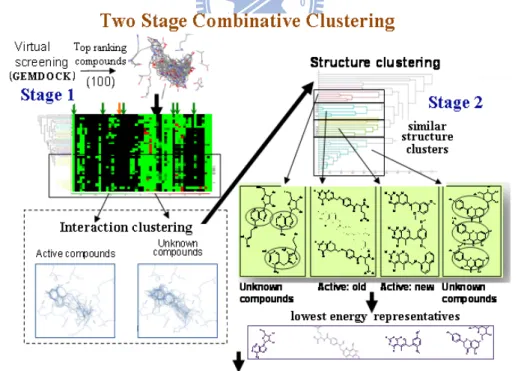
i
國
立
交
通
大
學
資訊管理研究所
博 士 論 文
A Study in Mining and Post Screening Methods for Compounds
Used in Various Biochemical Applications
資料探勘與篩選後分析方法於
多方面生化應用化合物之研究
研 究 生: Daniel L. Clinciu
指導教授: 羅濟群 教授 楊進木 教授
ii
A Study in Mining and Post Screening Methods for Compounds Used in Various Biochemical Applications
資料探勘與篩選後分析方法於 多方面生化應用化合物 之研究
Student: Daniel L. Clinciu Advisor: Dr. Chi-Chun Lo Co-Advisor: Dr. Jinn-Moon Yang
研 究 生: Daniel L. Clinciu 指導教授: 羅濟群 教授 楊進木 教授
A Thesis Submitted to the Institute of Information Management at National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of PhD in Information Management
June, 2011
iii
A Study in Mining and Post Screening Methods for Compounds Used in Various Biochemical Applications
Student: Daniel L. Clinciu Advisor: Dr. Chi-Chun Lo Co-advisor: Dr. Jinn-Moon Yang Institute of Information Management, Institute of Bioinformatics
Abstract
A phenomenal increase in the quality of human life is due to tremendous advancements and use of computer-aided methods in medicine and various biotechnological applications. Such technologies rely on the increasing availability of biochemical data and structural information which are highly significant for current advances. The solved crystal structures of 3D compounds stored in databases contribute greatly in bioinformatics as they are employed in studies and development of numerous lead compounds used in drug design and other industrial applications. However, screening and retrieving compounds for various applications presents a challenge for in retrieving and analyzing prospect targets. Therefore, a constant improvement of methods and tools is necessary for the proper classification, query, retrieval and analysis of available compounds data. With advances in computer technology, information management and data mining the developments of accurate, rapid and efficient algorithms enable studies in biotechnology to have significant improvements. However, mining appropriate candidates for various purposes by virtually screening thousands of docked protein-compound complexes is one of the biggest challenges. One of the main issues in virtual screening comes from an insufficient description of ligand binding mechanisms which results in the development of imprecise scoring functions.
In aiming to provide solutions to this issue we studied various docking algorithms and post screening methods used in mining and investigating specific compounds. Comparing different virtual screening and post screening analyses we observed that interaction profiles (e.g. van der Walls, hydrogen bonding) are highly relevant in the overall performance of compound mining. Moreover, this study concluded that a method which uses two combined stages of cluster analysis can be more efficient than one-stage clustering methods in selecting appropriate candidates for drug design and other biotechnological applications. Our study of interaction profiles also provided evidence of the possibility of mining novel compounds for potential uses in cosmetics, industry and agriculture in addition to pharmaceutics using similar virtual screening and post screening analysis.
The above findings and observations contributed to the development of our method, Two Stage Combinative Clustering (TSCC) where we combine virtual screening and two stages of cluster analyses (interaction and physico-chemical). The methodology of TSCC has contributed to combinatorial computation approaches used to indentify tetracycline derivatives for inhibiting Dengue virus neuraminidases and inhibitors for flaviviruses.
iv
TSCC, similar to other post screening analysis methods starts with the virtual screening of compounds obtained from various databases e.g., Available Chemical Directory (ACD) or Comprehensive Medical Chemistry (CMC) using GEMDOCK. Top ranking compounds are then clustered based on their protein-ligand binding interactions and grouped into clusters with distinct binding interactions. Compounds are also clustered based on physico-chemical features using atom composition and are grouped in similar structure clusters. Compounds with lowest energy from each interaction cluster are selected as representatives while active compounds and similar to active compounds are chosen as representatives from each structure cluster. Lastly, final representatives from both interaction and structure clustering are chosen based on energy and structure similarity respectively and can be verified trough bioassays for proper function and application. TSCC’s novel feature is the use of two clustering stages to better filter and accurately retrieve the final representative compounds. Another key feature is to represent interactions at the atomic-level for including measures of interactions strength, enabling better descriptions of protein-ligand interactions to achieve a more specific analysis of virtual screening. The proposed two-stage clustering method enhanced our post-screening analysis by revealing more accurate performances than a one-stage clustering in visualizing and mining compound candidates and improving the virtual screening enrichment while being used successfully to identify novel inhibitors and functions of some proteins.
Keywords: cluster analysis, data mining, docking, GEMDOCK, lead compound, post
screening analysis protein-ligand interaction profiles, target, compound database, virtual screening.
v
Acknowledgement
I want to thank God for all the great things and for this opportunity I was given to study at such a great and prestigious university. Special thanks go to Dr. Jinn-Moon Yang, Dr. Chi-Chun Lo and Dr. Simon J. T. Mao for their great help, direction and support in this research and my overall study. I’m also thankful to my colleagues in the Bioinformatics laboratory, especially Marcco C.N. Ko, Piki, C.W. Huang and Shen-Rong. I’m also thankful to the Ministry of Education in Taiwan for making possible international programs at universities in Taiwan where people from around the world can come and study and in the same time contribute to the wonderful world of science and research so that the quality of human life and all important aspects of humanity can benefit. Lastly, I want to emphasize that what we do for others is the most important thing because together with others our contribution and investment in the future of this world will make it a better place for us and future generations of humans.
