Comparative Investigation of Ultrafast Photoinduced Processes in
Salicylidene-Aminopyridine in Solution and Solid State
†Michel Sliwa,*,‡Nicolas Mouton,‡Cyril Ruckebusch,‡Ste´phane Aloı¨se,‡ Olivier Poizat,‡ Guy Buntinx,‡ Re´mi Me´tivier,§Keitaro Nakatani,§Hiroshi Masuhara,| and Tsuyoshi Asahi*,⊥ Laboratoire de Spectrochimie Infrarouge et Raman (CNRS UMR 8516), Centre d’Etudes et de Recherches Lasers et Applications (FR 2416 du CNRS), UniVersite´ des Sciences et Technologies de Lille, Baˆt C5, 59655 VilleneuVe d’Ascq Cedex, France, ENS Cachan, PPSM CNRS UMR 8531, IFR d’Alembert, 61 AV. du Pre´sident Wilson, 94230 Cachan, France, Graduate School of Materials Science, Nara Institute of Science and Technology, Ikoma, Nara 630-0192, Japan, Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung UniVersity, Hsinchu 30010, Taiwan, and Department of Applied Physics, Osaka UniVersity, Yamadaoka 2-1, Suita, Osaka 565-0871, Japan
ReceiVed: February 27, 2009; ReVised Manuscript ReceiVed: April 14, 2009
Photodynamics and excited-state intramolecular proton transfer (ESIPT) of N-(3,5-ditert-butylsalicylidene)-4-aminopyridine (4P) and N-(3,5-ditert-butylsalicylidene)-2-aminopyridine (2P) are studied by steady-state and transient optical spectroscopy in solution and solid state (microcrystalline powder). These two compounds are representative of anils Classes A and B, respectively. The results confirm that in solution both compounds have a similar dynamic behavior: upon excitation at 390 nm, ESIPT leads to different cis-keto* fluorescent species which relax to the photochromic trans-keto photoproduct within a few tens of picoseconds. Furthermore, for the first time, picosecond data are recorded for the two classes of anils in the solid state: in 2P, two cis-keto* fluorescent excited state species (23 and 250 ps lifetime) are relaxing exclusively to the cis-keto ground state, whereas in 4P, a single cis-keto* fluorescent species is observed which leads to the trans-keto photoproduct in 250 ps via an intermediate metastable species.
Introduction
Photochromic materials1-4have been studied intensively for several years for their potential applications in optical devices, from ophthalmic lenses to optical memories or fluorescent switches.5-9Our research is currently focused on materials in which nonlinear optical (NLO) properties can be controlled by light.10-13Such research to target switchable NLO systems has been increasing during the past decade.14-20 Especially our interest is centered on second harmonic generation (SHG) in solid state materials. However, a few of the molecules exhibiting photochromism in solution or in matrices keep this property in the crystalline state.21,22 Among all of the families of photo-chromic molecules, those for which the photochromism involves an excited state proton transfer are of great interest because the fast kinetics of this transfer is particularly valuable for fast switching applications and very attractive from a fundamental point of view.23 In this respect, we pay special attention to salicylidene-anilines, salicylidene-aminopyridines, and related Schiff bases, also called anils. Schiff bases and inter/intra molecular proton transfer are also of notable interest due to their extensive implication in many natural processes.23
In the past decade the photochemistry of anils was extensively studied by means of several ultrafast spectroscopic techniques
in solution24-30in addition to gas31,32and crystalline phases.33,34 These results complete the general mechanism of anils estab-lished from data obtained during almost half a century.21,22,35-41 Scheme 1 represents the generally accepted mechanism. The photoreaction is based on an excited-state intramolecular proton transfer (ESIPT) between an enol (-imine) form and a keto (-amine) form. The enol form is usually colorless or slightly yellow with an absorption band in the near UV. The keto-form is usually red due to an additional absorption band around 500 nm. Time resolved experiments show that the proton transfer occurs within 1 ps in the solid state and less than 50 fs in acetonitrile.26,34In solution, it is assumed that the species created after the ESIPT is a hot cis-keto* excited state which relaxes to the cold S1 fluorescent cis-keto* state in a few hundred femtoseconds.27The S
1fluorescent cis-keto* relaxes in a few tens of picoseconds by two competitive processes: radiative deactivation to the cis-keto ground state via a strongly Stokes-shifted (vs absorbance of enol form) fluorescence and nonra-diative deactivation toward the final trans-keto photoproduct.26,27,34 The cis-keto form returns rapidly (picosecond scale) to the initial enol form whereas the trans-keto photoproduct is stable at least a few tens of microseconds. Thus the trans-keto photoproduct is the photochromic form. In reality, the existence of many structural forms of the enol, cis-keto, and trans-keto isomers, with either zwitterionic or quinoidal configurations depending of the environment, complicates the general mechanism.31,32,42-45 Several excited cis-keto* states can arise from ESIPT with different possible geometries (twisted, planar, etc.), and it is generally not clear which of these excited states is the precursor of the final photoproduct46or whether the reaction involves the hot or the cold cis-keto* excited state.26
†Part of the “Hiroshi Masuhara Festschrift”.
* To whom correspondence should be addressed. (M.S.) Fax: + 33 3 20 43 67 55. Phone: + 33 3 20 33 63 53. E-mail: michel.sliwa@ univ-lille1.fr. (T.A.) Fax: +81 6 6879 7840. Phone: +81 6 6879 7839. E-mail: asahi@ap.eng.osaka-u.ac.jp.
‡UMR 8516 du CNRS. §PPSM CNRS UMR 8531.
|Nara Institute of Science and Technology and National Chiao Tung
University.
⊥Osaka University.
10.1021/jp901849a CCC: $40.75 2009 American Chemical Society Published on Web 05/15/2009
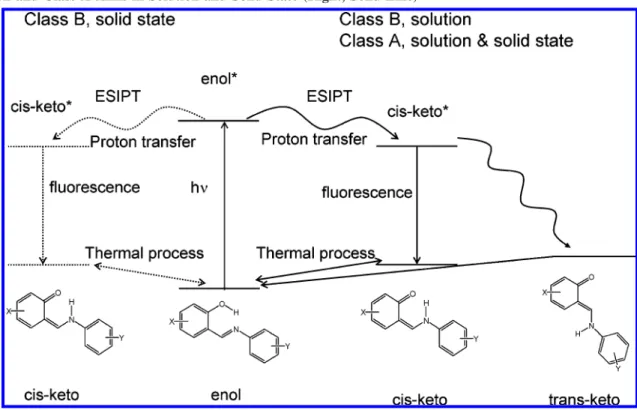
According to their solid-state photoreactivity, anils can be divided in two classes.21,22,35,38-40 Class A molecules are photochromic both in solution and in the crystalline state. They present a twist angle between the two rings in the enol form, are not fluorescent and crystallize generally in a noncompact arrangement where there is enough free space to allow molecules evolving toward the trans-keto form upon photoexcitation (Scheme 1). Back reaction from the keto to the enol form can occur both by irradiation with visible light or by thermal conversion. The rate determining step is the trans-keto to cis-keto isomerization, and the kinetics ranges from a few micro-seconds in solution to several months in solid state. Class B molecules behave like Class A molecules in solution (photo-chromic and non fluorescent), but are exclusively thermo(photo-chromic in the crystalline state: cis-keto and enol isomers coexist in the crystal via a temperature-dependent equilibrium. These anils are planar and crystallize in a compact way, which does not allow the final cis-trans rotation step to take place after ESIPT. Therefore, upon light excitation, ESIPT is followed only by relaxation to the cis-keto ground state form by a strong fluorescence, and rapid back proton transfer to the enol form to recover the initial equilibrium.
