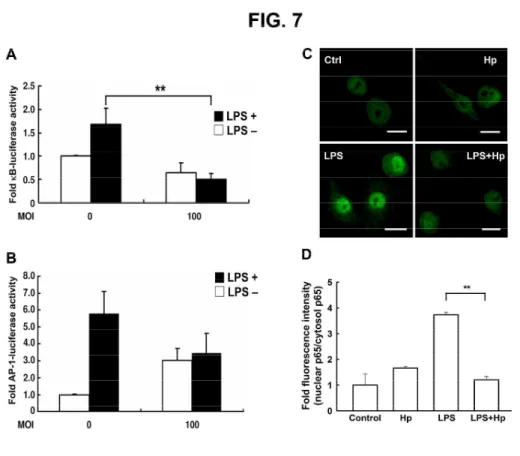
Suppression of LPS-induced NF-κB activation in macrophages by H. pylori
We next investigated the effects of H. pylori on the levels of two transcription factors, NF-κB and
AP-1, which play essential roles in the regulation of iNOS expression.29 We first examined the
effects of H. pylori on NF-κB expression using the luciferase assay in RAW 264.7 cells transfected
with the NF-κB-luciferase reporter. H. pylori inhibited LPS-stimulated activation of the NF-κB
promoter (Fig. 7A). H. pylori only slightly inhibited LPS-stimulated AP-1 activity, but the change
was not statistically significant (Fig. 7B). We next examined p65 localization and observed that it
was primarily located in the cytosol before LPS treatment. After 1 h of stimulation with LPS, p65
translocated into the nucleus of RAW 264.7 cells. When cells were co-cultured with LPS and H.
pylori, however, p65 remained largely in the cytosol (Fig. 7C). The quantitative data showed that
the inhibition of p65 translocation into the nucleus in LPS-treated macrophages upon H. pylori
infection (Fig. 7D). These data suggested that H. pylori alone did not alter the distribution of p65
but rather prevented LPS-induced translocation of p65 into the nucleus.
DISCUSSION
iNOS and NO are two well known factors that serve important roles in the antimicrobial response of
macrophages.30 Our results showed that infection of H. pylori elicited a small amount of NO
production by macrophages (Fig. 1). Not only live H. pylori but also heat-killed H. pylori and
bacterial lysate also slightly stimulated NO synthesis in the absence of bacterial LPS (Fig. 2). In
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
addition, without LPS treatment, H. pylori stimulated, in an MOI-dependent manner, the expression
of iNOS mRNA and protein (Fig. 5). Our results confirm the results of a previous study that showed
that H. pylori can stimulate iNOS expression and activity in murine macrophages.5 The release of
NO appears to be stimulated by H. pylori–derived components because even heat-killed H. pylori
can stimulate NO production.31 LPS derived from H. pylori produces only low biological activity as
a stimulator of NO,32 with estimates that it is 2,000- to 30,000-fold less potent than LPS derived
from E. coli.33 Wilson et al. suggested that H. pylori induces iNOS expression through both
LPS-dependent and -independent mechanisms.5 The expression of iNOS and the accumulation of
NO have been linked to H. pylori–associated gastritis.6, 34 These results demonstrate that NO
production plays an important role in gastric inflammatory responses elicited by H. pylori.
Apart from infection with H. pylori, the stomach is colonized with non-Helicobacter
microorganisms. The most common non-Helicobacter bacteria of the gastric microflora are
Streptococcus, Staphylococcus, Neisseria, Pseudomonas, and Enterobacteriaceae.15 In addition,
endoscope-transmitted infections have been reported and include Salmonella spp., E. coli, K.
pneumoniae, and P. aeruginosa.35 Long-term infection with H. pylori 15 or acid-suppressive therapy
16
enables non-Helicobacter bacteria to colonize the host stomach. A diverse community of 128
phylotypes was identified, using large-scale 16S rRNA sequencing, in 23 gastric endoscopic biopsy
samples, suggesting that the human stomach may be host to a distinct microbial ecosystem.18 Both
LPS and peptidoglycan (PGN), which is found in the cell wall of Gram-negative and Gram-positive
bacteria, have been implicated in the production of NO and proinflammatory cytokines by
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
macrophages.36-39 Patients with both H. pylori and non-Helicobacter bacteria in their gastric mucosa
have higher levels of proinflammatory cytokines than patients without bacterial infection.19 The
issue that these findings raise, therefore, is how H. pylori can survive in a hostile gastric
environment surrounded with profound inflammatory responses provoked by other microorganisms.