vi
Contents
Cover page
………...iAbstract ………iii
Acknowledgement……….………v
List of Figures ……….vii
List of Tables ………ix
Chapter 1. Introduction ………1
1.1 Background ………..1
1.2 Motivation ………4
1.3 Organization of Thesis ……….5
Chapter 2. Related Studies and Review of Related Methods ………7
2.1 The Emergence of Post Screening Analysis………...7
2.1.1 Interaction-Based Accuracy Classification (IBAC)………....8
2.1.2 Structural Interaction Fingerprint (SIFt)……….9
2.1.3 Visualized Cluster Analysis of Protein-Ligand Interaction………9
2.1.4 A New Hierarchical Clustering Approach for Large Compound Libraries..10
2.2 The Use of Protein-Ligand Interaction Profiles in the discovery of Molecular Mechanisms and Lead Compounds ……….………..…..12
CHAPTER 3. The Relevance of Protein-Ligand Interaction Profiles in Computer-Aided Lead Compound Discovery, Functions and Applications…………..….….13
3.1 INTRODUCTION………..………13
3.2 The Significance of Protein–Ligand Interaction Profiles in Methods of Retrieval and Analysis of Compounds………..……….…15
vii
3.2.1 Post Screening Analysis………..16
3.2.2 SIFt (Structural Interaction Fingerprint) ………17
3.2.3 VISCANA (Visualized Cluster Analysis of Protein-Ligand Interaction)…..19
3.2.4 iGEMDOCK: A Graphical Environment for Recognizing Pharmacological Interactions and Virtual Screening………..20
3.3 Summary………...22
CHAPTER 4. TSCC: A Two- Stage Combinative Clustering for Virtual Screening Using Protein-Ligand Interactions and Physico-Chemical Features……….…24
4.1 Introduction………...24
4.2 Materials and Methods………..26
4.2.1 Preparation of Target Protein and Compound Databases……….…28
4.2.2 Preparation of Virtual Screening Result for Cluster Analysis………….….29
4.2.3 Testing and Verifying Datasets……….30
4.3 Results………...33
4.4 Verifying the TSCC method using β-lactoglobulin ……….….…...44
4.4.1 Introduction ………..44
4. 4.2 Materials and methods ………...……….45
4.4.3 Molecular Docking and Post Screening Analysis……….……46
4.5 Results………46
4.5.1 Virtual Screening results………...46
4.5.2 Cluster Analysis Results………...49
viii 4.7 Summary………...……….53 CHAPTER 5. Conclusion……….55 5.1 Summary………...55 5.2 Future Works………56 APPENDIX A………..……….………57 APPENDIX B……….……..61 REFERENCES……….………62
ix
List of Figures
Figure 1. Crystal structure of β-lactoglobulin complexed with vitamin D-3. ……….…2 Figure 2. The overall research process in investigating of interaction profiles and their role in identifying suitable methods for lead compounds retrieval and their applications. ………...6 Figure 3. The biased compound ranking in virtual screening (molecular docking). Ergocalciferol (purple color) has a lower binding energy than Riboflavin (yellow) and Celestine blue (blue) and it is ranked higher by the docking program. However, riboflavin is the active compound known for binding the to cavity of the target protein………….………...8 Figure 4. View of protein-ligand binding interactions in Betalactoglobulin …………...……….13 Figure 5. SIFt, VISCANA and iGEMDOCK; three post screening analyses based on binding interactions were investigated in our study of interaction profiles and in the designing of Two Stage Combinative Clustering (TSCC). ………17 Figure 6. The concept of SIFT. 3D binding site of protein with an inhibitor (ligand) revealed as a sequence of positions in the binding site in contact with the ligand and their location in the structure of the protein (loop and β). ………..…….….18 Figure 7. a) The overall approach of VISCANA (from VS to the selection of representatives)...19 Figure 8. The virtual screening and post screening analysis processes in iGEMDOCK………..21 Figure 9. The linear energy functions of the pairwise atoms for the steric interactions and Hydrogen bonds in GEMDOCK (bold line) with a standard Lennard-Jones potential (thin line). Figure obtained from a previous study by Yang et al. [26].………...25 Figure 10. Overall process of TSCC in our first study (a) First stage clustering using P-L interactions generated via GEMDOCK. (b) Second stage clustering of first stage results using physico-chemical features. (Figure obtained from our previous study [48])……….……26 Figure 11. Designing a reference threshold of P-L interaction and atom-pair descriptors. …….27 Figure 12. Cluster analysis of hDHFR. ……….39 Figure 13. (a) Overlay of all 53 docked poses of known active compounds in the vicinity of the target protein Thimidine Kinase (PDB id: 1kim). (b) Hierarchical clustering of 53 TK docked poses’ protein-ligand interactions. Each docked pose is one line in the heat map, the red being the lowest P-L interaction energy and the green being the highest. The left side of the heat map shows the hierarchical clustering results of TK. The hot spots identified from known overlapping active compounds are shown at the top. (c) Overlay of docked poses of the cluster with most number of known active compounds and important h-bonds between protein and ligand. (d) Overlay of docked poses of the cluster with most number of unknown compounds and important
x
h-bonds between protein and ligand. The blue frames in the heat map were the major interaction difference among clusters c and d. ……….…………...40 Figure 14. The hierarchical clustering dendrogram for the 61 known compound structures showing the three major clusters. ……….……….41 Figure 15. The detail of hDHFR binding interactions of new drugs and old drugs on the verifying dataset. ………..42 Figure 16. The process and results of second stage cluster analysis on hDHFR testing dataset...43 Figure 17. The overall approach of TSCC in our second study using iGEMDOCK and interaction clustering and atomic composition clustering for two-stage combinative clustering..45 Figure 18. Active compounds used in the validation of the TSCC method………..…46 Figure 19. Conserved residues (LEU 39 and VAL 41) showing interaction through hydrogen bonding between β-LG cavity and the three active compounds..………..48 Figure 20. The dendrogram showing the occurrence of important residues between the three active compounds and β-LG cavity and also the top VS ranking compounds and β-LG cavity...49 Figure 21. Clustering analysis results. The ranking results from TSCC, two methods of one-stage clustering (Rank-IC and Rank-AC) and Virtual Screening for the three active compounds and four unknowns are shown in the four separate columns. ………..50 Figure 22. The three active compounds and four highest VS ranking unknown compounds; their structures, molecular weight and atom composition. Atom similarity: adenosine triphosphate and unknown mfcd00013358 (brown circles), adenosine triphosphate and mfcd00010114 (light blue circles), adenosine triphosphate and mfcd00012401 (dark blue circles) and adenosine triphosphate and mfcd00013358 (purple circles)………. 51
xi
List of Tables
Table 1. Popular docking tools and evolutionary algorithms currently used in VS. …………...15 Table 2. The RMSD between docked poses and crystal ligands. ……….34
Table 3. T-test of distance between similar and non-similar binding mode generated by converting the docked pose into protein-ligand interaction profile (α=0.01). ………..35 Table 4. T-test of distance between similar and non-similar structure generated by atom-pair representation (α=0.01). ………36 Table 5. T-test of distance between similar and non-similar compounds on each target protein. Descriptor was generated by converting the docked pose into protein-ligand interaction profile (α=0.01). ………...37 Table 6. Virtual screening results and ranking of the three active compounds (riboflavin: 575, adenosine triphosphate: 591 and calcitriol: 816). The shaded area (bottom of table) shows the highest ranking compounds (1 – 4) based on interaction energies generated by the docking program………..47
