Ying-Chieh Weng
Department of Chemical Engineering, National Taiwan University, Taipei 106, Taiwan, Republic of China
I.A. Rusakova and Andrei Baikalov
Texas Center for Superconductivity and Advanced Materials, University of Houston, Houston, Texas 77204-5932
J.W. Chen
Department of Physics, National Taiwan University, Taipei 106, Taiwan, Republic of China Nae-Lih Wua)
Department of Chemical Engineering, National Taiwan University, Taipei 106, Taiwan, Republic of China
(Received 28 April 2004; accepted 21 September 2004)
Nanocrystalline magnetite powders were synthesized by an electrocoagulation technique, in which an electric current was passed across two plate electrodes of carbon steel immersed in NaCl(aq) electrolyte, and the microstructure of the oxide powder was found to evolve in roughly three stages. The first stage involves formation and growth of severely defective colloidal crystallites. This is followed by
agglomeration among the oxide crystallites to form mesoporous agglomerates containing predominantly inter-crystallite pores, and the average crystallite size was found to reach a plateau. Finally, coarsening of the crystallites within the agglomerates leads to another rapid increase in crystallite size and reduction in pore opening. The synthesized powders typically showed a saturation magnetization of ∼75 emu/g and a coercivity Hcof ∼118 Oe. A mechanism involving competition between nucleation and growth of free colloids and coarsening of the skeletal framework was proposed to explain the temporary level-off in crystallite size during the synthesis.
I. INTRODUCTION
Magnetite (Fe3O4) has long been a material of both industrial and scientific interests. It has been used, for instance, as a black pigment,1a recording media,2a mag-netic component in magmag-netic fluids,3and a magnetic car-rier for bioseparation and drug delivery.4,5 Photocatalyst-coated magnetite has also been suggested for decompo-sition of organics in wastewater treatment.6 Different applications may prefer different microstructural proper-ties, in addition to the magnetic ones. For instance, the magnetic fluid and recording applications favor nonpo-rous nanocrystalline particles, while catalysis and bio-logical and medical applications prefer large surface area and mesoporosity. As a result, many synthesis methods, such as precipitation,7microemulsion,8hydrothermal,9,10 solventothermal,11,12 and microwave hydrothermal,13,14
have been developed to give magnetite powders of particular microstructures to meet the application requirements.
Tsouris et al.15–17 recently reported an electrocoagu-lation (EC) method for producing nanocrystalline mag-netite powder, where Fe ions are produced at the anode through electrochemical oxidation and then react with water to form the oxide. The oxide particles are then coagulated due to adsorption of ions on particle surface to neutralize the charge of the particles. Compared with other solution synthesis methods,7–14the EC method has the advantages of low-cost, continuous-process flexibil-ity, and in particular, not producing wastewater that con-tains high concentrations of anions, such as Cl− and NO3−, associated with the starting Fe-containing salts. We also recently demonstrated that the EC magnetite powders show promise in supercapacitor application due to its high surface area and superficial electrochemical activity.18
Although there have been reports on the electrochemi-cal behaviors of the process,16,17
the microstructures of
a)
Address all correspondence to this author. e-mail: [email protected]
the resulted magnetite powder have not been well estab-lished. In this work, the microstructural properties, in-cluding crystallite size, surface area, and porosity, of the EC magnetite powder have been characterized as a func-tion of synthesis time, current and temperature. Magne-tite in the form of colloidal crystallites and mesoporous skeletal agglomerates of varied compactness have been synthesized, and a microstructural evolution mecha-nism that accounts for the microstructural variations is proposed.
II. EXPERIMENTAL
The eletrocoagulation process was carried out by im-mersing two plates of carbon steel in a 0.04 M NaCl(aq) solution and applying a constant current to the electrodes. The carbon steel plates have an 8 × 8 cm dimension, and the process was conducted in a 2-liter beaker. During the process, the electrolyte solution was vigorously agitated by constantly bubbling with synthetic air (N2:O2⳱ 79: 21). A constant current of either 4.0 or 8.0 A (corre-sponding to a current density of 62.5 and 125 mA/cm2) was used, while two synthesis temperatures, 30 and 70 °C, have been used. Magnetite powders were col-lected by precipitation assisted with a magnet, washed three times with de-ionized water, and finally dried under vacuum at ambient temperature.