To mimic the hostile gastric environment, with activated macrophages and co-infection of H. pylori
and non-Helicobacter bacteria, purified LPS or PGN were added to our macrophage culture system
to determine the effects on NO production. In a preliminary study, we found that 24 h of treatment
with PGN (10 µg/ml) resulted in an approximately 4-fold increase in nitrite (8 µM) over basal levels
in RAW 264.7 cells (Supplementary Fig. 1). After 24 h of treatment with LPS (2 µg/ml), there was
an approximate 8-fold (22 µM) and 150-fold (158 µM) increase in nitrite over basal levels in RAW
264.7 cells and PEMs, respectively (Fig. 1). Murine macrophages were also more sensitive to LPS
than PGN. Because of the results of our preliminary studies, we chose LPS for use in the current
study.
In the present study, we used in vitro and ex vivo murine model systems to reveal how H.
pylori evades LPS-dependent killing by macrophages. To demonstrate that the rational design of our
murine model systems mimic a real-life in vivo situation, we used wild-type C3H/HeN and
TLR4-deficient C3H/HeJ mice to study the role of LPS in killing gastric H. pylori in vivo. As
shown in Supplementary Fig. 2, H. pylori was more significantly eradicated from the stomachs of
LPS-administered C3H/HeN mice than from the stomachs of LPS-administered C3H/HeJ mice. The
response of LPS-administered C3H/HeN mice was approximately 11-fold greater than that of
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
LPS-administered C3H/HeJ mice. This result confirmed that LPS enhanced in vivo anti-H. pylori
activity in LPS-responsive mice (C3H/HeN). In addition, the results of this experiment proved our
assumption from the in vitro and ex vivo murine models since the responses were mimicked in the
in vivo setting.
In this study, we demonstrated that LPS-induced NO production by macrophages was
suppressed by H. pylori and that this effect was dependent on the presence of live bacteria (Fig. 2).
Neither heat-killed bacteria nor crude extracts of H. pylori suppressed LPS-induced NO production.
The work of von Bothmer et al. likewise demonstrated that H. pylori water extract and
whole-bacterial suspension produced an L-arginine-sensitive inhibition of NO synthesis.40 These
data raise the issue as to why the bacteria need to be alive. One possibility is that the effect is
mediated through the cag-pathogenicity island (cag-PAI) or VacA. Our results using isogenic
mutants of ∆cagA or ∆vacA indicated that mutant H. pylori strains also inhibited LPS-induced NO production in a manner similar to the effects observed with wild-type H. pylori (Fig. 3). Despite our
results suggesting that the suppressive effect is mediated through direct interaction of H. pylori with
macrophages, the extent to which virulence factors are associated with the inhibition of NO remains
unknown. Future studies also are needed on the genetic analysis of the virulence factors in different
H. pylori strains.
Several reports have shown that NO can kill H. pylori in cell culture systems.22, 41 Our data
indicated that H. pylori could survive when H. pylori was co-cultured with LPS-activated
macrophages in a trans-well system (Fig. 4). This effect might be due to the suppressive effects of H.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
pylori on LPS-induced NO production by macrophages. This possibility is consistent with the
results of previous studies that demonstrated that arginase produced by H. pylori 22 and arginase II
released from macrophages 42 suppress NO production and lead to immune evasion by the bacteria.