1
Chapter 1
Introduction
1.1 Background
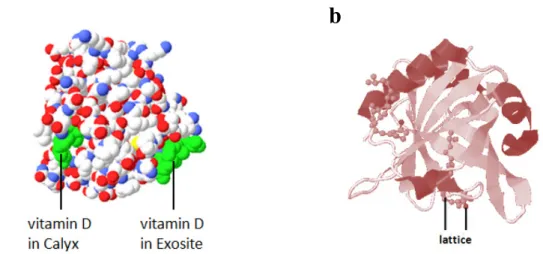
The transition of many preliminary biochemical studies from the wet to virtual laboratories propagated by computer-aided methods and an increase in technology has brought new insights and perspectives. Specifically, significant progress in development of novel compounds for pharmaceutics, agriculture, cosmetics, nutrition and other industries has been mediated by computational techniques and approaches in preliminary steps. In this transition process many principles from other disciplines were adopted into the field of biotechnology. Applications of information management to aid with compound database management and of data mining to successfully retrieve and mine compounds from databases [1] are just a few examples of the constantly used, researched and developed applications in the field of biotechnology. Data mining, especially, has been given a lot of attention lately because of the rapid increase in number of virtual compounds available in databases. Mining of compounds from databases involves a series of steps but nowadays it can be done much faster and easier using combined methods of virtual screening and post screening analysis. Virtual screening (VS) [2, 3] is the first step towards the retrieval of prospect compounds. It is important to note that in a virtual setting the key to research and studies of biochemical compounds is the relevance of their crystal structures [4, 5] for practical applications in preliminary results which will be further confirmed by bioassays [6 – 10]. A crystal structure is composed of a pattern, a set of atoms arranged in a particular way (Fig. 1a) and a lattice exhibiting long-range order and symmetry (Fig. 1b). Patterns are located upon the points of a lattice (Fig. 1b), which is an array of points repeating periodically in three dimensions. The points can be thought of as forming identical tiny boxes, called unit cells, that fill the space of the lattice. The lengths of the edges of a unit cell and the angles between them are called the lattice parameters. The symmetrical properties of the
crystal are embodied in its space group. The crystal structure of a compound and its symmetry play a role in determining many of its physical properties, such as cleavage, electronic band structure, and optical transparency. Various computer generated tools and programs are developed to “visualize and interpret” the characteristics of crystal structures and their
2
interaction with other crystal structures in specific studies of protein-protein complexes or protein-ligand complexes.
a b
Figure 1. Crystal structure of β-lactoglobulin (β-LG) complexed with vitamin D-3. a) Space-filling model showing specific ligand binding sites (calyx and exosite) and b) Ribbon-lattice crystal structure of β-lactoglobulin. Crystal structures of compounds can provide many clues of binding sites and interactions between various proteins and or ligands.
Virtual screening of molecular libraries to mine compounds with an available crystal structures has emerged as a practical and inexpensive method in the discovery of novel lead compounds especially for drug design and discovery. This current increase in use of VS accounts for the following valid reasons: its enrichment and speed, the reduced cost and time of studies when using VS, increasing numbers of compounds with crystal structures and the advent of structural proteomics technologies. Computational techniques in VS involve two essential elements: efficient molecular docking (a technique to predict the preferred orientation of one molecule to a second when bound to each other to form a stable complex) and a reliable scoring
method [11]. VS scoring methods must discriminate between non-native docked conformations and correct binding states of compounds during molecular docking phase to distinguish active compounds (usually a small number) from non-active compounds (an extremely large number) during the post-docking analysis. Scoring methods use three main classes of scoring functions that calculate the free binding energy: knowledge-based [12], physics-based [13] and empirical-based [14]scoring functions.
3
Inconsistencies in performance of scoring functions result in inadequate prediction of true binding affinity of a ligand to a receptor, thus, combining various scoring methods in VS may improve performance than in the average individual scoring functions. Similar inconsistencies have been noticed in information retrieval (IR) and Charifson et al. [15] proposed a study in which they used an interaction-based consensus approach to combine scoring functions which revealed enrichment in discrimination between active and inactive enzyme inhibitors. Later studies by Bissantz et al., Stahl and Rarey and Verdonk et al. [3, 11, 16] showed consensus scores which further improved VS enrichment. Although researchers attempt to bring out the benefit of combining methods with consensus scoring, the remaining issue for VS users rather than researchers is when and how these scoring functions should be combined. Furthermore, certain VS methods can identify important interactions or binding-site hot spots obtained from known active ligands and target proteins [17]. Because most docking programs [18-20] use energy-based scoring methods which are often biased towards selection of high molecular weight compounds and charged polar compounds they have problems identifying key features (e.g. hot-spots) essential to target protein responses. Thus, methods for post-screening analysis employing clustering to identify key features through docked compounds and understanding binding mechanisms are of great use in bioinformatics. As VS encounters increasingly large databases, post screening analysis is an essential step in drug design and discovery.
The first attempt at a post screening analysis was done by Kroemer et al [21] in their work “Interactions-Based Accuracy Classification (IBAC)”, an approach which aimed to determine the best way to assess correctness of docking conformations. Their study showed that RMSD values alone are insufficient to predict correct poses; therefore, binding modes should be closely inspected for specific interactions when assessing pose prediction accuracy. Through this study the relevance of interaction profiles emerged as the basis in studies of interaction and bindings among protein-protein and protein-ligand complexes.
Amari et al and Deng et al [22, 23] followed the lead of IBAC and developed post screening analysis methods called Visualized Cluster Analysis of Protein−Ligand Interaction (VISCANA) and Structural Interaction Fingerprint (SIFt) respectively. Deng et al made a pioneering attempt by developing the first method based on binding interactions in order to facilitate the visualization, organization, analysis and data mining of virtually screened
4
compounds which all other post screening analysis employed. Amari et al devised a different approach for post screening analysis, a method based on the ab Initio Fragment Molecular Orbital Method (FMO) [24] to be used for analysis of virtual ligand screening also using the binding interactions generated from VS.