Average crystallite size was determined by x-ray dif-fraction (XRD; Mac-Science/MXP, Tokyo, Japan) based on the Scherrer equation19
d⳱ 0.9 /(B2− b2)1/2cos ,
where d is the average crystallite size (nm);, the wave-length of x-ray ( ⳱ 0.15418 nm); , the Bragg angle of the (311) reflection of magnetite; B, the full width of the peak at half-maximum intensity; and b, the line broad-ening width of the equipment. Transmission electron microscopy (TEM) analysis was carried out on a Hitachi H7100 electron microscope (Tokyo, Japan), while the high-resolution (HR) TEM micrographs were taken on a JEOL 2000 FX (Tokyo, Japan), which operates at 200 kV and is equipped with energy dispersive analysis of x-ray (EDX) analyzer. Nitrogen adsorption (ASAP2010, Mi-cromeritics, Norcross, GA) was conducted to determine the Brunauer–Emmett–Teller (BET) surface area and the pore characteristics. Magnetization analysis was carried out on a SQUID (Quantum Design MPMS-5S, San Diego, CA).
III. RESULTS AND DISCUSSION
As the electrocoagulation process proceeded, the so-lution, which was initially transparent, was found to gradually turn first yellowish, then brownish, and finally black. The last two colors actually originated from the suspended particles, and powders exhibiting the same
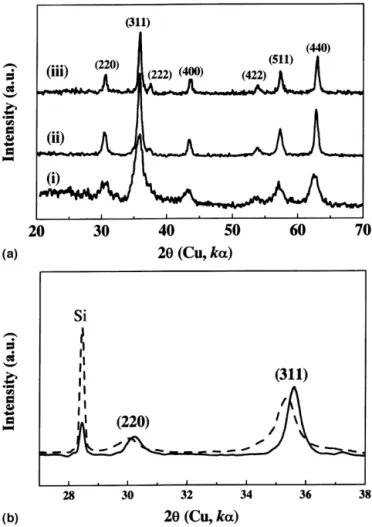
colors could be obtained when the solution was allowed to settle. Concurrently, the solution pH increased from being nearly neutral, then to 7–8 (brownish) and to the saturation at approximately 11 (black). The time needed for the color and pH to reach their final states was found to decrease with increasing current, being approximately 20 and 10 min for the 4.0 and 8.0 A-runs, respectively. XRD of both the brownish and black powders showed reflections typical of magnetite [Fig. 1(a)], suggest-ing that they possess the same fundamental structure. However, the brownish one exhibits larger d-spacings [Fig. 1(b)]. Based on a face-centric-cubic (fcc) structure, the least-square fitting of the d-spacing data acquired from the 2 range between 20° and 70° gives the brown-ish powder a lattice parameter ao⳱ 0.846 nm, in contrast with ao⳱ 0.839 nm for the black powder. The brownish phase is to be distinguished from ␥–Fe2O3, which has almost the same sets of XRD reflections as magnetite but with smaller d-spacings. The exact composition of the
FIG. 1. (a) XRD patterns for the powder synthesized under different conditions: (i) 4.0 A, 10 min; (ii) 4.0 A, 50 min; and (iii) 8.0 A; 50 min. (b) Comparison between the black (solid line) and brownish (dashed line) powders, with Si internal standard, showing that the brownish powder has larger d-spacings than the black one.
