Femtosecond Dynamics on Excited-State Proton/ Charge-Transfer Reaction in
4
′
-N,N-Diethylamino-3-hydroxyflavone. The Role of Dipolar Vectors in Constructing a
Rational Mechanism
Pi-Tai Chou,*,†Shih-Chieh Pu,†Yi-Ming Cheng,†Wei-Shan Yu,†Yueh-Chi Yu,† Fa-Tsai Hung,*,‡and Wei-Ping Hu *,§
Department of Chemistry, National Taiwan UniVersity, Taipei, Taiwan, ROC, National Hu-Wei UniVersity of Science and Technology, Yunlin, Taiwan ROC, and Department of Chemistry, The National Chung-Cheng UniVersity, Chia Yi, Taiwan, ROC
ReceiVed: December 21, 2004; In Final Form: February 24, 2005
The excitation behaviors for 4′-N,N-diethylamino-3-hydroxyflavone (Ia) have been investigated via femtosecond fluorescence upconversion approaches to gain detailed insights into the mechanism of the proton/charge-transfer coupling reaction. In polar solvents such as CH2Cl2and CH3CN, in addition to a slow, solvent-polarity-dependent rate (a few tens of picoseconds-1) of excited-state intramolecular proton transfer (ESIPT) reported previously, early femtosecond relaxation dynamics clearly reveal that the proton-transfer tautomer emission consists of a rise component of a few hundred femtoseconds. The temporal spectral evolution at the time domain of zero to a few hundred femtoseconds further resolves two distinct emission bands consisting of a proton-transfer tautomer emission and a time-dependent Stokes shifted emission. The results, in combination with ab initio calculations on the dipolar vectors for normal and tautomer species, lead us to unveil the importance of the relationship of the dipolar vectors among various states, and hence the corresponding solvation energetics in the overall ESIPT reaction. We conclude a similar dipolar character between ground-state normal (N) and excited proton-transfer tautomer (T*) species, whereas due to the excited-state intramolecular charge transfer (ESICT), the normal excited state (N*) possesses a large dipolar change with respect to N and T*. ESIPT is thus energetically favorable at the Franck-Condon excited N*, and its rate is competitive with respect to the solvation relaxation process. After reaching the solvent equilibration, there exists an equilibrium between N* and T* states in, for example, CH3CN. Due to the greatly different equilibrium polarization between N* and T*, both forward and reversed ESIPT dynamics are associated with a solvent-induced barrier. The latter viewpoint of the equilibrium type of ESIPT in Ia is in agreement with the previous reports based on steady-state,8picosecond,9,13and femtosecond14,15dynamic approaches.
Introduction
One of the most fundamental processes involved in chemical reactions as well as in living systems should be ascribed to the proton-transfer reaction.1,2Among a large number of proton-transfer relevant studies, the excited-state intramolecular proton transfer (ESIPT) has received considerable attention.3Typical ESIPT molecules possess a strong intramolecular hydrogen bond (HB) between O-H (or N-H) and CdO (or pyridinic nitrogen) groups, in which the intrinsic ESIPT4is essentially barrierless in nonpolar solvents5and may occur either during the periods of low-frequency, large-amplitude vibrational motions associated with the hydrogen bond6or ballistically so that the coherent motion can be resolved.7In polar media, perturbation via solvent polarity interaction may play an important role in modifying the ESIPT dynamics. Considering a significant difference in dipolar vector between excited normal (µ*N) and tautomer species (µ*T), the normal and tautomer equilibrium polarizations should be far separated. Since the large dipolar change in solution is normally coupled to solvent polarization effects, one expects the relative energetics between normal and tautomer to
be dependent on the solvent polarization coordinate. Thus, ESIPT may also be governed by solvent dynamics in a way similar to the nonadiabatic proton transfer shown in Scheme 1. One of the prototypical systems exhibiting remarkable sol-vent-polarization-dependent ESIPT reaction dynamics should be credited to 4′-N,N-dialkylamino-3-hydroxyflavones (system I) (see Scheme 2).8-10In polar, aprotic solvents, in contrast to an ultrafast ESIPT in their corresponding parent molecules, that is, 3-hydroxyflavone (<35 fs),11which give rise to a unique proton-transfer tautomer emission,12dual emissions were ob-served in system I. In the early work, based on the picosecond time-resolved experiment for system I in benzene titrated with CH3CN, Kelly and co-workers9a were able to resolve the relatively slow reaction dynamics with forward and reverse rates of a few tens of picoseconds-1, depending on the polarity of the benzene/CH3CN mixture. Recently, several other picosecond experiments were carried out for system I in various polar solvents. The results rendered both forward and reverse rates of ESIPT and hence the solvent-dependent equilibrium constant between N* and T*.13More recently, femtosecond dynamics were performed with an aim to gain finer insights into the charge/proton-transfer coupled reaction. On the basis of the fluorescence upconversion technique, Douhal et al.14were able to resolve the early (< a few picoseconds) decay dynamics of the normal emission of system I that had previously been
* Corresponding author. E-mail: [email protected].
†National Taiwan University.
‡National Hu-Wei University of Science and Technology. §The National Chung-Cheng University.
10.1021/jp044205w CCC: $30.25 © 2005 American Chemical Society Published on Web 04/07/2005
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
inaccessible and, accordingly, ascribed the fast decay compo-nents to a possible combination of intramolecular vibrational redistribution (IVR), solvation dynamics, and the formation time of a charge-transfer (CT) state that can be distinguished from the locally excited (LE) state. Moreover, a few hundred-femtosecond rise component was observed for the proton-transfer tautomer emission in, for example, CH3CN and was proposed to originate from several possible pathways. It might be due to the proton motion in the nonequilibrated CT state. Alternatively, the direct reaction of the solvent-relaxed and favorably twisted LE state to give T* is also possible. In another approach, Brown and co-workers,15on the basis of femtosecond transient absorption measurements, obtained similar dynamic results, while the interpretation is slightly different in that the early fast decay components of the excited normal state were rationalized mainly by the solvent relaxation dynamics. The early rising dynamics of the T* state were not reported, possibly due to the strongly spectral overlap among excited normal, tautomer transient absorbance and the stimulated emission. In brief, all recent pico-femtosecond approaches13-15are in mutual
agreement in that the results reconfirmed the solvent-dependent, slow (few tens of picoseconds) ESIPT9 with an appreciably large energy barrier in polar solvents (for example,∼4.6 kcal for 4′-N,N-(dimethylamino)-3-hydroxyflavone in CH2Cl213b). Particularly, in stronger polar media such as CH3CN and short carbon chain alcohols, there exists an excited-state equilibrium, so reversible proton transfer also takes place.