Attempts to cluster large numbers of compounds from VS by Bocker et al [25] resulted in
a post screening method for clustering large datasets of compounds in a high dimensional space. The key feature of NIPALSTREE is its ability to handle more than 800 000 data points in high-dimensional descriptor space in less than an hour computation time.
The above studies implemented post screening analysis in an attempt to enrich the screening results of various docking tools (e.g. GOLD, AUTODOCK, GEMDOCK,) [19, 20, 26] and to facilitate the visualization, organization, analysis and data mining of virtually screened compounds. However, there are two main issues with all post screening analyses including the ones mentioned: 1) if a docking tool is used for VS, which post screening analysis should it be joined with for the most overall efficiency and accuracy and 2) if a post screening analysis method was decided (IBAC, SIFt or VISCANA) [21 – 23] which docking tool or VS method is most suitable prior to a particular post screening analysis. In addition, the ideal combination of docking tool and post screening analysis should successfully obtain novel compounds following the screening of compound databases and post analysis of selection lists obtained from VS.
This research investigates the potential drawbacks of VS and of various post screening analyses in computer aided novel compound mining and discovery. It further investigates the role of interaction profiles and proposes an algorithm that specifically optimizes a docking tool (GEMDOCK) for screening database compounds which is combined with a new, two-stage clustering method for post screening analysis in an attempt to join VS and post screening analysis for faster and more efficient compounds mining and analysis.
1.2 Motivation
The importance of efficient data mining and analysis of potential lead compounds to be used in various industries is of high relevance in biotechnology. Compounds can be obtained from databases through virtual screening and post screening analysis and contribute greatly in
5
many applications (novel compounds for drug design and industrial uses). The availability of compounds found in databases enable studies to be conducted at much cheaper costs and faster paces than previously done in “wet” or traditional laboratory settings where the use of natural compounds and live specimen was a concern for many reasons (proper disposal of hazardous materials and the constant need of live cell cultures and animals). Thus, in the virtual laboratory settings, the basis for investigating biologically active compounds is the use of high resolution x-ray structures of protein-ligand or protein-protein complexes from which a crystal structure is developed and to which all its known natural properties assigned. New functions and roles of
existing compounds are always discovered; therefore compound databases must constantly be updated. Developments of high-throughput X-ray crystallography and advances in genomics [1-9] are constantly increasing the number of crystal structures available in protein databases [27, 28] leading to multiple therapeutic and industrial targets. Although the great number of available structures may present difficulties when retrieving compounds, the growing number of available methods aided by computer technology and principles from various disciplines (information management, data mining, consensus scoring) are rapidly evolving new and improved techniques to aid such studies.
This study investigates the significance of protein-ligand interaction profiles and compares various methods and tools used in virtual screening and post screening analysis for mining prospect compounds from databases and also expand their additional uses. It also shows the weakness of one-stage clustering methods in post screening analysis and why they have less success in identifying specific compounds and their various functions. Moreover, this study shows that a combined method of VS and a combined two-stage cluster analysis is more ideal for mining specific compounds and investigating their various functions.
1.3 Organization of Thesis
This thesis is organized as follows: In chapter 2 we describe related studies and similar methods of mining and analyzing prospect compound candidates from virtual databases along with their advantages and shortcomings. In Chapter 3 we perform an in-depth study of protein-ligand interaction profiles and present novel concepts obtained from our investigations in possible future work for additional applications of virtual screening and post screening analysis such as cosmetics, nutrition, industry and agriculture. In chapter 4 we describe our core work, the
6
development of Two-Stage Combinative Clustering (TSCC) and its improvement over one-stage post screening analysis methods. Chapter 5 concludes our studies and includes future work prospects. In Figure 2 below, the model for this research is presented.
Figure 2. The overall research process in investigating of interaction profiles and their role in identifying suitable methods for lead compounds retrieval and their applications
7
Chapter 2
2.1 Related Studies
The process of VS and post screening analysis is a common technique used in mining and analyzing compound candidates to be used in pharmaceutics or various other applications after their retrieval from databases. The VS technique involves docking tools (e. g. DOCK, GEMDOCK or GOLD) [19, 20, 26] to screen compound databases and rank compounds according to their binding energies. Compound databases store solved crystal structures (Figure 1) of chemically significant compounds which can be used in various studies (e.g. drug design, nutrition and other industries) [6, 9, 10, 29 – 31]. VS and docking is followed by post analyses using clustering (SIFt and VISCANA) [22, 23] which aim to reduce the number of false positives obtained from VS and propagate true positives to the top of the selection list.
2.1.1 The emergence of Post Screening Analysis
In the early days of computer-aided drug design, docking tools / programs were the only means of screening compounds for the possibility of drug design. Given the poor understanding of many critical factors at the time especially the incomplete knowledge of ligand binding mechanisms, VS was still a major accomplishment in moving forward a revolution in drug design and discovery with faster and more practical preliminary approaches than previously done through bioassays using biochemical methods. Traditional settings, in addition to requiring an extensive period of time to study various properties and make a drug ultimately available, had overwhelming expenses inquired through the use of conventional biochemical compounds, facilities and specimen. With the advent of computer aided drug design more of the preliminary work in drug design is done in virtual labs and when desired results are obtained, the stage requiring bioassays to confirm preliminary results is applied.
Most docking programs [19, 20, 26] use energy-based scoring methods which are often biased towards selection of high molecular weight compounds and charged polar compounds (Fig, 3). Therefore, they have problems identifying key features (e.g. hot-spots) essential to target protein responses resulting in the performance of these scoring functions to be mostly inconsistent when conducting a database search [3, 11]. The inaccuracy of various scoring methods inadequately predicting the true binding affinity of a ligand for a receptor is a major weakness for VS. Moreover, employing VS [2, 3] in computer-aided drug design usually results
8
in a high number of chosen compounds from which few are potential or suitable candidates. Thus, it is imperative that a post screening analysis is conducted in order to reduce the number of false positives in the selection lists generated from VS and to propagate true hits to the top of the selection lists.