Another explanation for the mechanisms through which H. pylori inhibits NO production is a
decrease in the concentration of L-arginine in culture medium, which interferes with the
L-arginine/NO pathway.40 A reduction in L-arginine availability inhibits iNOS expression and
attenuates NO-dependent bactericidal activity.43 Apart from competition or inhibition of NO
production by macrophages, previous studies showed that H. pylori induces apoptosis in both
macrophages 44 and T lymphocytes.45 In the present study, we did not add L-arginine to cultures. We
used a high dose of LPS to induce iNOS expression, and the effect of LPS was independent of the
concentration of L-arginine, as demonstrated previously.40 Cell viability was not influenced when
cells were incubated with H. pylori at a high MOI of 100 and in the presence of LPS (Fig. 1). H.
pylori appears to have intricate mechanisms to regulate the activity of macrophages and to maintain
bacterial survival under various infectious conditions.
LPS stimulates iNOS gene expression and NO production. The stimulation is positively
regulated by NF-κB, which is normally bound to its inhibitor, IκB in the cytoplasm.
Phosphorylation of IκB by IκB kinase results in the degradation of IκB, which dissociates NF-κB
and leads to the nuclear translocation of NF-κB and the up-regulation of downstream gene
expression. In addition to NF-κB, LPS can activate MAPK pathways in macrophages, including
p38, ERK-1/2, and JNK-1/2. A common strategy for pathogens to overcome host defense is
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
interference with the activity of NF-κB or MAPKs.10, 12, 13, 46 In the present study, H. pylori might
have suppressed LPS-induced NO production by macrophages by inhibiting LPS-stimulated NF-κB
activation. Our data demonstrated that the phosphorylation of p38 and ERK1/2 was attenuated by H.
pylori infection at an early stage of infection, whereas JNK-1/2 was not affected. These results
suggest that H. pylori targets p38 and ERK 1/2, but not JNK1/2. Our data are consistent with
previous findings that pathogens can exploit NF-κB to manipulate cellular responses.10, 13, 14, 47
Together, the findings support our hypothesis that H. pylori modulates host signaling to evade the
host immune system. In addition, our results provide insight into the molecular mechanisms through
which indigenous H. pylori survive commensurately in the stomach with non-Helicobacter
microorganisms that induce potent immune responses.
In conclusion, we demonstrate that infection with live H. pylori attenuates LPS-induced
iNOS gene transcription and NO production in a mouse macrophage model. We further find that H.
pylori inhibits LPS-induced MAPK signaling and NF-κB activation in macrophages. Collectively,
this study reveals a new mechanism through which H. pylori modulates host cell signaling to
protect itself from inflammatory responses and to survive in the harsh environment of the stomach.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
ACKNOWLEDGMENTS
This work was supported by the National Science Council (NSC 97-2313-B-039-003-MY3); China
Medical University (CMU98-S-09 and CMU99-S-09); and Tomorrow Medicine Foundation. The
authors thank Dr. Wen-Ching Wang (National Tsing-Hua University) for valuable suggestions and
comments on this work. We thank Dr. Ai-Li Shiau (National Cheng Kung University Medical
College) for providing of C3H/HeN and C3H/HeJ mice.
Declaration of conflicting interests: None Declared. 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
REFERENCES
1. Goodman KJ, Correa P. Transmission of Helicobacter pylori among siblings. Lancet 2000; 355:
358-362.
2. Rothenbacher D, Bode G, Berg G et al. Helicobacter pylori among preschool children and their
parents: evidence of parent-child transmission. J Infect Dis 1999; 179: 398-402.
3. Brandt S, Kwok T, Hartig R et al. NF-kappaB activation and potentiation of proinflammatory
responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A 2005; 102:
9300-9305.
4. Harris PR, Smythies LE, Smith PD et al. Inflammatory cytokine mRNA expression during early
and persistent Helicobacter pylori infection in nonhuman primates. J Infect Dis 2000; 181:
783-786.
5. Wilson KT, Ramanujam KS, Mobley HL et al. Helicobacter pylori stimulates inducible nitric
oxide synthase expression and activity in a murine macrophage cell line. Gastroenterology 1996;
111: 1524-1533.