brownish phase is not for certain at this point. In a pre-vious report by Dhage et al.14on the pH effect on crys-tallization of iron oxides, it has been shown that the composition of the magnetite crystallites depends on the pH. The pH conditions under which the brownish powder is formed in the present study lead to the formation of non-stoichiometric magnetite, which has Fe+3
vacancies at the octahedral sites in the spinel structure. The pres-ence of the cation vacancies is consistent with the ex-pansion in the lattice parameter aoobserved in this study. Figure 2 summarizes the crystallite size data calcu-lated based on the XRD results. It was found that, under the same current and temperature, the brownish powder in general has a smaller crystallite size and that, when the powder color turned from brownish to black with in-creasing synthesis time, the crystallite size first reached a plateau for certain period of time before finally increas-ing again. The plateau crystallite size (PCS) is larger for lower current, being approximately 10 and 6 nm for the 4.0 and 8.0 A at 30 °C, respectively. Increasing synthesis temperature tends to increase the PCS as well. These crys-tallite sizes are between those previously reported for mag-netite synthesized by a coprecipitation method (∼8 nm)8 and by a microwave hydrothermal method (∼34 nm).13
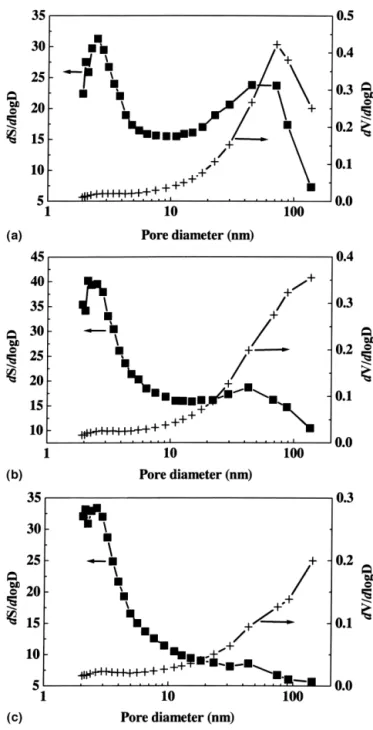
Nitrogen adsorption revealed that the powders consisted of predominantly meso- (2–50 nm) and macro- (<50 nm)
pores, while the mesopores contributed most of the sur-face area (Table I, Fig. 3). In contrast, the micropore volume is less than 2% of the total pore volume. It was also noticed that, while the total pore volume varied dra-matically with synthesis temperature, showing, for in-stance, reduction by approximately 50% between 30 and 70 °C, the variation in micropore volume is insignificant. The BET surface areas shown in Table I range from 25.5 to 37.7 m2
/g, which correspond to average crystallite sizes of 45.2–30.5 nm. The BET particle sizes are much larger than the crystallite sizes obtained from the XRD
TABLE I. Nitrogen adsorption data of selected powders.
Synthesis conditions BET surface area (m2/g) Micropore surface area (m2/g) Total pore volume (diameter <70 nm) (10−3cm3/g) Micropore volume (10−3cm3/g) 4.0 A, 30 °C, 30 min 36.9 4.09 148 1.57 8.0 A, 30 °C, 20 min 37.7 3.25 122 1.18 4.0 A, 70 °C, 30 min 25.5 3.14 70.7 1.25
FIG. 2. Average crystallite size versus synthesis time for different conditions.䊏䊐: black and brownish powders produced with 8.0A, 30 °C;䉱䉭: with 4.0 A, 30 °C; 䊉䊊: with 4.0 A, 70 °C.
FIG. 3. The surface area (S) and pore volume (V) distributions as a function of pore diameter (D): (a) 4.0 A, 30 °C, 30 min; (b) 8.0 A, 30 °C, 30 min; and (c) 4.0 A, 70 °C, 30 min.
studies, and this is consistent with the agglomerate nature of the crystallites.
TEM analysis showed that the black powders collected during the plateau stage have a skeletal structure of which the framework is composed of nonporous crystal-lites [Figs. 4(a) and 4(b)]. The crystalcrystal-lites exhibited dis-tributions in size ranging from one half of to 2–3 times the XRD crystallites sizes. The pores are predominantly inter-crystallite ones with sizes ranging from a few to more than a hundred nanometers. The nonporous nature of the framework may explain the lack of micropores in the structure as revealed by the adsorption analysis (Table I). Powders collected at synthesis time beyond the plateau showed similar skeletal structures except for a thicker backbone and smaller pores. Furthermore, in-creasing the synthesis temperature to 70 °C was found to significantly increase the compactness within the ag-glomerates and hence reduce the pore volume [Fig. 4(c)]. This is also consistent with the adsorption data (Table I). The brownish powders showed mainly spherical col-loids having sizes consistent with those deduced from the XRD data, along with some needle-shaped particles
[Fig. 4(d)]. However, the population of the needles ap-peared to decrease with increasing synthesis time, and they were not observed in most black powders. One plau-sible explanation is that the needles are formed only dur-ing the early stage of the synthesis, and the total amount of these particles in the black powder is typically too small to observe under TEM.