Despite the focus on the early time domain measurement, the correlation of dipolar factors among each reaction state, which is believed to be a factor key to describing how the solvent-polarity coordinate channels into the ESIPT/ESICT coupled reaction, is unfortunately still not clear. In the seminal studies of system I, both Chou et al.8and Kelley and co-workers9 have raised a core issue on the influence of dipolar vectors dictating the overall reaction. Particularly, applying the Marcus electron-transfer model, Kelley and co-workers9were able to describe the overall reaction dynamics through a 2-dimensional reaction profile incorporating solvent polarity and proton-transfer reaction coordinates depicted in Scheme 1. As shown in Scheme 1, the dipolar vector of N* in I was proposed to be drastically different from that of T*,9 so that ESIPT is energetically prohibited in the initially prepared N*. In other words, ESIPT will not proceed until it reaches the equilibrium polarization, followed by the thermal activation to a solvated configuration G+(see Scheme 1), where the solvent-polarization potentials between reactant (N*) and product (T*) cross so that proton transfer occurs. Furthermore, the proton-tunneling rate is concluded to be relatively slower than the rate of solvent relaxation. Thus, a nonadiabatic type of proton tunneling takes place essentially after each solvent equilibration is established in the excited state.
Comparing the proposed mechanism depicted in Scheme 1 with recent data based on pico-femtosecond experiments, one may promptly perceive some conflicts that await resolution. First of all, due to the large different equilibrium polarization between N* and T*, ESIPT is highly endergonic in polar, aprotic solvents16 and thus should be thermally prohibited upon Franck-Condon excitation, unless the excitation frequency is tuned to a much higher vibronic level, which is apparently not the case since most femtosecond approaches are probing at near the 0-0 onset of the S0-S1(ππ*) excitation (375-430 nm region, see Figure 1). Thus, the few-hundred-femtoseconds rise component of the tautomer emission, if it is, in part, attributed to the proton motion,14seems to conflict with the mechanism proposed in Scheme 1. Furthermore, modification of Scheme 1 is demanded if an intermediate CT state is incorporated in the reaction. The correlation of the reaction pathways among LE, CT*, and T* states may thus be rather complicated.
In this study, we have vigorously examined the early femtosecond dynamics on 4′ -N,N-diethylamino-3-hydroxyfla-vone (Ia). The results of femtosecond dynamics, in combination with theoretical approaches, reveal a previously unrecognized feature that cannot be fully rationalized by the currently adopted mechanism. Accordingly, a new proton/charge-transfer coupled reaction scheme for system I was proposed to accommodate the observed relaxation dynamics.
Experimental Section
Syntheses and purification of Ia and 4′ -N,N-diethylamino-3-methoxyflavone (Ib) were based on Claisen condensation reported in the literature.8 The various solvents used in the spectroscopic and dynamic measurements were of spectragrade quality (Merck Inc.) and used right after the fractional distil-lation, in which any contaminations from the protic-solvent impurity had been removed by adding suitable drying reagents. SCHEME 1: Considering the Possibly Large Differences
in Dipole Moment between Excited Normal (µ*N) and Tautomer Species (µ*T), the Normal and Tautomer Equilibrium Polarizations Might Be Far Separateda
aAccordingly, a solvent-induced barrier (∆G+) may channel into
the proton-transfer reaction. Note that the orientation of the dipole moment for both normal and tautomer species is arbitrarily picked.
SCHEME 2
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
Steady-state absorption and emission spectra were recorded by a Hitachi (U-3310) spectrophotometer and an Edinburgh (FS920) fluorimeter, respectively. For the pico-nanosecond lifetime measurements, the fundamental train of pulses from Ti-Sapphire oscillator (82 MHz, Spectra Physics) was pulse-selected to reduce its repetition rate to typically 0.8-8 MHz, and then used to produce second harmonics (375-425 nm) as an excitation light source. Signal was detected by a time-correlated single photon counting system (Edinburgh OB 900-L). The femtosecond fluorescence upconversion measurements were performed using a femtosecond optically gated system, details of which have been described in a previous report.17,18 For both pico- and femtosecond time-resolved measurements, the polarization of the pump laser was set at the magic angle (54.7°) with respect to that of the probe laser (or detecting system) to eliminate the fluorescence anisotropy.
Theoretical calculations were performed using the Gaussian 03 program.19The ground (S
0)-state molecular geometry and dipole moments of Ia and its proton-transfer tautomer were calculated using the Hartree-Fock (and B3LYP) theory with 6-31G(d′,p′) basis set. The corresponding molecular geometry and dipole moments at the S1 excited states were calculated using the CI Singles (CIS) theory20with the same basis set. The dipole moment of Ia at the ground-state geometry on the S1 state (vertical excitation) was also calculated at the CIS/6-31G(d′,p′) level. The nature of the stationary points was also ascertained by harmonic vibrational frequency analysis in both the ground and excited states.
Results and Discussion
1. Steady-State Approaches. Although the main focus of this study lies in the dynamic approaches, we feel that provision of some general background information is necessary to facilitate
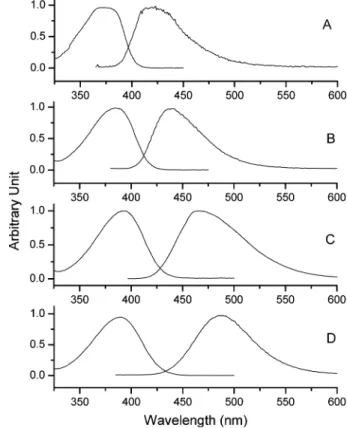
comprehension of the following discussion regarding time-resolved and theoretical approaches. Figures 1 and 2 depict the steady-state absorption and emission spectra of Ia and Ib, respectively, in various solvents. For a comparison in the same solvent, the absorption peak in Ia could bathochromically shift to as large as 12-15 nm with respect to Ib due to the intramolecular hydrogen bond enhancingπ electrons
delocal-ization. With an increase in the solvent polarity, the hypso-chromic shift in the absorption peak wavelength was observed for both Ia and Ib. In view of the low ionization energy for the diethylamino substituents, these results can be rationalized by the manifestation of the S0f S1excitation in Ia and Ib by a charge-transfer character incorporating diethylamine (electron donor) and carbonyl oxygen (electron acceptor). Franck-Condon excitation causes significant dipolar changes on Ia (or Ib), and hence the unfavorable solvated configuration in the excited state.