Due to technical complexity and difficulty to get photochro-mic anils in solid state, few reports exist on bulk crystal despite the fact that it is certainly the most interesting state regarding applications. Our research is thus now focused on the charac-terization at ultrafast time scale of photochromic materials like bulk crystals. In solid state, only femtosecond fluorescence studies were achieved on Class B salicylidene anilines which are not photochromic.33,34 For Class A anils, fluorescence experiments on the salicylidene-aniline (SA) in rigid matrix were done at low temperature and suggested a different mechanism involving two fluorescent cis-keto* species instead of one in solution.46 To the best of our knowledge, for Class A anils, excepted for our short preliminary report,47 no fast transient absorption studies of bulk crystals have been yet reported.
In this work, we present an investigation of two salicylidene aminopyridine isomers, N-(3,5-ditert-butylsalicylidene)-4-ami-nopyridine (4P) and N-(3,5-ditert-butylsalicylidene)-2-aminopy-ridine (2P) (Figure 1) because we have already reported studies of the stationary absorption properties of these compounds as polycrystalline thin films and evidenced by their potential utilization to photoswitch (4P)10,13or thermoswitch (2P)48 the SHG properties. 4P belongs to Class A, and its trans-keto form in polycrystalline film presents the longest lifetime in the dark among known anils (460 days), whereas 2P belongs to Class B. Temperature studies on polycrystalline film of 2P showed the existence of a mixture of enol and cis-keto forms at room temperature but the presence of exclusively the enol form at 120 K. The different behaviors of 2P and 4P upon photoexci-tation in solid state, contrasting with their similar molecular formulas, prompted us to undertake comparative investigations of their photodynamics both in solution and in the microcrys-talline powder state, with the final aim of getting a better overall understanding of anils photoreactivity. First, steady state and nanosecond spectroscopic studies will provide general informa-tion on the enol and photochromic trans-keto ground state forms which have a lifetime about a few µs for both compounds in solution. Then, ultrafast transient spectroscopy measurements in solution and in the solid state will inform on the photoinduced mechanisms in 2P and 4P and more generally on the photore-activity of Class A and Class B anils in solution and in the SCHEME 1: Overall Photoinduced Processes for Class B Anils in the Solid State (Left, Dashed Line); and Class B Anils in Solution and Class A Anils in Solution and Solid State (Right, Solid Line)
Figure 1. Molecular structures of N-(3,
5-ditert-butylsalicylidene)-2-aminopyridine (2P, Class B) and N-(3, 5-ditert-butylsalicylidene)-4-aminopyridine (4P, Class A).
solid state. Finally, previous X-ray structure results10,48joined to quantum chemical calculation data will complete the spec-troscopic data to lead to a comprehensive understanding of the photoinduced behavior of these compounds.
Experimental Section
Synthesis. All reagents (Aldrich) and solvents (SDS) are used as received. 2P and 4P are obtained by the condensation of equimolar amounts of 3,5-ditert-butylsalicylaldehyde and 2-ami-nopyridine and 4-ami2-ami-nopyridine respectively as mentioned elsewhere.10,48Purity of the product is checked by their melting point, FTIR spectra, NMR spectrum, and elemental analysis. Ground State Spectra. The ground state absorption spectra of microcrystalline powder samples are evaluated by Kubelka-Munk function49from the diffuse reflectance measured with a F-4500, Hitachi spectrophotometer. The photochromic trans-keto photoproduct is obtained by irradiation at 390 nm. Before measuring the spectrum of the enol form, the sample is irradiated with 500 nm light for a few minutes. Microcrystals of 2P and 4P are crushed to a powder using a mortar and dried. The average crystal size of the powder samples is about several tens of µm, confirmed by optical microscopy. Dry NaCl powder is used as diluent and puts in a quartz cell of 2 mm thickness with the compound. MgO powder which shows no absorption in the UV-visible region is used as a reference. The absorption is calculated using K/S ) (1 - R/R0)2/(2R/R0), where K and S represent respectively the absorption and scattering coefficients (cm-1) and R and R0are the diffuse reflectance of the compounds and MgO (cm-1), respectively.
UV-visible absorption spectra of solutions are recorded on a Varian Cary 5 in transmission mode. Spectrophotometric grade solvents are used. The fluorescence excitation spectra and the fluorescence quantum yields (φF), using quinine sulfate in 0.1 M sulfuric acid as the standard (φF ) 0.53), are recorded in F-4500 (Hitachi) and emission is corrected for the spectral sensitivity of the instrument. The absorbance of the solutions used to determine the fluorescence quantum yields and spectra do not exceed 0.1 at the excitation wavelength.
Nanosecond Transient Absorption Spectroscopy. Nanomi-crosecond transient absorption experiments are performed using a laser flash photolysis apparatus. Excitation pulses at 355 and (7-8 ns, 1 mJ) are provided by a 20-Hz Nd:YAG laser (DIVA II, Thales laser). The probe light is provided by a Xe lamp (XBO 150W/CR OFR, OSRAM). Samples are contained in a quartz cell (10× 10 mm2section) at a concentration adjusted (∼ 10-4 mol dm-3) to get an OD value of about 1.0 at the pump excitation wavelength. The transmitted light is dispersed by a monochromator (Horiba Jobin-Yvon, iHR320) and analyzed with a photomultiplier (R1477-06, Hamamatsu) coupled to a digitalized oscilloscope (LeCroy 454, 500 MHz).