6. Fu S, Ramanujam KS, Wong A et al. Increased expression and cellular localization of inducible
nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology
1999; 116: 1319-1329.
7. Baeuerle PA, Baltimore D. Activation of DNA-binding activity in an apparently cytoplasmic
precursor of the NF-kappa B transcription factor. Cell 1988; 53: 211-217.
8. Ajizian SJ, English BK, Meals EA. Specific inhibitors of p38 and extracellular signal-regulated
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
kinase mitogen-activated protein kinase pathways block inducible nitric oxide synthase and
tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide
and interferon-gamma. J Infect Dis 1999; 179: 939-944.
9. Carter AB, Monick MM, Hunninghake GW. Both Erk and p38 kinases are necessary for
cytokine gene transcription. Am J Respir Cell Mol Biol 1999; 20: 751-758.
10. Neish AS, Gewirtz AT, Zeng H et al. Prokaryotic regulation of epithelial responses by inhibition
of IkappaB-alpha ubiquitination. Science 2000; 289: 1560-1563.
11. Chen X, Kokkotou EG, Mustafa N et al. Saccharomyces boulardii inhibits ERK1/2
mitogen-activated protein kinase activation both in vitro and in vivo and protects against
Clostridium difficile toxin A-induced enteritis. J Biol Chem 2006; 281: 24449-24454.
12. Lad SP, Li J, da Silva Correia J et al. Cleavage of p65/RelA of the NF-kappaB pathway by
Chlamydia. Proc Natl Acad Sci U S A 2007; 104: 2933-2938.
13. Orth K, Palmer LE, Bao ZQ et al. Inhibition of the mitogen-activated protein kinase kinase
superfamily by a Yersinia effector. Science 1999; 285: 1920-1923.
14. Neznanov N, Chumakov KM, Neznanova L et al. Proteolytic cleavage of the p65-RelA subunit
of NF-kappaB during poliovirus infection. J Biol Chem 2005; 280: 24153-24158.
15. Kato S, Fujimura S, Kimura K et al. Non-Helicobacter bacterial flora rarely develops in the
gastric mucosal layer of children. Dig Dis Sci 2006; 51: 641-646.
16. Sanduleanu S, Jonkers D, De Bruine A et al. Non-Helicobacter pylori bacterial flora during
acid-suppressive therapy: differential findings in gastric juice and gastric mucosa. Aliment
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
Pharmacol Ther 2001; 15: 379-388.17. Monstein HJ, Tiveljung A, Kraft CH et al. Profiling of bacterial flora in gastric biopsies from
patients with Helicobacter pylori-associated gastritis and histologically normal control
individuals by temperature gradient gel electrophoresis and 16S rDNA sequence analysis. J Med
Microbiol 2000; 49: 817-822.
18. Bik EM, Eckburg PB, Gill SR et al. Molecular analysis of the bacterial microbiota in the human
stomach. Proc Natl Acad Sci U S A 2006; 103: 732-737.
19. Sanduleanu S, Jonkers D, De Bruine A et al. Double gastric infection with Helicobacter pylori
and non-Helicobacter pylori bacteria during acid-suppressive therapy: increase of
pro-inflammatory cytokines and development of atrophic gastritis. Aliment Pharmacol Ther
2001; 15: 1163-1175.
20. Lai CH, Chang YC, Du SY et al. Cholesterol depletion reduces Helicobacter pylori CagA
translocation and CagA-induced responses in AGS cells. Infect Immun 2008; 76: 3293-3303.
21. Lai CH, Kuo CH, Chen PY et al. Association of antibiotic resistance and higher internalization
activity in resistant Helicobacter pylori isolates. J Antimicrob Chemother 2006; 57: 466-471.
22. Gobert AP, McGee DJ, Akhtar M et al. Helicobacter pylori arginase inhibits nitric oxide
production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci U S A 2001;
98: 13844-13849.