In cyclohexane, Ia exhibits a dominant 560-nm emission band, the peak frequency of which is Stokes shifted by∼8000 cm-1with respect to the S0f S1(ππ*) absorption. Conversely, Ib, which is generally treated as a non-ESIPT model due to the lack of the O-H proton, reveals a normal emission maximized at 420 nm. Accordingly, the 560-nm emission in Ia originating from the proton-transfer tautomer is unambiguous. However, note that for Ia in cyclohexane, the appearance of a very small, but nonnegligible, normal emission with a peak wavelength at ∼425 nm (see insert of Figure 1) indicates a finite rate of ESIPT (vide infra).
In polar, aprotic solvents, while the absorption feature of Ia shows slight solvent independence with increases in the solvent polarity, dual emission, consisting of a short (Fct)- and a long (Fpt)-wavelength band, becomes more obvious, as the Fctband exhibits strong solvent-polarity dependence, being shifted from 460 nm (in benzene) to 520 nm (in CH3CN). The entire dual emission originating from a common ground-state species is Figure 1. Static absorption and fluorescence spectra of Ia in (A)
cyclohexane, (B) benzene, (C) dichloromethane, and (D) acetonitrile at 298 K. Dashed curves express the spectra fit for Ia in cyclohexane. The sign * shown in (A) denotes a Raman signal, which becomes obvious after magnifying the spectrum by∼100 folds.
Figure 2. Static absorption and fluorescence spectra of Ib in (A)
cyclohexane, (B) benzene, (C) dichloromethane, and (D) acetonitrile at 298 K.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
ascertained by the same fluorescence excitation spectra through-out the monitored wavelengths of 400-700 nm, which are also effectively identical with the absorption spectrum, indicating that the entire emission results from a common Franck-Condon excited state. For comparison, Ib reveals a single emission band, the peak wavelength of which is strongly dependent on the solvent polarity as well. In contrast to the occurrence of ultrafast ESIPT in 3-hydroxyflavone, giving rise to an exclusive ∼525-nm proton-transfer tautomer emission, the steady-state dual emission for Ia in polar, aprotic solvents simply implies the existence of either an appreciable barrier or an excited-state equilibrium associated with ESIPT.
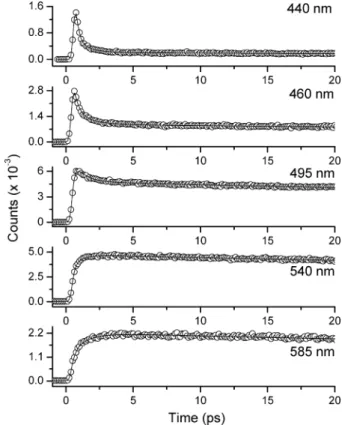
2. Femtosecond Dynamics of Ib. To simplify the discussion, the relaxation dynamics of Ib, in which only excited-state charge transfer takes place, will be first presented, followed by a clear delineation of charge/proton-transfer coupled reaction for compound Ia. The time-resolved fluorescence of Ib in cyclo-hexane was straightforward, and it was established by a response-limited (<150 fs) rise and a single-exponential popula-tion decay component of τf ∼ 0.3 ns at any monitored wavelengths of the emission band. Figures 3 and 4 show the time-resolved fluorescence spectra of Ib in dichloromethane and acetonitrile, respectively. Similar trends were observed in both solvents, in which the emission dynamics were non-singly exponential. Detailed fitting parameters for both early-time relaxation dynamics and population decay rate at selected wavelengths are listed in Table 1. Upon monitoring at short wavelengths, the relaxation dynamics consist of a fast, but resolvable (∼ a hundred femtoseconds to a few picoseconds) decay and rather long (.100 ps) decay components that can be treated as a constant value within the fitted range of <20 ps. The fast decay components require at least two, and for some medium wavelengths even three, exponential terms to achieve good convoluted fits for Ib. For simplicity, Table 1 lists no more than two decay components. The >100-ps decay
com-ponent, measured by the pico-nanosecond single photon count-ing technique, was resolved to be 2.68 and 2.05 ns in CH2Cl2 and CH3CN, respectively, and was then attributed to the population decay of the solvent-equilibrated S1 state. Upon increasing the monitored wavelength, while the population decay remains unchanged, the decay time of the early (fast) compo-nents gradually increases, accompanied by a decrease in the magnitude, and finally becomes a rise component at the very red edge of the emission. In this region, the best fitted time constants for the fast decay and/or rise components also vary by the probed emission wavelengths.
To clarify the observed relaxation dynamics, a temporal spectral evolution of the emission in CH2Cl2was performed, and the results depicted in Figure 5 reveal a continuous spectral shift in fluorescence peak maxima, being red shifted from the 420 nm at∼250 fs to ∼470 nm at e.g. 3 ps and remaining nearly unchanged at > 10 ps. Moreover, the spectra acquired at > 10 ps, within experimental error, are the same as those obtained from the steady state approach. We thus ascribe the early temporal evolution of Ib in dipolar solvents to a continu-ously evolving solvent relaxation rather than an interconversion between two excited species. In summary, upon Franck-Condon excitation, the instant dipolar change in Ib should be quite large and can thus be treated as a charge-transfer process. The coupling between locally excited and charge-transfer states must be fairly strong so that the solvent-polarization-induced barrier is negligible. Under the absence of a reaction barrier, i.e., a reaction ascribed to the optical electron transfer,21common adiabatic solvation energetics and hence the associated relaxation dynamics of Ib are mainly governed by the solvation process. 3. Femtosecond Dynamics of Ia. Due to the additional ESIPT pathway, the relaxation dynamics of Ia are expected to be relatively complicated. In cyclohexane (see Figure 6), the temporal resolution at the very blue side, for example, 440 nm, which is supposed to be in the normal emission that is very Figure 3. Time-resolved sum frequency signal of fluorescence and
gate pulse (800 nm) for Ib in dichloromethane. The solid lines express the corresponding best-fitted curves.