Femtosecond Transient Absorption Spectra. The femto-second transient absorption set up for solution studies has already been described elsewhere.50,51 Briefly, in our experi-ments, an 1-kHz Ti:sapphire laser system (Coherent oscillators and a BM Industries regenerative amplifier) delivers 780 nm, 100 fs (0.8 mJ), pulses. Pump pulse is set at 390 nm by frequency doubling the fundamental beam (∼150 fs pulse duration). The pump pulse energy at the sample is 6 µJ with a pump diameter about 0.5 mm (3 mJ/cm2). The white light continuum probe beam is generated by focusing the fundamental beam in a 1 mm CaF2 rotating plate. The pump-probe polarization configuration is set at the magic angle (54.7°) and the probe pulse is delayed in time relative to the pump pulse using an optical delay line (Microcontrol Model, precision (0.1
µm). The white light continuum is split into a probe (with pump) and a reference beam (without pump). The transmitted light of the probe and reference beam is recorded on 2 different channels of a multichannel spectrograph equipped with a CCD camera (Princeton Instrument) and the transient spectra are computed. The transient absorption measurement covered a 400-750 nm spectral range and a 0-5 ns temporal range. The optical density variation accuracy is (0.001 (before averaging) in the spectral range of the experiments. Sample solution (absorbance of 0.6 at 390 nm∼10-3M) is circulating in a flow cell equipped of 1 mm thick CaF2windows and characterized by a 2 mm optical path length. The transient absorption intensity is displayed as absorption and the spectral data at one time delay are measured 4500 times and averaged. All experiments are carried out at 294 ( 2 K. All spectra analyzed are corrected for group velocity dispersion effect (GVD) using a method described by Nakayama et al. and completed by Ziolek et al.26,52,53The chirp of white light continuum is obtained by measuring two-photon absorption (TPA) in a thin BK7 microscope coverslip and in a cyclohexane neat solution taking into account cell windows dispersion. The overall time resolution is estimated to 200 fs in cyclohexane. The real instrumental response function used for the convolution with the kinetic exponential functions is determined separately for each wavelength, taking into account the cell thickness as proposed by Ziolek et al.26,53 The spectrokinetic data are corrected from nonlinear transient absorption signals and artifacts from the pure solvent using soft-modeling approach based on Multivariate curve resolution - alternating least-squares method (MCR-ALS). As described elsewhere, non parametric description of the solvent signal can be achieved, and used in a multiexperiment analysis to correct the measured data.54
The details of the femtosecond diffuse reflectance spectro-scopic system have also already been reported.55,56Briefly, the femtosecond light source consists of a self-mode-locked Ti:sapphire laser (Mira 900 Basic, Coherent), pumped by an Ar+ laser (Innova 310, Coherent), and a 10 Hz Ti:sapphire regenerative amplifier system (TR70, Continuum) with a Q-switched Nd:YAG laser (Surelight I, Continuum). The funda-mental output from the regenerative amplifier (780 nm, 3-4 mJ pulse-1, 170 fs fwhm, 10 Hz) is frequency doubled (390 nm) and used as a femtosecond excitation pulse. The excitation fluence dependence is examined by adjusting the pulse intensity using a polarizer and a λ/2 plate. The residual of the fundamental output is focused into a 1 cm quartz water cell in order to generate a white-light continuum as a probe light. In the case of powder samples, the transient absorption intensity is displayed as percentage absorption (%absorption), given by %absorption ) 100 × (1 - R/R0), where R and R0represent the intensity of the diffuse reflected white-light continuum of a probe pulse with and without excitation, respectively. In the case of 2P which presents no stable photoproduct, transient absorption spectra at one delay time are averaged over 30 times. On the contrary 4P does not allow measurements in usual conditions because the trans-keto photoproduct has a lifetime of about 460 days. So the spectral data changes are measured with a single shot excitation. The sample position is changed between two shots. For each time delay, 5 measurements are averaged. Therefore measurements for 4P in solid state have a rather bad signal-to-noise ratio. To avoid nonlinear effects occurring in the solid state (multiphoton absorption, ionization)57 and to confirm a linear relation between %absorption and the concentration of absorbing species, excitation fluence is limited to 3 mJ/cm2, as for solution experiments. This value is chosen by checking the linearity of %absorption at 10 ps vs fluence. The temporal
resolution of transient absorption measurements with this spectroscopic system is about 1 ps. The experimental decay curves are analyzed with one, two or three exponential functions without correction. The results are judged by the value of the reduced chi square parameter and the appearance of the residuals. All experiments are carried out at 294 ( 2 K.
Theoretical Calculations. Quantum chemical calculations are performed using Gaussian 03 suite of programs with the 6-31G** basis set.58The ground state geometries are optimized by DFT/B3LYP method and calculations of the transitions energies are made by TD/DFT method.
Results and Discussion
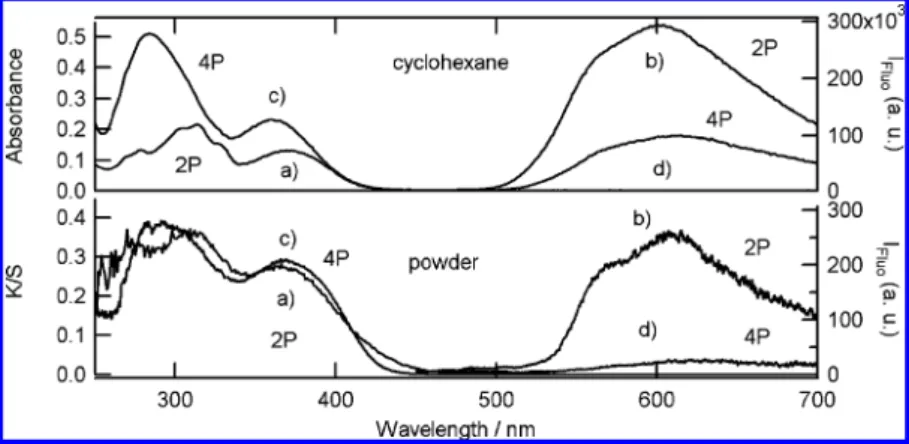
Ground State Enol Absorption and Fluorescence Spectra. In order to allow comparison of experiments in solution and solid state, a nonpolar solvent without hydrogen bonding is considered and cyclohexane is chosen regarding the compounds solubility. Absorption spectra and fluorescence emission excited at 390 nm (pump wavelength for femtosecond transient mea-surements) in cyclohexane (∼10-6M) and for microcrystalline powder (Kubelka-Munk function) of 2P and 4P at room temperature are shown in Figure 2.
In cyclohexane two main absorption bands are observed for 2P and 4P in the UV region (around 365 and 300 nm, Table 1). Similar bands were reported for others anils.59,60These two broad absorption bands are assigned to the enol tautomer. The fluorescence spectra of 4P and 2P show a weak and broadband from 500 to 700 nm with a strong Stoke shift characteristic of the cis-keto* form. For 2P, this broad fluorescence shows one maximum at 602 nm and a shoulder (Table 1) at 562 nm (∆ν ) 1180 cm-1) that are slightly red-shifted for 4P (∆ν ) 1240 cm-1). The fluorescence quantum yield is extremely weak (ΦF is equal to 2.5× 10-4and 7× 10-5for 2P and 4P, respectively), which is consistent with general results on the fluorescence of
both Classes of anils in solution (Class A and Class B anils are considered as non fluorescent in solution).