23. Babb JL, Kiyono H, Michalek SM et al. LPS regulation of the immune response: suppression of
immune responses to orally administered T-independent antigen. J Immunol 1981; 127:
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
1052-1057.
24. Chi DS, Qui M, Krishnaswamy G et al. Regulation of nitric oxide production from macrophages
by lipopolysaccharide and catecholamines. Nitric Oxide 2003; 8: 127-132.
25. Rao YK, Fang SH, Tzeng YM. Evaluation of the anti-inflammatory and anti-proliferation
tumoral cells activities of Antrodia camphorata, Cordyceps sinensis, and Cinnamomum
osmophloeum bark extracts. J Ethnopharmacol 2007; 114: 78-85.
26. Tang CH, Chuang JY, Fong YC et al. Bone-derived SDF-1 stimulates IL-6 release via CXCR4,
ERK and NF-kappaB pathways and promotes osteoclastogenesis in human oral cancer cells.
Carcinogenesis 2008; 29: 1483-1492.
27. Tang CH, Yang RS, Chen YF et al. Basic fibroblast growth factor stimulates fibronectin
expression through phospholipase C gamma, protein kinase C alpha, c-Src, NF-kappaB, and
p300 pathway in osteoblasts. J Cell Physiol 2007; 211: 45-55.
28. Lai CH, Fang SH, Rao YK et al. Inhibition of Helicobacter pylori-induced inflammation in
human gastric epithelial AGS cells by Phyllanthus urinaria extracts. J Ethnopharmacol 2008;
118: 522-526.
29. Marks-Konczalik J, Chu SC, Moss J. Cytokine-mediated transcriptional induction of the human
inducible nitric oxide synthase gene requires both activator protein 1 and nuclear factor
kappaB-binding sites. J Biol Chem 1998; 273: 22201-22208.
30. Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat
Rev Microbiol 2004; 2: 820-832. 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
31. Shapiro KB, Hotchkiss JH. Induction of nitric oxide synthesis in murine macrophages by
Helicobacter pylori. Cancer Lett 1996; 102: 49-56.
32. Muotiala A, Helander IM, Pyhala L et al. Low biological activity of Helicobacter pylori
lipopolysaccharide. Infect Immun 1992; 60: 1714-1716.
33. Perez-Perez GI, Shepherd VL, Morrow JD et al. Activation of human THP-1 cells and rat bone
marrow-derived macrophages by Helicobacter pylori lipopolysaccharide. Infect Immun 1995;
63: 1183-1187.
34. Mannick EE, Bravo LE, Zarama G et al. Inducible nitric oxide synthase, nitrotyrosine, and
apoptosis in Helicobacter pylori gastritis: effect of antibiotics and antioxidants. Cancer Res
1996; 56: 3238-3243.
35. Lee RM, Kozarek RA, Sumida SE et al. Risk of contamination of sterile biopsy forceps in
disinfected endoscopes. Gastrointest Endosc 1998; 47: 377-381.
36. Wang ZM, Liu C, Dziarski R. Chemokines are the main proinflammatory mediators in human
monocytes activated by Staphylococcus aureus, peptidoglycan, and endotoxin. J Biol Chem
2000; 275: 20260-20267.
37. Boulet I, Ralph S, Stanley E et al. Lipopolysaccharide- and interferon-gamma-induced
expression of hck and lyn tyrosine kinases in murine bone marrow-derived macrophages.
Oncogene 1992; 7: 703-710.
38. Bhat NR, Zhang P, Lee JC et al. Extracellular signal-regulated kinase and p38 subgroups of
mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci 1998; 18:
1633-1641.
39. Bone RC. Gram-positive organisms and sepsis. Arch Intern Med 1994; 154: 26-34.
40. von Bothmer C, Bolin I, Pettersson A et al. Stimulated murine macrophages as a bioassay for H.
pylori-related inhibition of nitric oxide production. Scand J Gastroenterol 2003; 38: 380-386.