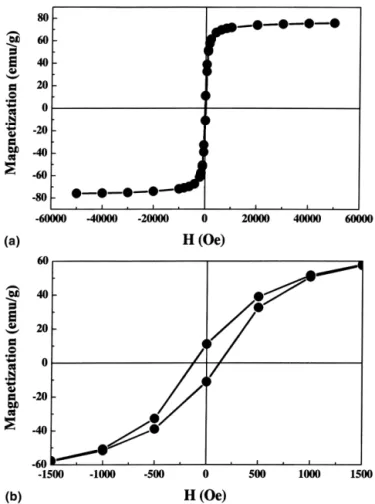
Magnetization analysis of all the synthesized powders gave hysteresis magnetization loops, typical of the ferri-magnetic material. An example, obtained from the pow-der synthesis unpow-der 4.0 A and 30 °C, is shown in Fig. 5. Table II summarizes the magnetic properties of four dif-ferent powders. The three black powders which resulted from prolonged synthesis time exhibited very similar magnetic properties, showing a saturation magnetization of approximately 75 emu/g and a coercivity Hcwithin the range of 118 ± 11 Oe. On the other hand, the brownish powder, which was obtained within shorter synthesis time, exhibited significantly lower values. These mag-netic properties are comparable to those obtained by other solution chemistry routes. For instance, Khollam et al.13reported a saturation magnetization of 70 emu/g
FIG. 4. TEM micrographs: (a, b) black powder synthesized at 30 °C (4.0 A, 30 min); (c) the black powder at 70 °C; and (d) the brownish powder at 30 °C (8.0 A, 3 min). Scale bar: (a) 50 nm, (b) 2 nm, (c) 50 nm, and (d) 25 nm.
and a coercivity Hc of 178 Oe for microwave-hydrothermally derived magnetite powder having XRD crystallite size of approximately 34 nm.
In a previous study by Ying et al.17
on the reaction mechanism of the process, it has been shown that the process involves release of ferrous ions from the anode
Fe↔ Fe+2 + 2 e−
. (1)
In the case of oxygenated solution, part of the ferrous ions can be further oxidized to Fe+3
Fe+2↔ Fe+3+ e− . (2)
On the other hand, the cathodic reaction
4 H++ O2(g)+ 4 e−↔ 2H2O , (3) continues to consume H+
, causing solution pH to in-crease along the course of the process. It was also sug-gested that the magnetite phase was formed through a serial reaction scheme consisting of first the formation of Fe(OH)3(s), which was subsequently reduced at cathode to form magnetite
3 Fe(OH)3(s)+ H +
+ e−↔ Fe3O4(s)+ 5 H2O . (4) However, both Fe(OH)3 and Fe3O4 are insoluble in water and should precipitate right out as they are formed. It is not clear how a colloid (nucleus) can be reduced at cathode and how reaction (4) can result in crystallites with a size over 10 nm as observed in this study.
On the other hand, it has been shown in a precipitation method7that nanometer-sized magnetite crystallites can be synthesized in one step from the solution containing both ferrous and ferric ions under alkaline condition. It is suggested that magnetite nuclei are formed in the elec-trocoagulation process via the similar reaction, namely
Fe+2+ 2Fe+3+ 8OH−↔ Fe3O4(s)+ 4H2O . (5) Coarsening of the existing nuclei may proceed by con-tinuous incorporation of ferrous and ferric ions via sur-face adsorption and reaction. Coarsening by oriented at-tachment of nano-crystallite20,21
as observed for iron oxyhydroxyide by Banfield et al.22
is also possible under high concentration of nuclei. Coarsening of the existing nuclei accounts for the increase in crystallite size during the initial stage of the synthesis.