Figure 4. Time-resolved sum frequency signal of fluorescence and
gate pulse (800 nm) for Ib in acetonitrile. The solid lines express the corresponding best-fitted curves.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
small in the steady-state measurement (see insert of Figure 1A), can be best fitted by two single-exponential decay components with lifetime of 0.7 ps (24%) and 2.1 ps (75%) (see Table 1). However, since these two values are very close, considering the experimental error, a single exponential fit of 2.0 ps is also acceptable. This decay component eventually disappears when the emission wavelength is monitored at, for example, 620 nm. Instead, a finite rise component of∼1.95 ps is followed by a long population decay, which can be treated as a constant in an acquisition period of∼15 ps. The long population decay was further resolved to be ∼1.28 ns with the picosecond time-correlated single photon counting method. The rise of the tautomer emission, within experimental error, correlates well
with the decay dynamics of the normal species, suggesting a precursor-sater type of relationship between normal and tautomer emission, with an ESIPT rate of 2.1 ps-1. This value, TABLE 1: Photophysical Properties of Ia and Ib in Various Solvents
solvent emissiona(Φ) early dynamics ps population decay ns
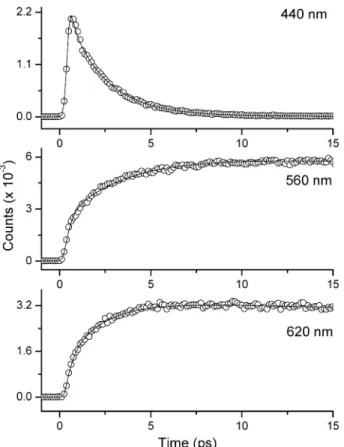
Ia cyclohexane N: 425 nm 440 nm [τ1: 0.70 (0.24),τ2: 2.1 (0.75)] 1.28 T: 560 nm 560 nm [τ1: 0.28 (-0.20),τ2: 3.3 (-0.30)] (for T only) (0.21) 620 nm [τ1: 0.32 (-0.17),τ2: 1.95 (-0.33)] benzene N: 460 nm 440 nm [τ1: <0.1 (0.18),τ2: 2.72 (0.57),τ3: 9.86 (0.24)] 0.9 T: 570 nm 480 nm [τ1: 3.94 (0.52),τ2: 9.86 (0.46)] (for T only) (0.19) 620 nm [τ1: 0.68 (-0.09),τ2: 7.32 (-0.37)] 650 nm [τ1: 1.46 (-0.14),τ2: 10.26 (-0.32)] dichloromethane N: 495 nm 440 nm [τ1: 0.65 (0.91),τ2: 34.7 (0.08)] 0.72 T: 570 nm 470 nm [τ1: 1.5 (0.54),τ2: 41.2 (0.39)] (0.17) 625 nm [τ1: 1.4 (-0.17),τ2: 24.9 (-0.26)] 660 nm [τ1: 0.67 (-0.23),τ2: 19.4 (-0.27)] acetonitrile N: 510 nm 460 nm [τ1: <0.1 (0.42),τ2: 0.44 (0.51),τ3: 30.8 (0.05)] 0.43 T: 575 nm 485 nm [τ1: 0.81 (0.57),τ2: 33.6 (0.27)] (0.096) 610 nm [τ1: 0.88 (-0.18),τ2: 19.3 (-0.18)] 650 nm [τ1: 0.71 (-0.17),τ2: 23.4 (-0.22)] 720 nm [τ1: 0.83 (-0.26),τ2: 20.1 (-0.16)] 1b cyclohexane 420 nm (0.226) 0.3 benzene 440 nm (0.599) 420 nm [τ1: 0.34 (0.06),τ1: 3.17 (0.24)] 0.9 485 nm [τ1: 0.15 (-0.36),τ2: 2.5 (-0.11)] 525 nm [τ1: 0.12 (-0.34),τ2: 1.9 (-0.16)] dichloromethane 465 nm (0.205) 425 nm [τ1: 0.17 (0.66),τ2: 1.62 (0.23)]) 2.68 440 nm [τ1: 0.38 (0.18),τ2: 1.27 (0.57)] 515 nm [τ1: 1.31 (-0.32) 560 nm [τ1: 1.68 (-0.38) acetonitrile 490 nm (0.847) 440 nm [τ1: 0.23 (0.65),τ2: 1.1 (0.25)] 2.05 460 nm [τ1: 0.49 (0.65),τ2: 3.3 (0.08)] 540 nm [τ1: 0.15 (-0.35),τ2: 1.1 (-0.05)] 585 nm [τ1: 0.82 (-0.34),τ2: 3.5 (-0.03)]
aN: “normal” or “charge-transfer” state emission maxima, T: tautomer state emission maxima. Data listed in the early dynamics are the rise (negative preexponential factor), and short decay components. The much longer population decay components are not shown in this column. Data in () are the fitted preexponential factor. Note that the sum of preexponential factors for some data is not equal to 1.0 due to the exclusion of the population decay component.
Figure 5. The temporal emission spectra of Ib in CH2Cl2acquired at a delay time of: ]: 250 fs, 0: 375 fs, O: 500 fs, 4: 625 fs, g: 750 fs, b: 1 ps, 9: 1.5 ps, 2: 3 ps, f: 15 ps. The thick solid lines express the steady-state fluorescence.