41. Bussiere FI, Chaturvedi R, Cheng Y et al. Spermine causes loss of innate immune response to
Helicobacter pylori by inhibition of inducible nitric-oxide synthase translation. J Biol Chem
2005; 280: 2409-2412.
42. Lewis ND, Asim M, Barry DP et al. Arginase II restricts host defense to Helicobacter pylori by
attenuating inducible nitric oxide synthase translation in macrophages. J Immunol 2010; 184:
2572-2582.
43. Chaturvedi R, Asim M, Lewis ND et al. L-arginine availability regulates inducible nitric oxide
synthase-dependent host defense against Helicobacter pylori. Infect Immun 2007; 75:
4305-4315.
44. Gobert AP, Cheng Y, Wang JY et al. Helicobacter pylori induces macrophage apoptosis by
activation of arginase II. J Immunol 2002; 168: 4692-4700.
45. Zabaleta J, McGee DJ, Zea AH et al. Helicobacter pylori arginase inhibits T cell proliferation
and reduces the expression of the TCR zeta-chain (CD3zeta). J Immunol 2004; 173: 586-593.
46. Maresca M, Miller D, Quitard S et al. Enteropathogenic Escherichia coli (EPEC)
effector-mediated suppression of antimicrobial nitric oxide production in a small intestinal
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
epithelial model system. Cell Microbiol 2005; 7: 1749-1762.
47. Tato CM, Hunter CA. Host-pathogen interactions: subversion and utilization of the NF-kappa B
pathway during infection. Infect Immun 2002; 70: 3311-3317.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
FIGURE LEGENDS
Fig. 1. H. pylori–mediated inhibition of LPS-induced NO production in the RAW 264.7 cell line (A)
and in murine primary peritoneal exudated macrophages (PEMs; C). Cells were treated without or
with LPS (2 µg/ml) and infected with H. pylori at various MOI from 0 to 100. After 24 h (RAW
264.7 cells) or 48 h (PEMs) incubation, the culture supernatants were collected for determination of
nitrite levels using the Griess reagent. The MTT assay showed that there was no loss of cell
viability in RAW 264.7 cells (B) or murine PEMs (D) during the incubation period. The data
represent the mean ± standard deviation derived from three independent experiments. Statistical
significance was determined using the Student's t-test (*P < 0.05; **P < 0.01). LPS:
lipopolysaccharide.
Fig. 2. Live H. pylori are essential for inhibition of LPS-induced NO production by macrophages.
Cells were treated without or with LPS and were either un-infected or infected with live H. pylori
(live Hp), heat-killed H. pylori (boiled Hp), crude extracts prepared from H. pylori (Hp lysate). Live
and heat-killed H. pylori were used at an MOI of 100. After RAW 264.7 cells were cultured for 24 h
(A) or murine PEMs were cultured for 48 h (B), culture supernatants were collected to measure
nitrite levels. Nitrite release from control cells treated with LPS alone was set as 100%. Results are
expressed as the mean ± standard deviation derived from three independent experiments. Statistical
significance was determined using the Student's t-test (**P < 0.01). Hp: H. pylori, LPS:
lipopolysaccharide. 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
Fig. 3. Wild-type and isogenic mutants of H. pylori produce the same inhibitory effect on
LPS-induced NO production by macrophages. RAW 264.7 cells were treated without or with LPS
and infected with wild-type (WT) or isogenic mutants of H. pylori at an MOI of 100. After
incubation for 24 h, culture supernatants were collected to determine levels of nitrite. The data
represent the mean ± standard deviation derived from three independent experiments. Statistical
significance was determined using the Student's t-test (*, P < 0.05). Hp: H. pylori, LPS:
lipopolysaccharide.
Fig. 4. Bactericidal effects of NO on H. pylori. Murine PEMs from C57BL/6 wild-type (A) or
iNOS–/– (B) mice were grown on the bottom layer of trans-well culture plates and infected with H.
pylori at MOI of 0 or 100 for 48 h. The PEMs were then co-incubated with H. pylori in the
trans-well insert membrane (0.1 µm) for another 6 h, and colony forming units (CFU) were counted.