To explain the plateau in average crystallite size, we consider the competition between coarsening of the large crystallites that constitute the framework of the agglom-erate and formation and coarsening of smaller free nu-clei. That is, as the population of colloidal crystallites increases, crystallites start to collide and fuse together to form skeletal agglomerates. Coarsening of the connected crystallites is expected to slow down due to loss in sur-face area in contact with the solution, and hence forma-tion and growth of nuclei prevail. As a result, while the total number of crystallites continuously increases with increasing synthesis time, the average size does not change significantly. This explains the existence of the plateau. On the other hand, when the amount of the skel-etal agglomerates increases to certain point when the ferrous and ferric ions are rapidly consumed by the in-terfacial reaction, the homogeneous nucleation rate is re-duced (due to low Fe+2 and Fe+3 concentrations), and coarsening of connected crystallites within the agglom-erates becomes predominant. This results in the increase in average crystallite size at the later stage of the process after the plateau.
FIG. 5. Hysteresis magnetization loop of the powder synthesized un-der 4.0 A for 30 min at 30 °C; (a) and (b) are drawn with different x-axis scale to show the saturation magnetization and coercivity, respectively.
TABLE II. Magnetization properties of selected powders.
Synthesis conditions Saturation magnetizationa (emu/g) Coercivity Hc (Oe) Appearance 4.0 A, 30 °C, 30 min 76 129 black 8.0 A, 30 °C, 20 min 75 109 black 4.0 A, 70 °C, 30 min 73 107 black 4.0 A,. 30 °C, 10 min 60 8 brownish aThe saturation magnetization data were obtained under a field of 5 tesla
Accordingly, the plateau is determined by the compe-tition between nucleation and interfacial reaction, which leads to the coarsening. The plateau would occur at a smaller crystallite size in the case of a relatively higher nucleation rate and a lower interfacial reaction rate. This may explain the observation that, as shown in Fig. 2, the plateau crystallite size (PCS) is reduced as a result of an increase in the electric current. On the other hand, an increase in temperature from 30 to 70 °C led to a larger PCS. This may be interpreted as a result of a rate-enhancement in the interfacial reaction greater than that in nucleation. From the thermo-kinetic point of view, this finding suggests that the interfacial reaction has a higher activation energy and hence is more temperature-sensitive than nucleation.
IV. CONCLUSION
In summary, microstructural evolution of the magne-tite powder synthesized in an electrocoagulation process was studied. Severely defective magnetite colloids were synthesized at the early stage of the process under pH approximately 7–8. This is followed by agglomeration among the oxide crystallites to form mesoporous skeletal agglomerates of which the framework is constituted of nonporous magnetite crystallites under pH > 9. This stage is also characterized by reduced crystal defects and a plateau in average crystallite size. At later stage, coars-ening of the crystallites within the agglomerates prevails, leading to rapid increase in average crystallite size and reduction in pore opening. The meso-/macroporous na-ture in conjunction with a high specific surface area makes the EC magnetite powder suitable for applications that require not only large surface area but also rapid access by pore diffusion to the surface.
ACKNOWLEDGMENT
This work is supported by National Science Council under Contract No. NSC90-2214-E-002-009. Part of the TEM and magnetic studies are supported by the State of Texas through funding for the Texas Center for Super-conductivity and Advanced Materials at the University of Houston.
REFERENCES
1. J. Bohacek, J. Subrt, T. Hanslik, and J. Tlaskal: Preparing particu-late magnetites with pigment properties from suspensions of basic iron(III) sulfates with the structure of Jarosite. J. Mater. Sci. 28, 2827 (1993).
2. R.L. Comstock: Modern magnetic materials in data storage. J. Mater. Sci. Mater. Electron. 13, 509 (2002).
3. K.T. Wu, P.C. Kuo, Y.D. Yao, and E.H. Tsai: Magnetic and op-tical properties of Fe3O4 nanoparticles ferrofluids prepared by
coprecipitation technique. IEEE Trans. Magn. 37(4) 2651–2653 (2001).