For microcrystalline powder, 2P and 4P absorbance spectra are similar to those in cyclohexane, with only a slight hypso-chromic shift of the first maximum for 2P and a small bathochromic shift for 4P (Table 1). In addition for 2P, an absorption tail above 450 nm is observed, which is typical in Class B anils of the coexistence of the cis-keto form in the crystal at room temperature. Fluorescence spectra for microc-rystalline powder show two different features for 2P and 4P. 2P, which belongs to Class B anils, shows a strong fluorescence, whereas 4P, which belongs to Class A anils, is almost nonfluorescent. One maximum and a shoulder are clearly observed for 2P as in cyclohexane but are slightly red-shifted by about 5 nm (Table 1). For 4P only one red-shifted maximum (∆ν ) 20 nm) remains for the powder sample.
Only one maximum is generally reported for the fluorescence of anils in solution. Different origins can be considered for the two-component emission observed for 2P and 4P. The about 1200 cm-1 energy difference between the shoulder and the maximum could be attributed to a vibronic structure or to the existence of two cis-keto* species named cis-keto1* and cis-keto2* in the following (longest and shortest wavelength, respectively). The later hypothesis is in agreement with results reported by Higelin et al. for the SA (N-salicylidene-aniline) in dibenzyl host crystals who found two fluorescent species in matrix at low temperature that they assumed to have planar and partially twisted structures, respectively, but did not have any evidence about their nature.46 This dual fluorescence is more pronounced for 2P in solid state for which cis-keto form already exists at room temperature. Thus one hypothesis for this dual fluorescence is a combination of fluorescence from a small amount of cis-keto excited directly and from enol form after ESIPT. However emission spectra for 2P and 4P in cyclohexane Figure 2. Steady-state absorption and fluorescence (excitation wavelength 390 nm) spectra for 2P (a and b) and 4P (c and d) in cyclohexane (top)
and microcrystalline powder (bottom).
TABLE 1: First Two Transitions and Inter-Ring Torsion Angle Calculated for the 2P and 4P Ground State Enol Using the TD/DFT B3LYP/6-31G** Method from the ab Initio DFT/6-31G** Optimized Geometries, As Compared to Experimental Absorption, Fluorescence, and X-ray Data Obtained in Cyclohexane Solution and for Powder Samples
2P 4P
DFT/6-31G** cyclohexane crystal DFT/6-31G** cyclohexane crystal
absorption spectrum maxima (nm) 380 370 366 365 361 368 313 313 309 305 284 293 fluorescence maxima (nm) 562 569 567 602 608 610 630 φF 2.5× 10-4 7× 10-5 torsion angle 0° 6.9° 37.8° 41.8°
for different excitation wavelengths are identical. Moreover extinction spectra for the shoulder and fluorescence maximum are identical to the enol absorption spectra of 2P and 4P (Supporting Information). Recent quantum modelings were done for SA and they showed that ESIPT occurs from a planar enol* which gives a planar cis-keto*. This planar cis-keto* is not stable and undergos out-of-plane torsion which is the precursor of cis-trans isomerization to the trans-keto photoproduct.61Thus one hypothesis for the two fluorescent cis-keto* is a planar and a nonplanar isomer or different nonplanar isomers. The bulky tert-butyl group can stabilize them and favor the existence of two fluorescent cis-keto* species in comparison with SA. The difference in fluorescence intensity between 2P and 4P in solid state can be well explained by their different Classes: as mentioned above, the cis-keto* in Class B anils does not lead to the trans-keto photoproduct and relaxes by non radiative decays (internal conversion) and fluorescence to cis-keto ground state. In contrast, fast formation of the trans-keto photoproduct in Class A anils quenches the fluorescence of the cis-keto*. In solution, however, both Class A and Class B anils lead to the trans-keto photoproduct after the ESIPT, which explains the weak fluorescence of the cis-keto*. Our previously reported X-ray data10,48confirm that 2P is planar and has a crystallization more compact than 4P which is twisted (torsion angle given in Table 1). Ab initio studies of the molecular geometries (DFT/ 6-31G**) and electronic transitions (TD-DFT) of the enol ground state were achieved and compared with absorption and X-ray experimental values. Both the DFT/6-31G** and X-ray structures agree to point out a twisted conformation for 4P enol with similar angles (37.8° and 41.8°, respectively) and a planar conformation for 2P enol (0° and 6°, respectively). Considering the absorption bands, TD-DFT calculations are also reasonably consistent with the experimental data (Table 1). Therefore, good agreement between experiments and calculations is found not only for structural but also spectral data. The enol ground state seems thus to have similar geometries in the gas phase, cyclohexane solution, and crystal state.
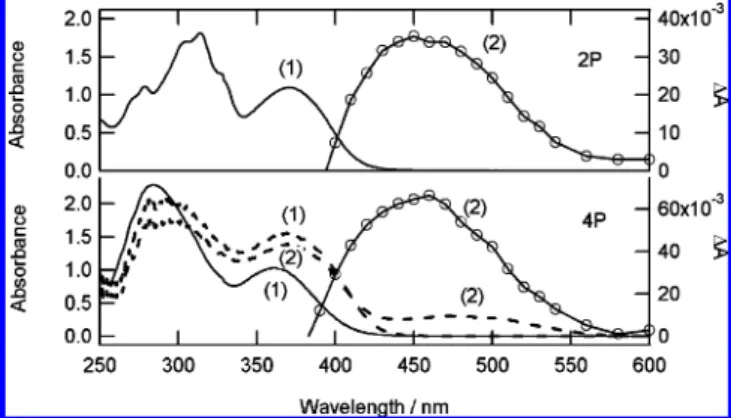
Absorption Spectra of the Trans-Keto Photoproduct. The trans-keto photoproduct of 4P is very stable in the solid state and the back reaction to the enol form can be fitted mainly by a monoexponential kinetics (time constant of 460 days), plus an additional minor component (0.1%, time constant 8 h).102P which belongs to Class B shows no trans-keto photoproduct for microcrystalline powder. In solution, the trans-keto photo-product can be detected only by flash photolysis. For both 2P and 4P compounds, transient spectra in cyclohexane show a positive band from 400 to 600 nm with a maximum at about 460 nm (Figure 3) and a negative band below 400 nm (not shown). Both positive and negative bands relax with a biexpo-nential decay with similar time constants and relative amplitude at all wavelengths: 122 µs (45%) and 19 µs (55%) for 4P and 186 µs (62%) and 35 µs (38%) for 2P. These decay kinetics are insensitive to the presence of oxygen and thus cannot be related to any triplet state. By analogy with other anils, the absorption is unambiguously assigned to the trans-keto photo-product and the negative band to the ground state depletion of the initial enol. For 4P, this assignment is consistent with the fact that the transient absorption in cyclohexane (λmax) 460 nm) is similar to the absorption spectrum of the stable trans-keto photoproduct in microcrystalline powder (λmax) 475 nm). Differences in the trans-keto absorption maxima between solution and solid state should refer to different conformations stabilized by the solvent and the environment in the crystal. The decay of the trans-keto photoproduct of SA in solution is
monoexponential with a time constant of few ms.26 The biexponential decay observed for 2P and 4P in cyclohexane can be assigned to the presence of different trans-keto isomers having similar spectral signatures. Similarly to the possible existence of two fluorescent cis-keto* species, bulky tert-butyl group can favor the existence of different trans-keto isomers in solution. In the solid state, the observation of only one trans-keto isomer for 4P can be tentatively accounted for to the existence of strong intermolecular interactions in the rigid matrix.