Bactericidal activity is expressedas the mean ± standard deviation derived from three independent
experiments. Statistical significance was determined using the Student's t-test (**P < 0.01). LPS:
lipopolysaccharide, MOI: multiplicity of infection.
Fig. 5. H. pylori–mediated inhibition of iNOS expression in LPS-treated macrophages. Murine
PEMs were treated without or with LPS and infected with H. pylori at different MOI. (A) Cell
lysates were prepared after 48 h of incubation to measure iNOS protein expression by western
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
blotting. Protein expression levels were quantified with densitometric analysis and normalized to
β-actin (B) iNOS mRNA expression was measured by quantitative real-time PCR after 6 h of
incubation, and GAPDH was used as an internal control. The data are presentedas the mean ±
standard deviation of three independent experiments. Statistical significance was determined using
the Student's t-test (*P < 0.05; **P < 0.01). LPS: lipopolysaccharide, MOI: multiplicity of
infection.
Fig. 6. Signaling pathways involved in the inhibition of LPS-induced macrophage activation by H.
pylori. Murine PEMs were incubated without LPS (A) or with LPS (B) and infected (or not infected)
with H. pylori at an MOI of 100 for the indicated times. Expression levels of phosphorylated p38
(p-p38), p-ERK, and p-JNK were determined by western blotting. Representative western blot
results from one of three independent experiments are shown. (C) Protein expression levels were
quantified with densitometric analysis, normalized to β-actin, and presentedas the mean ± standard
deviation derived from three independent experiments. Statistical significance was determined
using the Student’s t test (*P < 0.05; **P < 0.01 as compared with LPS-stimulated cells in the
absence of H. pylori infection [LPS+, Hp–], lower panel). (D) Murine PEMs were pretreated for 30
min with SB203580 (p38 inhibitor), PD98059 (ERK inhibitor), or SP600125 (JNK inhibitor),
followed by incubation with LPS and infection with H. pylori at an MOI of 100 for 48 h.
Representative western blot results from one of three independent experiments are shown. The data
are expressedas the mean ± standard deviation determined from three independent experiments.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
Statistical significance was determined using the Student's t-test (**P < 0.01). Hp: H. pylori, LPS:
lipopolysaccharide.
Fig. 7. H. pylori–mediated attenuation of LPS-induced NF-κB activation. RAW 264.7 cells
transfected with a reporter gene for NF-κB (A), AP-1 (B), or β-gal-lacZ (1 µg each) were treated
without or with LPS and infected (or not infected) with H. pylori at an MOI of 100. Luciferase
activity was normalized to the expression level of β-gal-lacZ. The data are expressedas the mean ±
standard deviation derived from three independent experiments. Statistical significance was
determined using the Student's t-test (**P < 0.05). (C) The nuclear translocation of p65 after 1 h
treatment without or with LPS and infection of H. pylori at an MOI of 100. Cells were washed and
treated with anti-p65 followed by fluorescein isothiocyanate–conjugated anti-mouse IgG (green).
Cells were co-stained with propidium iodide to visualize the nucleus (red). Cells were analyzed by
confocal fluorescence microscopy. Regions of p65 nuclear localization appear in yellow in the
overlay. Representative images from one of three independent experiments are shown. Scale bar, 10
µm. Ctrl: control, Hp: H. pylori, LPS: lipopolysaccharide, MOI: multiplicity of infection.
Supplementary Fig. 1. H. pylori–mediated inhibition of PGN-induces NO production by RAW
264.7 cells. Cells were treated with or without PGN (10 µg/ml) and infected with H. pylori at an
MOI ranging from 0 to 100 for 24 h. Culture supernatants were collected for determination of nitrite
release. The data are expressed as the mean ± standard deviation derived from three independent
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
experiments. Statistical significance was determined using the Student's t-test (*P < 0.05). PGN:
peptidoglycan.