Figure 6. Time-resolved sum frequency signal of fluorescence and
gate pulse (800 nm) for Ia in cyclohexane. The solid lines express the corresponding best-fitted curves.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
within experimental error, is in agreement with that reported by Douhal et al. (1.8 ps-1in methylcyclohexane)14and Brown and co-workers (1.6 ps-1in methylcyclohexane).15
Figures 7 and 8 depict the time-resolved traces of Ia in dichloromethane and acetonitrile, respectively. Detailed fitting parameters for early-time relaxation dynamics and population
decay rate are listed in Table 1. In comparison to that of Ib (see Figures 3 and 4), drastically different relaxation dynamics were resolved for Ia. First of all, in CH2Cl2for example, upon monitoring at the short wavelength region ascribed to the charge-transfer emission, the relaxation dynamics of Ia consist of an ultrafast (a few hundred femtoseconds) decay component (1), Figure 7. Time-resolved sum frequency signal of fluorescence and gate pulse (800 nm) for Ia in dichloromethane. The solid lines express the
corresponding best-fitted curves.
Figure 8. Time-resolved sum frequency signal of fluorescence and gate pulse (800 nm) for Ia in acetonitrile. The solid lines express the corresponding
best-fitted curves.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
which requires at least two, and for some medium wavelengths even three, exponential terms to achieve good convoluted fits, a fast decay (∼35 ps, component (2)), accompanied by a long population decay of 720 ps (component (3)) acquired by the ps/ns time-correlated system. Both components (1) and (2) gradually become the rise components upon increases of the monitoring wavelength, especially toward the red edge of the proton-transfer tautomer emission (vide infra).
Similar results were obtained for the case of Ia in CH3CN, consisting of a very fast decay (few hundred femtoseconds) component (1), which requires at least two, and for some medium wavelengths even three, exponential terms to achieve good convoluted fits, a fast (∼33 ps, wavelength-dependent rise/ decay) component, and a population decay time of∼430 ps throughout the monitored wavelengths of 460-720 nm. It is also noteworthy that while the quantitative correlation between ultrafast decay (short wavelength) and rise (long wavelength) in component (1) is difficult mainly due to their wavelength dependence, the time constant of component (2) (few tens ps) and population decay components (3), within experimental error, remains constant throughout the monitored dual emission of 460-720 nm. The results of components (2) and (3) led Kelley and co-workers9 as well as recent pico-femtosecond experi-ments13-15 to conclude the establishment of an excited-state equilibrium between normal (that is, charge-transfer) and proton-transfer tautomer states, in which the rates of both forward and reverse ESIPT are much faster than that of the population decay rate for both states. ESIPT and its associated relaxation dynamics at >10 ps9, for example, can thus be expressed by the coupling reaction scheme shown here:
where k1and k-1denote the forward and reverse rates of ESIPT, respectively. Keqrepresents the equilibrium constant between N* and T* species. In other words, after a Franck-Condon excitation, the intrinsic rate of proton transfer is proposed to be much slower than that of the solvent relaxation. Thus, followed by the solvent relaxation, the excited normal species N* under-goes two decay pathways with time constants ofλ1andλ2, while the overall dynamics of the tautomer species T* should theo-retically reveal rise and decay components with rate constants ofλ2andλ1, respectively.λ1andλ2can be resolved via pico-second time-resolved measurements. Accordingly, the forward, reverse rate of ESIPT, and hence the equilibrium constant Keq between N* and T*, can be deduced. This, in combination with temperature-dependent study, leads to the resolution of the solvent-induced energy barrier for both forward and reverse
ESIPT. Details of relevant analyses have been elaborated on in previous reports9,13and thus were not pursued in this study.
According to the above coupling reaction scheme, the solvent-dependent, few hundred femtoseconds decay component (1) resolved at short wavelengths of the charge-transfer emission and the corresponding ultrafast rise component monitored at longer wavelengths may spontaneously be ascribed to the dynamics of solvent relaxation. However, several salient dif-ferences can be pointed out via careful comparison with respect to Ib in this early time domain. First, for Ia, despite the wavelength-dependent decay rate of the major constituent of component (1), its time constant, under the same monitoring wavelength and detection condition, is always faster than the solvation dynamics obtained for Ib. As shown in Table 1, the ultrafast decay component at 440 nm was fitted to be 645 fs for Ia in CH2Cl2, which is nearly twice as fast as that of 1.27 ps measured in Ib (see Table 1). Upon monitoring at 485 nm for Ia, in which, as indicated by the spectrum and dynamics of Ib in the same solvent, the contribution from charge-transfer emission is negligible, a great portion of the ultrafast rise components was still observed. Upon monitoring at a very red side of the tautomer emission of 720 nm, which is free from the charge-transfer emission, an ultrafast rise component of ∼830 fs was still resolved, with an initial contribution of ∼26%. Obviously, in addition to a channel of solvent relaxation and a pathway of solvent-polarity-induced ESIPT that takes place in the equilibrium polarization, another ultrafast deactivation pathway in Ia, possibly incorporating intrinsic proton transfer, must play a key role to account for the observed early relaxation dynamics in polar, aprotic solvents. Perhaps the most convincing support for this viewpoint is given by the spectral temporal evolution of Ia. As shown in Figure 9, the time-dependent spectral evolution of Ia in CH2Cl2, acquired at an earlier time domain of <1.0 ps, has revealed two distinct bands, the short wavelength band of which gradually shifted from 450 to∼500 nm upon increases in the pump-probe delay time, accompanied by the increase of the peak intensity. Conversely, while the other band, with a peak wavelength at 570 nm, also gradually increases, its peak wavelength seems to be independent of the temporal evolution. Comparing the steady-state spectra, the 570-nm band is unambiguously assigned to a proton-transfer tautomer emission. After a delay time of >10 ps, for example, the 570-nm band continuously increases, together with the decrease of the 500-nm band. At a delay time of >180 ps, for example, the ratio of the dual emission remained nearly constant, and the spectral feature was identical with that obtained from the steady-state approach.