Femtosecond Transient Absorption in Solution. Transient absorption spectra of 4P and 2P are measured in cyclohexane following excitation at 390 nm. Spectra at different time delays are shown in Figure 4. 4P and 2P transient absorption spectra are quite similar, in agreement with the fact that Class B and Class A anils have usually the same behavior in solution. At short time, two main bands are observed, the intensity of which decreases with time: a positive absorption band covering the 400-600 nm domain which has one maximum around 430 nm and a shoulder around 510 nm (Table 2) and a weak negative band (600-750 nm). A rather clear crossover point is observed around 600 nm. After complete decay of this initial spectrum (50-100 ps), the spectral features change totally and show a single, very broad positive absorption band in the 400-550 nm range that remains nearly constant until 5 ns.
Taking into account the fact that it is notably truncated by the presence of the partly overlapping transient absorption band on its high energy side, the position of the short-time negative band corresponds satisfactorily to that of the steady-state emission related to cis-keto* fluorescent species. This band is thus assigned to the stimulated emission of the cis-keto* species formed after the ESIPT reaction. Kinetics are fitted taking into Figure 3. Steady-state absorption spectra before (1) and after (2)
irradiation of 2P (top) and 4P (bottom) in 10-4M cyclohexane solution (λexc) 355 nm, solid line, right scale) and in 0.1 wt % microcrystalline powder in NaCl powder (λexc) 390 nm, dashed line, left scale).
TABLE 2: Fitted Time Constants for 2P and 4P Kinetics in Cyclohexane (in ps)a positive absorption band maximum/ps positive absorption band shoulder/ps negative absorption band maximum/ps 435 nm 510 nm 630 nm 2P 4.3 (0.03) 3 (0.01) 5 (-0.003) 25 (0.15) 24 (0.06) 25 (-0.02) 440 nm 515 nm 650 nm 4P 0.850 (0.01) 1 (0.003) 4.5 (0.08) 4.5 (0.03) 4.5 (-0.008) 14.5 (0.06) 14.5 (0.02) 14.5 (-0.008)
aValues are within (0.5 ps for time constants below 5 ps and within (1 ps for longer ones. Values in parentheses indicate pre-exponential factors associated with each time constant.
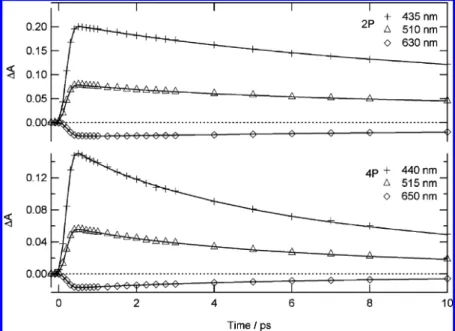
account the instrumental response function (Figure 5, Table 2). For both 2P and 4P, the stimulated emission can be well fitted with the convolution of the instrumental response function and two exponential decay components. Thus the negative band assigned to cis-keto* has the same apparent risetime as the 200 fs instrumental response function, which means that the ESIPT process is much faster and cannot be resolved with our experimental setup. This conclusion is in agreement with the 50 fs found for ESIPT in the case of SA in solution.26 The biexponential decay of the stimulated emission is characterized, at the maximum, by time constants of 14.5 ps (50%) and 4.5 ps (50%) for 4P and 25 ps (84%) and 5 ps (16%) for 2P. This biexponential kinetics differs from the 6.8 ps monoexponential decay reported for SA in solution26and strongly suggests the existence of two cis-keto* isomers in the case of 2P and 4P. This suggestion can be related to our above observation that two components (one maximum and one shoulder respectively) are present in the steady-state fluorescence spectra of 2P and 4P in solution, and supports the assignment of these components to two distinct fluorescent cis-keto* species, cis-keto1* and cis-keto2*, having different lifetimes, rather than to some vibronic structure. If we assume similar transition moments for these two species, the longest-lived one can be tentatively correlated to the most intense spectral component (∼600 nm maximum) in the steady-state fluorescent spectrum. Furthermore the longer decay time found for 2P is in agreement with its higher fluorescence quantum yield.
For the positive absorption band, the kinetics measured for 2P can also be well fitted with the convolution of the instrumental response function and two exponential decays of time constants similar to those found for the stimulated emission band with different relative contributions at all wavelengths (Table 2). This absorption band can thus be unambiguously considered as the signature of the two fluorescent cis-keto* species. For 4P, a third shorter component (<2 ps) is needed to fit the absorption kinetics in addition to the two components also characterizing the stimulated emission decay. Such a short
component has been already observed for different anils and two possible origins were proposed:24,26(a) a relaxation of the enol* species from which the ESIPT occurs and/or (b) a vibrational cooling of a hot S1cis-keto* formed after ESIPT. From femtosecond time-resolved fluorescence measurements of SA in solution, the decay of the hot S1 cis-keto* has been evaluated to a few hundred femtoseconds and the relaxation of the enol* to less than 100 fs.27For 4P in cyclohexane, the short absorption decay component being notably longer than the ESIPT process, found to be faster than 200 fs, a contribution of the enol* seems excluded. We thus propose to assign this short component predominantly to vibrational cooling of the hot cis-keto*. The fact that this short-time decay component is not observed for 2P can be due to a faster vibrational relaxation of the hot cis-keto* or smaller energy difference between the enol* and fluorescent cis-keto* species. As mentioned previously (Table 1) different geometry between 2P (planar) and 4P (twisted) for the enol ground state were found. Thus for 2P and 4P different energy and isomer (planar, nonplanar) can be safely considered for enol* and cis-keto* after the ESIPT and might explain that no hot-cis-keto* component were observed.
The residual broad positive absorption in the 400-550 nm range looks like the spectrum of the final trans-keto photoproduct measured at 355 nm excitation with the nanosecond transient absorption experiment for both 4P and 2P (Figure 3). It is not possible to establish which one of the cis-keto* isomers (or both) are the precursor of the final trans-keto photoproduct. From our results, the general scheme of the photodynamics of salicylidene-aminopyridine in cyclohexane is similar (nature of the processes involved and time constants) to that already reported for SA (Scheme 1) except the coexistence of two fluorescent cis-keto* forms.