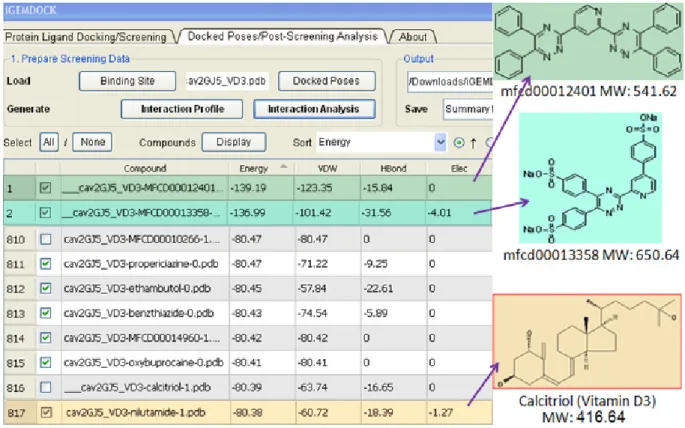
Figure 3. The biased ranking of compounds in virtual screening (molecular docking). Unknown compounds MFCD00012401 (green color) MFCD00013358 (teal green color) are ranked much higher than Vitamin D3 (ranked 816) due to their energy and molecular weight. However, only vitamin D3 is known for its ability to bind to the target protein (β-LG) [66, 67] among all compounds listed in this table.
2.1.2 Interaction-Based Accuracy Classification (IBAC)
IBAC is an approach developed by Kroemer et al [21] which determines the best way to assess correctness of docking conformations. It first calculates the RMS deviation of the predicted pose from the crystal structure and then it compares the predicted pose to the pose experimentally observed. In simple terms, using IBAC, Kroemer et al optimized the binding site definitions and docking protocols for 6 VS programs used in their studies (FlexX [32], GOLD [20], ICM [33], LigandFit [34], NWU [35, 36] and QXP [37]). They executed docking runs and
9
reported details of the ligand tautomeric forms and bond orders and how RMSDs from crystal structures correlated with interactions-based accuracy classifications. Kroemer et al. concluded that RMSD values alone lack the ability to predict correct poses and binding modes should be investigated further for specific interactions when assessing pose prediction accuracy. Through the work of Kroemer et al. the relevance of interaction profiles emerged as the foundation of interaction and bindings studies for protein-protein and protein-ligand complexes.
2.1.2 Structural Interaction Fingerprint (SIFt)
SIFt [23] uses a simple, generic and robust approach for representing and analyzing 3D protein-ligand interactions. Its key feature is the generation of an interaction fingerprint that converts 3D structural binding information into a one-dimensional (1D) binary string (Figure 9). The fingerprint representation of the interaction patterns is compact, and allows for rapid clustering and analysis of large numbers of complexes. The SIFt is calculated on a set of input 3D protein–small molecule complexes. To analyse SIFTs the Tanimoto coefficient (Tc) [38] is used as the quantitative measure of bit string similarity.
This representation of interactions as fingerprints using the SIFt method enables clustering, filtering and profiling of large docking results libraries and crystal structures of the protein kinase family in complexes with various inhibitors. Although SIFt opened a broad road for post screening analysis, much of the road is still unpaved and difficult to travel in terms of methods used currently in post screening analysis.
2.1.3 Visualized Cluster Analysis of Protein-Ligand Interaction
VISCANA [15], a method which stands for Visualized Cluster Analysis of Protein-Ligand Interaction based on the ab Initio Fragment Molecular Orbital Method (FMO) [24] used for virtual ligand screening was proposed by Amari et al. They developed a cluster analysis using the dissimilarity defined as the squared Euclidean distance between interfragment interaction energies (IFIEs) of two ligands. In VISCANA a clustering method is combined with a graphical representation of the IFIEs by representing each data point with colors that quantitatively and qualitatively reflect the IFIEs. This method claims to classify structurally different ligands into functionally similar clusters according to the interaction pattern of a ligand and amino acid residues of a receptor protein. VISCANA also estimates the docking
10
conformation by analyzing patterns of the receptor-ligand interactions of some conformations through the docking calculations.
However, as stated by Amari et al. in their study, VISCANA lacks sufficient descriptions of van der Waals forces and hydrogen bond interactions which play an important role in receptor-ligand binding [39, 40]. This may account for selection of false positives instead and the failure to select true hits or active compounds. This method is aiming to increase VS enrichment; however, it doesn’t provide significant improvements over SIFt or extend further uses into drug design and discovery or other possible applications.
2.1.4 A New Hierarchical Clustering Approach for Large Compound Libraries: NIPALSTREE
NIPALSTREE, is an approach by Bocker et al [25] for clustering large datasets of virtual compounds in a high dimensional space. It uses the first Principle Component (PC) which employs NIPALS (non-linear iterative least squares) where the data set is split at point i or j (determined points where two neighbors exceed a predefined distance threshold T). The procedure is recursively applied on the resulting subsets until the maximal distance between cluster members exceeds a user-defined threshold. NIPALSTREE clustering employs PCA for hierarchical clustering algorithm as follows: A d-dimensional descriptor matrix is projected onto the first PC. Based on the scoring vector S, the given descriptor matrix is sorted in ascending order and split at the median position, i.e., two equally large descriptor sets-from now on termed “left” and “right” submatrix s are created. This is repeated for the new subsets until the maximum distance between the entries in a submatrix underscores a predefined similarity threshold (Θ). In order to judge the quality of a clustering result an index is introduced to assess whether molecules interacting with the same target (receptor or receptor family) lie in the same subtree. An enrichment factor (EF) is calculated for each cluster, which gives an estimate of how well compounds that bind to the same target (or target class) are clustered in a dendrogram node i expressed in the following equation:
11
entries in node i, Nc being the total number of entries of class c in the data set, and N being the
overall number of entries. EF > 1 indicates that more compounds belonging to the activity class c are clustered in a tree node than expected from an equal distribution. The EF value depends on the size of the dendrogram section under consideration: On the upper dendrogram levels, where clusters are large, EF values are usually smaller, whereas EF values on the lower dendrogram level scan get large without a statistical relevance. A possible way to overcome the cluster size dependency of the EF is to additionally divide it by the logarithm of the dendrogram level, assuming that at each cluster the data set is separated into equally large partitions. In this way, an adoption of the EF to the dendrogram level can be achieved.
Although NIPALSTREE is able to deal with more than 800 000 data points in high-dimensional descriptor space in less than an hour computation time it does not specify how false positives are addressed; this is a major concern for all methods performing compound retrieval and analysis. Besides a rapid clustering of compounds, NIPALSTREE cannot offer visualization and accurate data mining of compounds and it is impractical as a method of retrieval and analysis for specific compounds in either drug design or other industrial uses.