At the time domain of > 5 ps, for example, when solvent (CH2Cl2) relaxation is nearly complete, the decrease of charge-transfer emission, accompanied by the increase of the tautomer emission, can be rationalized by the slow ESIPT rate due to the different equilibrium polarization between charge-transfer (N*) and proton-transfer (T*) species, resulting in a solvent-induced barrier. This viewpoint has been addressed in several previous reports.9,13-15Thus, the reaction dynamics at this time domain can be well described by the coupling mechanism expressed in eq (a). However, at an early time domain of <3 ps, for example, our results of simultaneous increase for both charge-transfer and proton-transfer emission, in combination with the few hundred femtosecond rise component on the tautomer emission, drew a new reaction feature that demands the modification of the originally proposed mechanism shown in Scheme 1. The appearance of both normal and tautomer emission at <1 ps, together with a good correlation between [N*] ) [N*]0 λ2- λ1 {(λ2- X)e -λ1t+ (X - λ 1)e -λ2t} [T*] )k1[N*]0 λ2- λ1{e -λ1t- e-λ2t} (a) λ1∼kN+ kTKeq1 1 + Keq ,λ2) k1+ k-1, X ) kN+ k1, Y ) kT+ k-1
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
the spectral shift of the normal emission and the growth dynamics of the 570-nm emission (in CH2Cl2), suggest that upon Franck-Condon excitation, the rates of solvent relaxation and ESIPT must be compatible. However, such a process, according to the commonly adopted polarization equilibrium depicted in Scheme 1,9amust be highly energetically unfavorable. It should be noted that Scheme 1 is constructed on the basis of a large change in the dipole moment between ground (N) (assumed to be zero9a) and excited (charge-transfer) states of the normal species (N*). In addition, the dipole moment of the excited tautomer (T*) is quite different from that of N and N*. To rationalize the observed relaxation dynamics in both the picosecond and femtosecond time scales, we alternatively proposed that N and T* should possess a similar dipolar property, while the dipole moment for N* and T* are quite different due to the charge-transfer property in N*. Thus, ESIPT is energetically favorable at the Franck-Condon excited N* (vide infra). Support of this viewpoint is rendered by a theoretical approach detailed as follows.
4. Theoretical Approaches. In a recent study, to rationalize the dipolar functionality tuning ESICT/ESIPT coupled reaction, we have attempted to calculate the dipolar vector for Ia in various states with semiempirical approaches at an INDO/S-CI level based on AM1 geometry-optimized ground-state struc-tures.22The results qualitatively estimated that in view of dipole orientation, N and T* are similar, while N* and T* are quite different, although the corresponding magnitudes are somewhat scattered, with a lack of good correlation. Note that the relationship of a similar dipolar orientation between N and T* has been proposed by Kasha and co-workers in order to explain the observed static/dynamic perturbation upon applying Ia in
probing serum albumin.23To verify the dipolar vectors among various states proposed above in a more quantitative manner, we have thus performed theoretical approaches based on the 6-31G(d′,p′) basis sets at a Hartree-Fock level. The geometry-optimized structures of N and T for Ia with some critical bond distances and angles are depicted in Figure 10. As a result, the dipole moments for N and T were calculated to be 5.4 and 5.2 D, respectively, and the angle between these two dipole vectors was∼12°, indicating a similar dipolar vector between N and T. Experimentally, as supported by the nearly solvent-polarity-independent proton-transfer tautomer emission, it is also reason-able to assume a similar dipole moment in terms of both magnitude and orientation between T and T*. Thus, prior to the theoretical attempts toward the excited-state dipolar vectors, we can safely conclude that the excited proton-transfer tautomer (T*) possesses a similar dipolar character with respect to that of the normal ground state (N), supporting our proposed dipolar relationship. More rigorously, calculation based on the CIS/6-31G(d′,p′) method for N* estimated a dipolar vector with a large magnitude of 9.2 D and an orientation as large as ∼30° relative to N (see Figure 10). Conversely, the dipole moment for T* was calculated to be 5.5 D, with a vector orientation being 15°with respect to that of N. In addition, the calculation clearly revealed the same dipolar vector between T (5.2 D) and T* (5.5 D), consistent with the nearly solvent-polarity independent proton-transfer tautomer emission. Al-though the level and method used in current theoretical approaches are not high enough for us to ensure the quantitative values, the resulting correlation firmly supports the viewpoints in that N, T, and T* are subject to similar solvent-polarization free energy, while that of N* is quite different due to its charge-transfer property.
5. Constructing a Rational ESIPT Mechanism. The theoretical approaches, in combination with the results obtained from pico-femtosecond dynamics for Ia, lead us to propose the overall reaction pathways depicted in Scheme 3. Scheme 3, parts A and B, depict the potential energy surface as a function of a solvent polarization coordinate and a proton-transfer reaction coordinate, respectively. As shown in Scheme 3A, upon Franck-Condon excitation, Ia undergoes an instant, dipolar change due to the charge-transfer reaction. The unfavorable polarization configuration is subsequently subject to a rapid solvent relaxation/reorientation to reach an energetically more favorable solvated configuration. However, due to the same dipolar vectors and hence identical polarization environments between N and T* states, the solvent-polarization effect should be decoupled from the ESIPT pathway at early times right after the Franck-Condon excitation. If ESIPT is energetically favorable, proton transfer takes place free from the solvent-polarity perturbation (see polarization configuration at PF in Scheme 3B). The rate of intrinsic ESIPT, which may be governed by low-frequency, large-amplitude vibrational motions associated with the hydrogen bond,6 may be of the same magnitude as the rate of solvent relaxation, resulting in a competitive deactivation pathway. Because the fast decay component, that is, component (1), of N* in Ia, which is dominated by both rates of ESIPT and the solvation process, is on the order of a few hundred femtoseconds, the rate of ESIPT must be finite. For an oversimplified approach, the major decay component at a blue region of∼440 nm was fitted to be 0.65 and 1.27 ps-1 for Ia and Ib, respectively, in CH2Cl2. Taking the solvent relaxation rate for both Ia and Ib to be the same, the rate of ESIPT is thus estimated to be∼1.33 ps-1. Note that this value, though being very qualitative, is consistent with that Figure 9. The temporal spectral evolution of Ia in CH2Cl2acquired