Supplementary Fig. 2. LPS-dependent in vivo killing of gastric H. pylori. C3H/HeN and C3H/HeJ
mice were intragastrically inoculated with H. pylori and purified LPS. The amounts of living H.
pylori in the stomach were determined 7 days after infection. The data are expressed as the mean ±
standard deviation (n = 6 for each group). Statistical significance was determined using the
Student's t-test (**P < 0.01). CFU: colony forming units.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
For Peer Review
Fig. 1. H. pylori–mediated inhibition of LPS-induced NO production in the RAW 264.7 cell line (A) and in murine primary peritoneal exudated macrophages (PEMs; C). Cells were treated without or with LPS (2 µg/ml) and infected with H. pylori at various MOI from 0 to 100. After 24 h (RAW 264.7 cells) or 48 h (PEMs) incubation, the culture supernatants were collected for determination of nitrite
levels using the Griess reagent. The MTT assay showed that there was no loss of cell viability in RAW 264.7 cells (B) or murine PEMs (D) during the incubation period. The data represent the mean
± standard deviation derived from three independent experiments. Statistical significance was determined using the Student's t-test (*P < 0.05; **P < 0.01). LPS: lipopolysaccharide.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59
For Peer Review
Fig. 2. Live H. pylori are essential for inhibition of LPS-induced NO production by macrophages. Cells were treated without or with LPS and were either un-infected or infected with live H. pylori (live Hp), heat-killed H. pylori (boiled Hp), crude extracts prepared from H. pylori (Hp lysate). Live and heat-killed H. pylori were used at an MOI of 100. After RAW 264.7 cells were cultured for 24 h (A) or murine PEMs were cultured for 48 h (B), culture supernatants were collected to measure nitrite
levels. Nitrite release from control cells treated with LPS alone was set as 100%. Results are expressed as the mean ± standard deviation derived from three independent experiments. Statistical significance was determined using the Student's t-test (**P < 0.01). Hp: H. pylori, LPS:
lipopolysaccharide. 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59
For Peer Review
Fig. 3. Wild-type and isogenic mutants of H. pylori produce the same inhibitory effect on LPS-induced NO production by macrophages. RAW 264.7 cells were treated without or with LPS and infected with wild-type (WT) or isogenic mutants of H. pylori at an MOI of 100. After incubation for
24 h, culture supernatants were collected to determine levels of nitrite. The data represent the mean ± standard deviation derived from three independent experiments. Statistical significance was
determined using the Student's t-test (*, P < 0.05). Hp: H. pylori, LPS: lipopolysaccharide.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59
For Peer Review
Fig. 4. Bactericidal effects of NO on H. pylori. Murine PEMs from C57BL/6 wild-type (A) or iNOS-/- (B) mice were grown on the bottom layer of trans-well culture plates and infected with H. pylori at
MOI of 0 or 100 for 48 h. The PEMs were then co-incubated with H. pylori in the trans-well insert membrane (0.1 µm) for another 6 h, and colony forming units (CFU) were counted. Bactericidal
activity is expressed as the mean ± standard deviation derived from three independent experiments. Statistical significance was determined using the Student's t-test (**P < 0.01). LPS:
lipopolysaccharide, MOI: multiplicity of infection.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59
For Peer Review
Fig. 5. H. pylori–mediated inhibition of iNOS expression in LPS-treated macrophages. Murine PEMs were treated without or with LPS and infected with H. pylori at different MOI. (A) Cell lysates were prepared after 48 h of incubation to measure iNOS protein expression by western blotting. Protein expression levels were quantified with densitometric analysis and normalized to β-actin (B) iNOS mRNA expression was measured by quantitative real-time PCR after 6 h of incubation, and GAPDH was used as an internal control. The data are presented as the mean ± standard deviation of three independent experiments. Statistical significance was determined using the Student's t-test (*P <
0.05; **P < 0.01). LPS: lipopolysaccharide, MOI: multiplicity of infection.
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59