2.2. The Use of Protein-Ligand Interaction Profiles in the discovery of Molecular Mechanisms and Lead Compounds
Since protein-ligand and protein-protein complexes are components of a great number of pharmaceutical [5, 41], nutritional [10] and industrial compounds [29-31] it is reasonable to employ computer-aided lead compound design and discovery methods for other applications besides pharmaceutics. Due to its significant role and impact on the quality of human life, drug design was the main focus in early days of virtual screening and bioinformatics. However, as methods and studies in drug design reveal that VS and post screening analysis are relatively inexpensive and efficient we want to explore the other fields (nutrition, agriculture and industry) which were not given as much attention. Protein-ligand complexes of various compounds interact through similar properties [40] and necessitate similar methods of screening, retrieval and analysis of their crystal structures (Figure 1) regardless what their final application may be. Therefore, the first part of this research focuses to conduct comparative studies on features and properties of protein-ligand interaction profiles to better understand their relevance in the mining of novel compounds. Additionally, we investigate possibilities of employing interaction profiles in the mining of compounds to be used in other applications besides drug design such as
12
cosmetics, skin care, nutrition, safe fertilizers and pesticides, compounds for scents in perfumes and deodorants and safe detergents. Furthermore, we employ interaction profiles in investigating mechanisms of significant molecules for human health and nutrition (e.g. uptake of vitamin D in the human body by Betalactoglobulin).
Although the interest of researchers in mining novel compounds for other uses besides pharmaceutics is minimal at the present time, as computer-aided methods continue to improve and increase in use, other industries (e.g. cosmetics, agriculture, nutrition) look to employ their benefits. Therefore, the approaches and techniques used in computer-aided drug design can be of particular interest for different biotechnological approaches. VS combined with post screening analysis are seemingly efficient in investigating transporter proteins such as β-lactoglobulin (β-LG), their mechanisms and various functions in the human body. Many compounds having various functions and mechanisms in the body are protein-ligand complexes which can be investigated based on protein-ligand interactions and physico-chemical features.
13
CHAPTER 3
The Relevance of Protein-Ligand Interaction Profiles in Computer-Aided
Lead Compound Discovery, Functions and Applications
3.1 Introduction
Identification of protein-ligand interaction networks on a proteome scale is crucial in addressing a wide range of biological issues such as correlating molecular functions to physiological processes and designing safe and efficient target compounds which can be used in therapeutics, nutrition, cosmetics, skin care products, agriculture and industry. In order to understand the role and significance of protein-ligand interactions (Fig. 4) in various applications throughout the field of bioinformatics and biotechnology the properties and functions of a ligand [42, 43] must be well addressed. As seen previously, the ligand (vitamin D, Fig. 1) is a molecule, ion or atom which can bind to a specific location or the binding site of a protein [39, 44]. Currently, antibodies are the most commonly used ligands in biotechnology and life-science investigations, although protein scaffolds (protein regulators), nucleic acids and peptides (repeating structural units in amino acids) are also employed. Since protein-ligands complexes of various compounds are used in cosmetics, hair dyes, skin care products, fertilizers, detergents [29-31] and nutrition supplements [10], protein-ligand interaction profiles and physico-chemical features could be used in the identification of such lead compounds.
a
b
Figure 4. View of protein-ligand binding interactions in Betalactoglobulin (a transporter protein) complexed with vitamin D using Swiss PDB viewer. a) Electrostatic potential and molecular surface. b) Hydrogen bond interactions among atoms (green dotted lines).
14
The ligand binding site of the primary target is extracted or predicated from a 3D experimental structure or homology model of proteins [35, 45] and characterized by a geometric potential. Protein-ligand interactions occur when a ligand binds to a protein which is usually integral to the function of its cognate (assimilated or symbiotic) protein. In the binding of a ligand to a protein, the following interactions are of significance: electrostatic forces (interaction between electrically charged particles explained by Coulomb’s law), van der Walls forces (the sum of the attractive or repulsive forces between molecules or parts of the same molecule) and hydrogen bonding (the attractive interaction of a hydrogen atom with an electronegative atom which can occur inter or intramolecularly) [39, 40]. Based on these interactions, evaluations are made using ligand-based approaches employed commonly in pharmacophore modeling by using physical and chemical traits of known ligands to identify novel inhibitors. Another approach, the receptor-based, identifies ligands that use structural and other features on the target receptor to identify the best inhibitor.
Docking [18, 26, 32, 33, 46] is then used to identify the fit between a receptor and the potential ligand by screening a database of ligands against one or more target receptors via two distinct parts: docking (the search scheme to identify suitable conformations or poses) and scoring (a measure of the affinity of various poses). Scoring methods must discriminate between non-native docked conformations and correct binding states of compounds during molecular docking phase to distinguish active compounds (usually a small number) from non-active compounds (an extremely large number) during the post-docking analysis. Although there are over 60 docking programs and tools available [24], we present some of the most popular programs made publicly available (Table 1). DOCK [18], incremental construction (FlexX) [32] and evolutionary algorithms (GEMDOCK, GOLD, AutoDock) [26, 33, 46] are used to screen and downsize compound groups in order to select suitable candidates for post-screening analysis. However, inconsistencies in the performance of scoring functions results in inadequate prediction of true binding affinity of a ligand to a receptor; thus, combining various scoring methods in VS may improve performance than in the average individual scoring functions. Similar inconsistencies have been noticed in information retrieval (IR) and Charifson et al. [15] proposed a study in which they used an interaction-based consensus approach to combine scoring functions which revealed enrichment in discrimination between active and inactive enzyme inhibitors. Studies by Bissantz et al. [3], Stahl and Rarey [11] and Verdonk et al. [16]
15
showed works on consensus scores which further improved VS enrichment. However, the remaining issue for VS users rather than researchers is when and how these scoring functions should be combined in either drug design or industrial compounds design.
Docking programs URLs REFERENCES
DOCK http://dock.compbio.ucsf.edu/ 18 FlexX http://biosolveit.de/flexx/index.html?ct=1 32 AutoDock http://autodock.scripps.edu/ 46
GEMDOCK http://gemdock.life.nctu.edu.tw/dock/igemdock.php 26
GOLD http://www.ccdc.cam.ac.uk/products/life_sciences/gold/ 33
Table 1. Popular docking tools and evolutionary algorithms currently used in VS
Furthermore, certain VS methods can identify important interactions or binding-site hot spots obtained from known active ligands and target proteins [17]. However, due to biases towards higher molecular weight and charged polar compounds [18] docking alone is not sufficient to analyse, determine and retrieve the most adequate lead compounds therefore post screening analyses are emerging as useful methods to aid with further elimination of false positive hits obtained from VS.