Solid State Transient Absorption Spectra. As it was discussed previously, Class A and Class B anils show different behaviours after photoexcitation of powder samples. Class B anils is analyzed first as no trans-keto photoproduct is generated. 1. Class B Anil, 2P. Transient absorption spectra for micro-crystalline powder of 2P after excitation at 390 nm are shown in Figure 6. The poor quality of spectra below 500 nm is due to the existence of an absorbing ground state cis-keto population at room temperature. The time evolution can be divided into two steps: first, the rise of a positive absorption band between 450 and 580 nm and a strong negative band between 580 and 750 nm in a few ps and second, the decay of both positive and negative bands with a clear crossover point at 580 nm.
Only one maximum is observed for the positive absorption band in comparison with cyclohexane spectra which show two maxima. This is related to the poor quality of spectra below 500 nm. The shape and position of the maximum of the negative band are almost identical in cyclohexane and in the microcrys-talline powder. Furthermore a crossover point is also found around 580 nm. By analogy with solution results, the positive absorption and negative components are thus assigned to absorbance and stimulated emission of cis-keto* species. However, in solid state, no remaining absorption band is observed after the decay of the cis-keto* spectrum. This is consistent with the Class B character of 2P, for which the cis-keto* cannot evolve to the trans-keto form in the solid state. Therefore the transient absorption in solid state is essentially characteristics of cis-keto* species.
For 2P microcrystalline powder, like in solution, the ultrafast risetime of all the bands (positive and negative) implies that ESIPT arises within the time resolution of our experimental setup (<1 ps), which is in agreement with previous femtosecond Figure 4. Transient absorption spectra of 2P (top) and 4P (bottom) in
cyclohexane obtained for different time delays after 390 nm excitation (3 mJ/cm2).
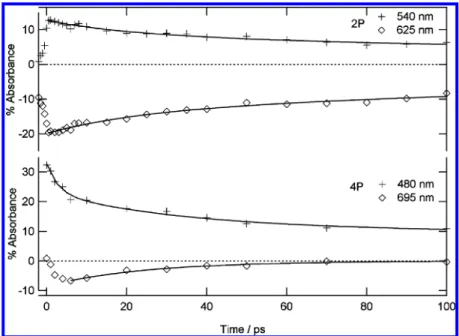
fluorescence spectroscopy studies of thermochromic crystalline salicylidene derivatives.33,34It is difficult to determine precisely an instrumental response function in diffuse reflectance experi-ments. Therefore only the decay part of the kinetics is fitted (Figure 7). The negative band assigned to the stimulated emission of cis-keto* decays at 625 nm with a biexponential kinetics of time constants 250 (70%) and 23 ps (30%). Similarly to solution results, the biexponential decay is attributed to the presence of two fluorescent cis-keto*, cis-keto1*, and cis-keto2*. For the positive absorption band, the 540 nm decay can be fitted by a biexponential function with 256 ps (60%) and 25 ps (40%) time constants. These two time constants are almost equal to the decay components of the stimulated emission band (Table 3). As shown above for 2P in solution, a shorter component assigned to vibrational cooling of hot cis-keto* is also not
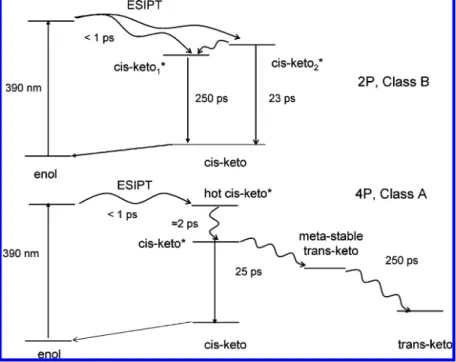
present. However for 4P in cyclohexane the hot cis-keto* components were found mainly below 500 nm (Table 2) and the poor quality of the data below 500 nm does not allow us to conclude on the existence or not of hot cis-keto* in the powder sample. In previous femtosecond fluorescence spectroscopy studies of thermochromic crystalline salicylidene aniline deriva-tives, only one decay of several hundreds ps time constant for cis-keto* was found, 33,34 which is in agreement with the presence of only one maximum found in their steady-state fluorescence spectrum. Like in solution, if we assume similar transition moment, the longest-lived cis-keto* species can be correlated to the most intense component in the steady-state fluorescent spectrum. Finally a scheme for the photodynamics of 2P in solid state can thus be proposed (Scheme 2). Figure 5. Kinetic traces of transient absorption at characteristic wavelengths for 2P (top) and 4P (bottom) in cyclohexane and best fits (full line).
2. Class A Anil, 4P. Figure 8 presents the transient absorption spectra for 4P microcrystalline powder after excitation with a femtosecond 390 nm laser pulse (3 mJ/cm2). Just after the excitation, the spectrum shows one broadband with two maxima at 445 and 550 nm. In a few picoseconds, this broad positive absorption starts its decay and a negative band covering the 630-750 nm spectral range appears. Both the positive and the negative absorption band decay in few tens of picoseconds and a broad positive absorption band at 520 nm remains. This band gradually shifts in few hundred picoseconds to yield a final broad absorption band peaking around 480 nm and stable until 5 ns. The initial short time absorption spectrum is similar to that found for cis-keto* in cyclohexane (although slightly red-shifted) except that all of the bands are rising within the instrumental time response function in cyclohexane (Figure 4). Therefore the positive and negative bands are assigned with confidence to cis-keto* species. We conclude that, like in cyclohexane, ESIPT in solid state is faster than the temporal resolution of our experiments and occurs within 1 ps. The final absorption spectrum (480 nm band) is also undoubtedly assigned to the trans-keto species by analogy with the steady-state absorption characterization of this stable photoproduct in microcrystalline powder. Although the bad signal-to-noise ratio makes difficult a rigorous quantitative analysis of the kinetics, we found that the decay of the stimulated emission band can be fitted by a single-exponential with 25 ps time constant whereas, as discussed above, two exponentials are needed in cyclohexane.
Such monoexponential fit is in agreement with the fluorescence steady-state spectrum which shows only one maximum. The time evolution of the positive transient absorption can be appropriately fitted by a three-exponential decay with lifetimes of 2, 30, and 250 ps and different relative contributions at all wavelengths (Table 3). By analogy with the results in cyclo-hexane, the shortest decay time is assigned to vibrational cooling of hot cis-keto*. Then the second component is equal to the decay constant of the stimulated emission band and assigned to fluorescent cis-keto* relaxation. Finally, the third kinetic component corresponds to the shift of the residual 520 nm absorption to the final trans-keto 480 nm absorption, with a time-constant of 250 ps. It reveals thus the existence of an intermediate species between the cis-keto* species and trans-keto photoproduct in the solid state. The nature of this intermediate is questionable. Its absorption spectrum shows notable analogy with that of the final trans-keto photoproduct. A hypothesis is that, in the microcrystalline state, steric constraints due to the solid environment prevent direct formation of the final trans-keto photoproduct and induce a two-step mechanism in the structural relaxation of the trans-keto structure involving an intermediate metastable trans-keto-type isomer in the ground state. On this basis, a tentative photodynamics scheme can be drawn for 4P (Scheme 2).