4. M. Shinkai: Functional magnetic particles for medical application. J. Biosci. Bioeng. 94, 606 (2002).
5. J.M. Perez, F.J. Simeone, Y. Saeki, L. Josephson, and R. Weissleder: Viral-induced self-assembly of magnetic nanoparticles allows the detection of viral particles in biological media. J. Am. Chem. Soc.
125,10192 (2003).
6. D. Beydoun, R. Amal, G.K.C. Low, and S. McEvoy: Novel pho-tocatalyst: Titania-coated magnetite. Activity and photodissolu-tion. J. Phys. Chem. B 104, 4387 (2000).
7. Y.S. Kang, S. Risbud, J.F. Rabolt, and P. Stroeve: Synthesis and characterization of nanometer-size Fe3O4and␥–Fe2O3particles.
Chem. Mater. 8, 2209 (1996).
8. H.S. Lee, W.C. Lee, and T. Furubayashi: A comparison of copre-cipitation with microemulsion methods in the preparation of mag-netite. J. Appl. Phys. 85, 5231 (1999).
9. L. Diamandescu, D. Mihaila-Tarabasanu, V. Teodorescu, and N. Popescu-Pogrion: Hydrothermal synthesis and structural char-acterization of some substituted magnetites. Mater. Lett. 37, 340 (1998).
10. Y. Li, H. Liao, and Y. Qian: Hydrothermal synthesis of ultrafine ␣–Fe2O3and Fe3O4powders. Mater. Res. Bull. 33, 841 (1998).
11. D.S. Bae, K.S. Han, S.B. Cho, and S.H. Choi: Synthesis of ultra-fine Fe3O4powder by glycothermal process. Mater. Lett. 37, 255
(1998).
12. S. Sun and H. Zeng: Size-controlled synthesis of magnetite nano-particles. J. Am. Chem. Soc. 124, 8204 (2002).
13. Y.B. Khollam, S.R. Dhage, H.S. Potdar, S.B. Deshpande, P.P. Bakare, S.D. Kulkarni, and S.K. Date: Microwave hydrother-mal preparation of submicron-sized spherical magnetite (Fe3O4)
powders. Mater. Lett. 56, 571 (2002).
14. S.R. Dhage, Y.B. Khollam, H.S. Potdar, S.B. Deshpande, P.P. Bakare, S.R. Sainkar, and S.K. Date: Effect of variation of molar ratio (pH) on the crystallization of iron oxide phases in microwave hydrothermal synthesis. Mater. Lett. 57, 457 (2002). 15. C. Tsouris, D.W. Depaoli, and J.T. Shor: Method and apparatus to
electrolytically produce high-purity magnetite particles, U.S. Patent No. 6 179 987 (2001).
16. C. Tsouris, D.W. DePaoli, J.T. Shor, M.Z.C. Hu, and T.Y. Ying: Electrocoagulation for magnetic seeding of colloidal particles. Colloids Surf. A 177, 223 (2001).
17. T.Y. Ying, S. Yiacoumi, and C. Tsouris: An electrocoagulation method for the formation of magnetite particles. J. Disp. Sci. Technol. 23, 569 (2002).
18. N.L. Wu, S.Y. Wang, C.Y. Han, D.S. Wu, and L.R. Shiue: Elec-trochemical capacitor of magnetite in aqueous electrolytes. J. Power Sources 113, 173 (2003).
19. F.W. Jones: The measurement of particle size by the x-ray method. Proc. Roy. Soc. (London) 166A, 16 (1938).
20. R. Lee Pan and J.F. Banfield: Morphology development and crys-tal growth in nanocryscrys-talline aggregates under hydrothermal con-ditions: Insight from titania. Geochimi. Cosmochimi. Acta 63, 1549 (1999).
21. Y. Yeadon, M. Ghaly, J.C. Yang, R.S. Averback, and J.M. Gibson: Contact epitaxy observed in supported nanoparticles. Appl. Phys. Lett. 73, 3208 (1998).
22. J.F. Banfield, S.A. Welch, H. Zhang, T.T. Ebert, and R.L. Penn: Aggregation-based crystal growth and microstructure develop-ment in natural iron oxyhydroxide biomineralization products. Science 289, 751 (2000).