Methods for post-screening analysis employing clustering to identify key features obtained via docked compounds and the understanding of binding mechanisms are of great use in bioinformatics. Therefore, computer-aided drug and industrial target design require VS as a primary step to generate interaction and structure profiles followed by post screening analysis for adequate filtering, visualization and mining of the final candidates.
3.2 The Significance of Protein-Ligand Interaction Profiles in Methods of Compound Retrieval and Post Screening Analysis
Interactions between molecules (Fig. 4) are important for understanding many biological phenomena. From gene expression to enzyme reactions, the activities are dictated by molecular interactions. Because of DNA microarray success, researchers are studying the protein counterpart in greater detail [47]. Protein microarray can be used for studying a variety of
16
biological phenomena such as interactions of protein-ligand, protein–protein, antibody–antigen, protein–DNA, analysis of subunits in protein complexes, screening of target proteins expressed from phage library, analysis of mutant proteins, quantitative assay, discovery of diagnostic markers, analysis of protein expression profiles, development of diagnostic microarray and development of microarray-based lead screening system. The interactions of significance in analysis and retrieval of lead compounds for drug design are intermolecular interactions such as van der Walls forces, electrostatic forces and Hydrogen bonds interactions [39, 40]. Also called interaction energies, they can be obtained from virtual screening of docked compounds calculations [13]. The calculations of interaction energies are organized into data sets of interaction profiles (IPFs) and can be used as one of the criteria in a cluster analysis to further filter out and select more specific or the final target compounds. Thus, cluster analysis of various compounds with similar interaction energies will group the various compounds into separate clusters from which a representative is chosen usually based on RMSD values while undergoing what is termed a post screening analysis.
3.2.1 Post Screening Analysis
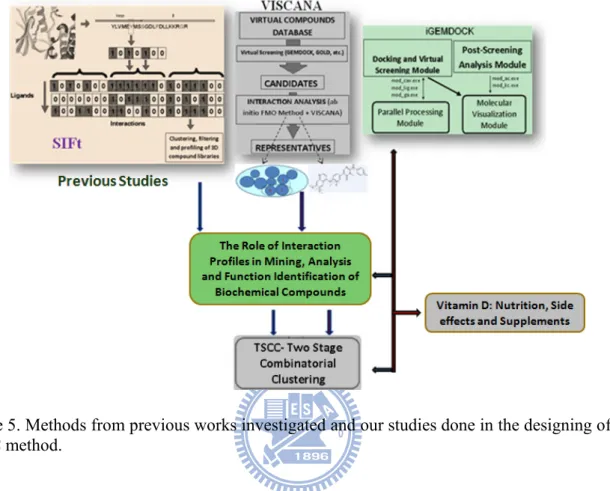
Methods of post screening analysis [21-23] are designed to facilitate the visualization (interpretation of binding interaction), organization (cluster and organize structures in a meaningful way), analysis (compare and profile the binding interactions of different structures) and data mining (search for structures containing key interactions or specific features) of virtually screened compounds. As mentioned earlier, binding interactions [39] (e.g. van der Walls forces, electrostatic forces and hydrogen bond interactions) of protein-ligand complexes are a critical part of mining and selecting the target representatives in post analysis methods. Descriptions of binding interactions and interaction strength measures for protein-ligand complexes are very important for better mining of appropriate candidates from selection lists generated by VS [48]. Thorough an in-depth study of protein-ligand interactions in various post screening analysis, we attempt to develop an integrated method of VS and post screening analysis in order to speed up the screening and analysis of compounds, generate better interaction-specific information and to obtain suitable representatives. The overall details of this study are shown in Figure 5.
17
Figure 5. Methods from previous works investigated and our studies done in the designing of our TSCC method.
Bellow we investigate and compare a few pioneering methods of post screening analysis which were all originally designed to enrich virtual screening. Later in our work we will perform some comparative studies and inductive analysis which provide a foundation for expanding the use of virtual screening and post screening analysis into the mining and analysis of targets used in various other applications besides pharmaceutics.
3.2.2 Structural Interaction Fingerprint (SIFt)
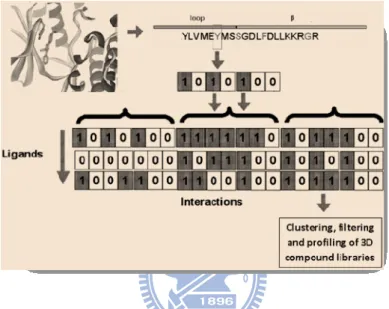
SIFt [23] uses a simple, generic and robust approach for representing and analyzing 3D protein- ligand interactions. Its key feature is the generation of an interaction fingerprint that converts 3D structural binding information into a one-dimensional (1D) binary string (Fig. 6). The fingerprint representation of the interaction patterns is compact, and allows for rapid clustering and analysis of large numbers of complexes. The SIFt is calculated on a set of input 3D protein–small molecule complexes. The protein structure may have been determined
18
experimentally by NMR or crystallography, or generated through homology modeling. The SIFt is generated by first defining the union of those residues that are in contact between the protein and the small molecule complex. The resulting panel of ligand binding site residues, which act as a mask covering all of the interactions occurring between the protein and the ligands, is then used as the common reference frame to construct the interaction fingerprints.
Figure 6. The 3D binding site of protein with an inhibitor (ligand) revealed as a sequence of positions in the binding site in contact with the ligand and their location in the structure of the protein (loop and β). Each binding site position is represented by a bitstring. The joining of all bitstrings end-to-end for each binding site residue is repeated for all ligands and is used in the selection process.
To analyse SIFTs the Tanimoto coefficient (Tc) [38] is used as the quantitative measure of bit string similarity. The Tc between two bit strings A and B is defined as:
Tc(A,
B)
=
A
IB
/
A
UB
where is the number of ON bits common in both A and B and is the number of ON bits present in either A or B. Tanimoto coefficients between random bit strings with a length of 400 bits adopt a near-Gaussian distribution centered at approximately 0.33, with a sigma of about 0.03. This representation of interactions as fingerprints using the SIFt method enables clustering, filtering and profiling of large libraries of docking results as well as crystal structures of the protein kinase family in complexes with various inhibitors.