Conclusions
The study presented here enabled us for the first time to undertake a solid state vs solution comparison of the dynamic behavior at the picosecond time scale of two neighboring salicylidene-aminopyridines, 4P and 2P, which are representative of the Class A and Class B anils, respectively. In both cases, ESIPT is faster than the temporal resolution of our experiments with time constants less than 200 fs in cyclohexane and less than 1 ps in microcrystalline powder. From the analysis of steady-state fluorescence spectra and stimulated emission decay kinetics, the existence of two fluorescent cis-keto* species (cis-keto1* and cis-keto2*) is evidenced except for 4P in micro-crystalline powder. This behavior is different from previous results on anils in solution24,26that report the formation of only one fluorescent cis-keto* species. Such a difference can probably be related to the presence of bulky tert-butyl substituents in 2P Figure 7. Kinetic traces of transient absorption at characteristic wavelengths for 2P (top) and 4P (bottom) powder samples and best fits (full line).
TABLE 3: Fitted Time Constants for 4P and 2P Kinetics in Powder Sample (in ps)a
positive absorption band wavelength/ps negative absorption band maximum/ps 540 nm 625 nm 2P 25 (5) 23 (6) 256 (7) 250 (14) 480 nm 545 nm 695 nm 4P 2.8 (9.6) 2.3(12) 31.4 (11) 29.5 (22) 25.0 250 (5.6) 251.5 (12) a
Values in parentheses indicate pre-exponential factors associated with each time constant.
and 4P that can favor the coexistence of several cis-keto* geometries. The exact nature (geometry) of these two cis-keto* species is not clear. For 4P both in solution and in powder sample, a fast decay component (a few ps) of the cis-keto* transient absorption is observed and attributed mainly to vibrational cooling. This short-time decay component is not observed for 2P and it is also attributed to different geometries between 2P and 4P in the excited state (observed already for enol ground state, 2P planar and 4P non planar) which can lead to faster vibrational relaxation of the hot cis-keto* or smaller energy difference between the enol* and fluorescent cis-keto* species after the ESIPT. In solution, the data confirm that photoexcitation of both Classes of anils leads to the final trans-keto photoproduct directly from the cis-trans-keto* in a few tens of
picoseconds. The general scheme in solution is similar to that reported for SA (Scheme 1) except that the ESIPT leads to two fluorescent cis-keto*. For 4P microcrystalline powder sample, an additional intermediate species assigned to a “metastable” ground state trans-keto form is clearly characterized for the first time following the cis-keto* decay and shown to relax to the stable trans-keto photoproduct in 250 ps. As expected, 2P microcrystalline powder, which belongs to Class B anils, does not show any trans-keto photoproduct in the solid state. A general scheme of the photodynamics of 2P and 4P in solid state has been proposed (Scheme 2). Even if this scheme does not apply with certainty to all anils (number and nature of cis-keto* and trans-keto isomers), it should be helpful for the understanding of Schiff bases photodynamics in solid state. SCHEME 2: Photodynamics of 2P (Top, Class B) and 4P (Bottom, Class A) Microcrystalline Powder Following 390 nm Excitation
Finally although the solid state kinetics are slowed down compared to the solution ones, the overall enol to trans-keto switching process appears to occur within 250 ps for crystalline 4P, which can thus be considered as a fast photoswitch for its implementation in devices. Future investigations involving calculations of excited state geometries for 2P and 4P and other experiments such as subpicosecond time-resolved IR are under progress and should provide more detailed information on the nature of the intermediate species, the nature of different cis-keto*, the absence of hot-cis-keto* relaxation for 2P and the dynamics of formation of the trans-keto photoproduct.
Acknowledgment. The authors thank the Groupement de Recherche GDRI 93 from CNRS and the Centre d′e´tudes et de Recherches Lasers et Applications (CERLA) for their help in the development of this work. CERLA is supported by the Ministe`re charge´ de la Recherche, Re´gion Nord/Pas de Calais, and the Fonds Europe en de De´veloppement Economique des Re´gions. The authors acknowledge Ecole Doctoral Physico-Chimie du Sud de Paris, and French-Japanese Doctoral College for financial support.
Supporting Information Available: Absorption, extinction and emission (for different wavelengths) spectra of 4P and 2P in cyclohexane. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) Photochromism; Brown, G. H., Ed.; Wiley-Interscience: New York, 1971.
(2) Photochromism: Molecules and Systems; Du¨rr, H., Bouas-Laurent, H., Eds.; Elsevier: Amsterdam, The Netherlands, 1990.
(3) Organic Photochromic and Thermochromic Compounds; Crano, J. C., Guglielmetti, R. J., Eds.; Plenum: New York, NY, 1998.
(4) Bouas-Laurent, H.; Durr, H. Pure Appl. Chem. 2001, 73, 639. (5) Irie, M. Chem. ReV. 2000, 100, 1683.
(6) Alfimov, M. V.; Fedorova, O. A.; Gromov, S. P. J. Photochem. Photobiol., A 2003, 158, 183.
(7) Raymo, F. M.; Alvarado, R. J.; Giordani, S.; Cejas, M. A. J. Am. Chem. Soc. 2003, 125, 2361.
(8) Sworakowski, J.; Nespurek, S.; Toman, P.; Wang, G.; Bartkowiak, W. Synth. Met. 2004, 147, 241.
(9) Raymo, F. M.; Tomasulo, M. Chem. Soc. ReV. 2005, 34, 327. (10) Sliwa, M.; Letard, S.; Malfant, I.; Nierlich, M.; Lacroix, P. G.; Asahi, T.; Masuhara, H.; Yu, P.; Nakatani, K. Chem. Mater. 2005, 17, 4727. (11) Spangenberg, A.; Sliwa, M.; Metivier, R.; Dagnelie, R.; Brosseau, A.; Nakatani, K.; Pansu, R.; Malfant, I. J. Phys. Org. Chem. 2007, 20, 992.
(12) Sliwa, M.; Spangenberg, A.; Metivier, R.; Letard, S.; Nakatani, K.; Yu, P. Res. Chem. Intermed. 2008, 34, 181.
(13) Sliwa, M.; Nakatani, K.; Asahi, T.; Lacroix, P. G.; Pansu, R. B.; Masuhara, H. Chem. Phys. Lett. 2007, 437, 212.
(14) Coe, B. J. Chem.--Eur. J. 1999, 5, 2464.
(15) Asselberghs, I.; Zhao, Y.; Clays, K.; Persoons, A.; Comito, A.; Rubin, Y. Chem. Phys. Lett. 2002, 364, 279.
(16) Bertarelli, C.; Gallazzi, M. C.; Lucotti, A.; Zerbi, G. Synth. Met.