at a delay time of (A) 9: 250 fs, 4: 375 fs, 3: 500 fs, ]: 625 fs, 0: 750 fs, O: 1 ps, and g: 3 ps. (B) O: 30 ps, 4: 60 ps, ]: 90 ps, g: 120 ps, x: 180 ps, 3: 240 ps, and 0: 300 ps. The thick solid lines express the steady-state fluorescence.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
of 2.0 ps-1measured in the cyclohexane. In this ESIPT channel, the subsequently relaxation should incorporate internal conver-sion and perhaps, in part, the solvent relaxation to reach the equilibrium polarization of T*. Regarding the solvent relaxation channel, after reaching a critical solvation configuration such as point P+depicted in Scheme 3A, proton transfer becomes energetically uphill due to the unfavorable solvent polarization (see Scheme 3B). Therefore, the subsequent deactivation should be dominated by the solvent relaxation, finally reaching a solvent-stabilized, equilibrium polarization PN*. Because of the different equilibrium polarization configuration between N* (PN*) and T* (PT*), the solvent polarity effect is expected to be coupled into the ESIPT reaction pathway, resulting in a nonnegligible barrier. In the equilibrium polarization, the N* h T* type of ESIPT mechanism is associated with solvent-polarity-induced barrier and has been extensively discussed in previous reports (vide supra).9,13-15
An intriguing issue regards the ESIPT rate of 1-2 ps-1 measured in cyclohexane. The results are in sharp contrast to the <50 fs-1of ESIPT for the parent compound 3-hydroxyfla-vone.11On one hand, assuming the dynamics of ESIPT in Ia to be free from solvent perturbation in cyclohexane, it may imply that the charge-transfer property in Ia leads to different distribution of the charge density from that of 3-hydroxyflavone, so that the driving force (that is, photoacidity and basicity, and so forth) for ESIPT is appreciably reduced in Ia. We also performed a deuterium isotope (that is, O-D) experiment of Ia in cyclohexane. The rate of proton transfer of 2.2 ps-1for deuterated Ia, within experimental error, is identical with that
of the nondeuterated (1) (2.1 ps-1). Due to the lack of deuterium isotope effect, the intrinsic proton transfer may occur through an adiabatic process. Accordingly, the results of Ia in polar solvents can be described as an adiabatic proton-transfer governed by solvent dynamics in a fashion similar to the nonadiabatic proton transfer. Note that similar viewpoints have been made by Hynes and co-worker24 on the basis of the theoretical approach of several proton-transfer systems. On the other hand, the relatively slow proton-transfer rate in cyclo-hexane may indicate that solvent polarization still plays a role for the ESIPT in cyclohexane. Due to the large dipolar change between N* and T*, the induced-dipole/dipole interaction is considered to be nonnegligible. It is also worthy to note that a dynamic polarization model in nonpolar solvents has recently been proposed by Hamaguchi and co-workers.25With either static or dynamic perturbation, a subtle difference in the equilibrium polarization between N* and T* may be expectable even in nonpolar solvents. Thus, a small but nonnegligible solvent-induced barrier might still be possible, reducing the rate of ESIPT for Ia in nonpolar solvents.
Conclusion
In conclusion, we report on the excitation behaviors for the charge-coupled proton-transfer reaction for Ia and in the femtosecond time scale. Due to the similar polarization effect between N and T*, upon 370-420 nm Franck-Condon exci-tation of Ia, ESIPT is energetically allowed, and the rates of ESIPT and solvent relaxation are competitive. Once the charge-Figure 10. The geometry-optimized structure and selected critical bond distances and angles for N, N*, T, and T* of Ia.. See text for details.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
transfer state is thermally populated, a relatively slow, solvent-polarity-dependent proton transfer takes place due to the greatly different equilibrium polarization between N* and T*, resulting in an appreciable barrier. As a result, the overall reaction dynamics can be well described by a mechanism incorporating both solvent polarization and proton-transfer reaction coordinates (Scheme 3). Recently, we have performed femtosecond dynam-ics on the ESICT/ESIPT coupled reaction in 2-hydroxy-4-(di-p-tolyl-amino) benzaldehyde.18The results also indicate that relative dipolar differences among various states are crucial to fine-tune the reaction dynamics, although, in this case, the solvent-polarity-induced barrier is concluded to be rather small. Thus, the reaction rate is approximately equal to the inverse of the solvent longitudinal relaxation time and is independent of the reactant/product coupling. Despite this difference, it is obvious that the correlation of dipolar vectors among N, N*, and T* plays a key role in describing the ESICT/ESIPT coupled reaction. It is thus of fundamental interest to examine if the dipolar tuning mechanism can be generalized to other ESICT/ ESIPT systems. Of particular interest is when T* possesses a charge-transfer character. In this case the energetics of T* should be fine-tuned via the solvent polarity, so that the corresponding ESIPT dynamics may serve as an ideal model to test Marcus theory.
Acknowledgment. We thank National Science Council (Grant Numbers 99-1989-2004) for the financial support.
References and Notes
(1) Here, we simply adopt the term “proton transfer” rather than “hydrogen atom transfer” throughout the text, because it is more suitable in describing the solvent polarity coupled dynamics.
(2) For example, see: Studies in Physical and Theoretical Chemistry; Mu¨ller, A., Ratajack, H., Junge, W., Diemann, E., Eds.; Electron and Proton
Transfer in Chemistry and Biology, Vol. 78; Elsevier: Amsterdam, The
Netherlands, 1992.
(3) For recent examples, see: (a) Scheiner, S. J. Phys. Chem. A 2000,
104, 5898. (b) Waluk, J. Conformational Aspects of Intra- and
Intermo-lecular Excited-State Proton Transfer. In Conformational Analysis of
Molecules in Excited States; Waluk, J., Ed.; Wiley-VCH: New York, 2000.
(c) Chou, P. T. J. Chin. Chem. Soc. 2001, 48, 651. (d) Wu, K. C.; Cheng, Y. M.; Lin, Y. S.; Yeh, Y. S.; Pu, S. C.; Hu, Y. H.; Yu, J. K.; Chou, P. T.
Chem. Phys. Lett. 2004, 382, 203. (e) Paterson, M. J.; Robb, M. A.;
Blancafort, L.; DeBellis, A. D. J. Am. Chem. Soc. 2004, 126, 2912. (f) Lochbrunner, S.; Wurzer, A. J.; Riedle, E. J. Phys. Chem. A 2003, 107, 10580. (g) de Vivie-Riedle, R.; De Waele, V.; Kurtz, L.; Riedle, E. J. Phys.