2003, 139, 933.
(17) Asselberghs, I.; Clays, K.; Persoons, A.; Ward, M. D.; McCleverty, J. J. Mater. Chem. 2004, 14, 2831.
(18) Sanguinet, L.; Pozzo, J. L.; Rodriguez, V.; Adamietz, F.; Castet, F.; Ducasse, L.; Champagne, B. J. Phys. Chem. B 2005, 109, 11139.
(19) Plaquet, A.; Guillaume, M.; Champagne, B.; Castet, F.; Ducasse, L.; Pozzo, J. L.; Rodriguez, V. Phys. Chem. Chem. Phys. 2008, 10, 6223. (20) Plaquet, A.; Guillaume, M.; Champagne, B.; Rougier, L.; Mancois, F.; Rodriguez, V.; Pozzo, J. L.; Ducasse, L.; Castet, F. J. Phys. Chem. C
2008, 112, 5638.
(21) Hadjoudis, E.; Mavridis, I. M. Chem. Soc. ReV. 2004, 33, 579. (22) Amimoto, K.; Kawato, T. J. Photochem. Photobiol., C 2005, 6, 207.
(23) Elsaesser, T.; Bakker, H. J. Ultrafast hydrogen Bonding Dynamic and Proton Transfer Processes in the condensed Phase; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2002.
(24) Mitra, S.; Tamai, N. Phys. Chem. Chem. Phys. 2003, 5, 4647. (25) Vargas, C. V. J. Phys. Chem. A 2004, 108, 281.
(26) Ziolek, M.; Kubicki, J.; Maciejewski, A.; Naskrecki, R.; Grabowska, A. Phys. Chem. Chem. Phys. 2004, 6, 4682.
(27) Rodriguez-Cordoba, W.; Zugazagoitia, J. S.; Collado-Fregoso, E.; Peon, J. J. Phys. Chem. A 2007, 111, 6241.
(28) Ziolek, M.; Burdzinski, G.; Filipczak, K.; Karolczak, J.; Macie-jewski, A. Phys. Chem. Chem. Phys. 2008, 10, 1304.
(29) Mitra, S.; Tamai, N. Chem. Phys. 1999, 246, 463.
(30) Ziolek, M.; Filipczak, K.; Maciejewski, A. Chem. Phys. Lett. 2008, 464, 181.
(31) Otsubo, N.; Okabe, C.; Mori, H.; Sakota, K.; Amimoto, K.; Kawato, T.; Sekiya, H. J. Photochem. Photobiol., A 2002, 154, 33.
(32) Okabe, C.; Nakabayashi, T.; Inokuchi, Y.; Nishi, N.; Sekiya, H. J. Chem. Phys. 2004, 121, 9436.
(33) Sekikawa, T.; Kobayashi, T. J. Phys. Chem. A 1997, 101, 644. (34) Sekikawa, T.; Kobayashi, T.; Inabe, T. J. Phys. Chem. B 1997, 101, 10645.
(35) Cohen, M. D.; Schmidt, G. M. J. J. Phys. Chem. 1962, 66, 2442. (36) Nakagaki, R.; Kobayashi, T.; Nakamura, J.; Nagakura, S. Bull. Chem. Soc. Jpn. 1977, 50, 1909.
(37) Barbara, P. F.; Rentzepis, P. M.; Brus, L. E. J. Am. Chem. Soc.
1980, 102, 2786.
(38) Hadjoudis, E. J. Photochem. 1981, 17, 355.
(39) Hadjoudis, E.; Vittorakis, M.; Moustakali-Mavridis, I. Tetrahedron
1987, 43, 1345.
(40) Hadjoudis, E. Mol. Eng. 1995, 5, 301.
(41) Harada, J.; Uekusa, H.; Ohashi, Y. J. Am. Chem. Soc. 1999, 121, 5809.
(42) Yuzawa, T.; Takahashi, H.; Hamaguchi, H. Chem. Phys. Lett. 1993, 202, 221.
(43) Knyazhansky, M. I.; Metelitsa, A. V.; Bushkov, A. J.; Aldoshin, S. M. J. Photochem. Photobiol., A 1996, 97, 121.
(44) Knyazhansky, M. I.; Metelitsa, A. V.; Kletskii, M. E.; Millov, A. A.; Besugliy, S. O. J. Mol. Struct. 2000, 526, 65.
(45) Zgierski, M. Z.; Grabowska, A. J. Chem. Phys. 2000, 112, 6329. (46) Higelin, D.; Sixl, H. Chem. Phys. 1983, 77, 391.
(47) Asahi, T.; Masuhara, H.; Nakatani, K.; Sliwa, M. Mol. Cryst. Liq. Cryst. 2005, 431, 541.
(48) Sliwa, M.; Spangenberg, A.; Malfant, I.; Lacroix, P. G.; Metivier, R.; Pansu, R. B.; Nakatani, K. Chem. Mater. 2008, 20, 4062.
(49) Kortu¨m, G. Reflectance Spectroscopy; Springer-Verlag: Berlin, Heidelberg, NY, 1969.
(50) Buntinx, G.; Naskrecki, R.; Poizat, O. J. Phys. Chem. 1996, 100, 19380.
(51) Moine, B.; Rehault, J.; Aloise, S.; Micheau, J. C.; Moustrou, C.; Samat, A.; Poizat, O.; Buntinx, G. J. Phys. Chem. A 2008, 112, 4719.
(52) Nakayama, T.; Amijima, Y.; Ibuki, K.; Hamanoue, K. ReV. Sci. Instrum. 1997, 68, 4364.
(53) Ziolek, M.; Lorenc, M.; Naskrecki, R. Appl. Phys. B: Lasers Opt.
2001, 72, 843.
(54) Ruckebusch, C.; Sliwa, M.; Re´hault, J.; Naumov, P.; Huvenne, J. P.; Buntinx, G. Anal. Chim. Acta 2008, 642, 228.
(55) Asahi, T.; Furube, A.; Fukumura, H.; Ichikawa, M.; Masuhara, H. ReV. Sci. Instrum. 1998, 69, 361.
(56) Suzuki, M.; Asahi, T.; Masuhara, H. J. Photochem. Photobiol., A
2006, 178, 170.
(57) Asahi, T.; Suzuki, M.; Masuhara, H. J. Phys. Chem. A 2002, 106, 2335.
(58) Frisch, M. J.; Gaussian 03, revision C.02; Gaussian, Inc.: Wall-ingford, CT, 2004.
(59) Cohen, M. D.; Flavian, S. J. Chem. Soc., B 1967, 321. (60) Hadjoudis, E.; Moustakalimavridis, I.; Xexakis, J. Isr. J. Chem.
1979, 18, 202.
(61) Ortiz-Sanchez, J. M.; Gelabert, R.; Moreno, M.; Lluch, J. M. J. Chem. Phys. 2008, 129.