Chem. A 2003, 107, 10591. (h) Cheng, C. C.; Chang, C. P.; Yu, W. S.;
Hung, F. T.; Liu, Y. I.; Wu, G. R.; Chou, P. T. J. Phys. Chem. A 2003,
107, 1459. (i) Lukeman, M.; Wan, P. J. Am. Chem. Soc. 2003, 125, 1164.
(4) ESIPT is prohibited for some 6, five-membered ring types of strong intramolecular hydrogen bonding molecules with an nπ* con-figuration as the lowest singlet manifold. For a recent review, see ref 3a.
(5) ESIPT incorporating phenol O-H to a β-carbon atom is not included in this category, see: Lukeman, M.; Wan, P. J. Am. Chem. Soc.
2002, 124, 9458. Also, a recent study on 5-(2-pyridyl) 1-H-pyrazoles in
nonpolar solvents reveals an intrinsic ESIPT incorporating an appreciably large energy barrier due to the skeletal reorganization, see: Yu, W. S.; Cheng, C. C.; Cheng, Y. M.; Wu, P. C.; Song, Y. H.; Chi, Y.; Chou, P. T.
J. Am. Chem. Soc. 2003, 125, 10801.
(6) Chudoba, C.; Riedle, E.; Pfeiffer, M.; Elsaesser, T. Chem. Phys.
Lett. 1996, 263, 622.
(7) Lochbrunner, S.; Wurzer, A. J.; Riedle, E. J. Chem. Phys. 2000,
112, 10699.
(8) (a) Chou, P. T.; Martinez, M. L.; Clements, J. H. J. Phys. Chem.
1993, 97, 2618. (b) Chou, P. T.; Martinez, M. L.; Clements, J. H. Chem. Phys. Lett. 1993, 204, 395.
(9) (a) Swinney, T. C.; Kelley, D. F. J. Chem. Phys. 1993, 99, 211. (b) Parsapour, F.; Kelley, D. F. J. Phys. Chem. 1996, 100, 2791.
(10) Ormson, S. M.; Brown, R. G.; Vollmer, F.; Rettig, W. J. Photochem.
Photobiol. A 1994, 81, 65.
(11) Ameer-Beg, S.; Ormson, S. M.; Brown, R. G.; Matousek, P.; Towrie, M.; Nibbering, E. T. J.; Foggi, P.; Neuwahl, F. V. R. J. Phys.
Chem. A 2001, 105, 3709.
(12) For example, see: (a) Sengupta, P. K.; Kasha, M. Chem. Phys.
Lett. 1979, 68, 382. (b) McMorrow, D.; Kasha, M. J. Phys. Chem. 1984, 88, 2235.
(13) (a) Shynkar, V. V.; Me´ly, Y.; Duportail, G.; Pie´mont, E.; Klym-chenko, A. S.; DemKlym-chenko, A. P. J. Phys. Chem. A 2003, 107, 9522. (b) Roshal, A. D.; Organero, J. A.; Douhal, A. Chem. Phys. Lett. 2003, 379, 53.
(14) Douhal, A.; Sanz, M.; Carranza, M. A.; Organero, J. A.; Santos, L. Chem. Phys. Lett. 2004, 394, 54.
(15) Ameer-Beg, S.; Ormson, S. M.; Poteau, X.; Brown, R. G.; Foggi, P.; Bussotti, L.; Neuwahl, F. V. R. J. Phys. Chem. A 2004, 108, 6938.
(16) In protic solvent, perturbation via intermolecular hydrogen bonding formation may complicate the ESIPT mechanism and thus is not considered in this study. For a recent study of system I in protic solvent, see: Shynkar, V. V.; Klymchenko, A. S.; Piemont, E.; Demchenko, A. P.; Mely, Y. J.
Phys. Chem. A 2004, 108, 8151.
(17) Chou, P. T.; Chen, Y. C.; Yu, W. S.; Chou, Y. H.; Wei, C. Y.; Cheng, Y. M. J. Phys. Chem. A 2001, 105, 1731.
(18) Chou, P. T.; Yu, W. S.; Cheng, Y. M.; Pu, S. C.; Yu, Y. C.; Lin, Y. C.; Huang, C. H.; Chen, C. T. J. Phys. Chem. A 2004, 108, 6487.
(19) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.;
SCHEME 3: (A) Proposed Relaxation Dynamics of Ia (in CH3CN, for example) as a Function of Solvent Polaritiesa(B) Relative Energetics between N* and T* at PN*, PT*, P+, and PFb
aP
N*and PT*are equilibrium polarization for N* and T*, respectively. P+ denotes the critical solvation configuration. PF represents the polarization configuration upon Nf N* Franck-Condon excitation. PT and SR stand for proton transfer and solvent relaxation, respectively.
bThe free-energy scale is arbitrary. Thus, the comparison of energetics
among various polarization configurations is meaningless.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03,
ReVision C. 02; Gaussian, Inc.: Pittsburgh, PA, 2003.
(20) Foresman, J. B.; Head-Gordon, M.; Pople, J. A.; Frisch, M. J. J.
Phys. Chem. 1992, 96, 135.
(21) Paddon-Row, M. N. Electron Transfer in Chemistry, Volume 3; Balzani, V., Ed.; Wiley-VCH: Weinheim, 2001; p 179.
(22) Chou, P. T.; Huang, C. H.; Pu, S. C.; Cheng, Y. M.; Liu, Y. H.; Wang, Y.; Chen, C. T. J. Phys. Chem. A 2004, 108, 6452.
(23) Sytnik, A.; Gormin, D.; Kasha, M. Proc. Natl. Acad. Sci. U.S.A.
1994, 91, 11968.
(24) (a) Kiefer, P. M.; Hynes, J. T. J. Phys. Chem. A 2002, 106, 1834. (b) Kiefer, P. M.; Hynes, J. T. J. Phys. Chem. A 2002, 106, 1834. (c) Kiefer, P. M.; Hynes, J. T. J. Phys. Chem. A 2002, 107, 9022.
(25) Iwata, K.; Ozawa, R.; Hamaguchi, H. J. Phys. Chem. A 2001, 105, 3709.
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009