19
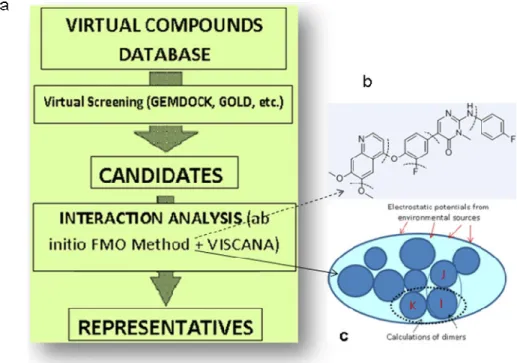
3.2.3 VISCANA (Visualized Cluster Analysis of Protein-Ligand Interaction)
VISCANA [22] (Fig. 7) is a method based on the ab Initio Fragment Molecular Orbital Method (FMO) [24] used for analysis of virtual ligand screening. The ab initio FMO method at the Hartree-Fock level is shown in the details following the method figure.
Figure 7. a) The overall approach of VISCANA (from VS to the selection of representatives).
b) The fragmentation of a polypeptide at different bonds. c) Division of biomolecules into a
collection of small fragments in the molecular orbital calculations (FMO method).
First, biomolecules or molecular clusters are divided into small fragments, and the ab initio MO calculations on the fragments (monomers) under the electrostatic potential from surrounding fragment pair as seen in Fig 7b and c. This is then solved repeatedly until all monomer densities become self-consistent. Finally, through the use of the total energies of the monomer EI and the dimer EIJ, the total energy of the system E is calculated by the following equation:
20
The FMO method has the advantage of describing the charge-transfer between a receptor and a ligand in comparison to a conventional force field method using fixed atomic charges. Based on this principle Amari et al. developed a cluster analysis using the dissimilarity defined as the squared Euclidean distance between interfragment interaction energies (IFIEs) of two ligands. VISCANA combines a clustering method with a graphical representation of the IFIEs by representing each data point with colors that quantitatively and qualitatively reflect the IFIEs. This method classifies structurally different ligands into functionally similar clusters according to the interaction pattern of a ligand and amino acid residues of a receptor protein. VISCANA also estimates docking conformation by analyzing patterns of the receptor-ligand interactions of some conformations through the docking calculations. VISCANA could be applied not only to the FMO method but also any molecular interaction system which can provide interaction energies or other properties of interest such as charge distribution.
3.2.4 iGEMDOCK: A Graphical Environment for Recognizing Pharmacological Interactions and Virtual Screening
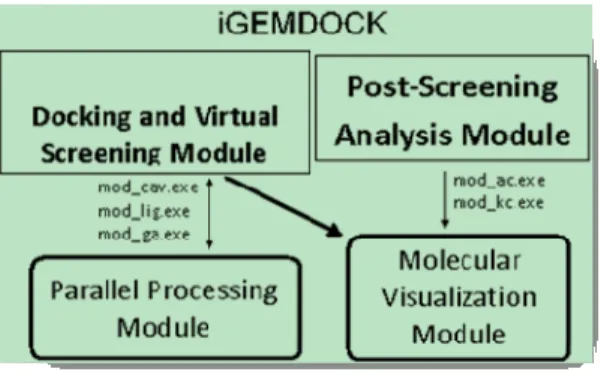
iGEMDOCK (Fig. 8) is an extension of the original docking tool GEMDOCK developed by Yang et el. [26] which adds a post screening analysis method to the original docking algorithm (http://gemdock.life.nctu.edu.tw/dock/igemdock.php). GEMDOCK’s two key
functions for VS are used: 1) the searching algorithm [49] and 2) the scoring function [50] which is based on an empirical energy function:
ligpre pharma bind tot E E E E = + +
where Ebind is the empirical binding energy, Epharma is the energy of binding site pharmacophores
(hot spots), and Eligpre is a penalty value if a ligand does not satisfy the ligand preferences. Epharma
and Eligpre are especially helpful in selecting active compounds from hundreds of thousands of
non-active compounds by excluding ligands that violate the characteristics of known active ligands, thereby improving the selection of true positives.
21
Figure 8. The virtual screening and post screening analysis processes in iGEMDOCK
The integration of different-stage programs of VS environments into GEMDOCK constituted the emergence of iGEMDOCK for docking, virtual screening and post screening analysis of database compounds using a friendly interface. In post-screening analysis iGEMDOCK enriches the hit rate and derives pharmacological interactions from screened compounds to provide biological insights. The pharmacological interactions represent conserved interacting residues which form binding pockets with specific physico-chemical properties expressing the essential functions of the target protein.
This new algorithm provides both virtual screening and post screening analysis as well as a more detailed and complete understanding of ligand binding mechanisms which makes the study and discovery of lead compounds much easier and less time consuming than other similar post screening analyses. iGEMDOCK is based on the efficiency of GEMDOCK which was able to mine various inhibitors such as aurintricarboxylic acid tetracycline derivatives which inhibit flaviviruses [6] and influenza virus neuraminidase inhibitors [8].
3.3 Summary
Methods of post screening analysis that enhance virtual screening enrichment and retrieve target compounds more accurately are of great use and interest in current bioinformatics. In this review we summarized and compared methods of VS and post screening analysis of lead compounds which emphasize the relevance of interaction profiles in mining suitable candidates.
22
SIFt (structural interaction fingerprint) is one of the pioneer methods in post screening analysis to include interaction-specific information into the real number strings. This enables the visualization, organization, analysis and retrieval of structures containing key interactions or specific features. A combination of SIFt and ChemScore (an empirical scoring function) contributed to a modest increase in the enrichment factor (EF) which was calculated based on the ability to recover known inhibitors. The enrichment increased from 37.0 EFa (SIFt) to 42. 3 EFa
(SIFt + ChemScore) [23].
VISCANA (Visualized Cluster Analysis of Protein-Ligand Interaction) uses a different approach through the FMO method. It has the advantage of describing the charge-transfer between a receptor and a ligand in comparison to a conventional force field method using fixed atomic charges. The difference between VISCANA and other conventional screening methods is that most methods choose the higher rank of a docking score on a point. In VISCANA a compound with a low docking score may belong to the same cluster that contains active compounds and the compound could be a suitable candidate. However, Amari et al. affirmed in their study VISCANA needs further development of quantum mechanical methods (the second-order Møller-Plesset perturbation theory based on the FMO method) to obtain more reliable descriptions of van der Walls interactions and hydrogen bonds which are important in determining receptor-ligand binding [22]. Other post screening studies reveal that unreliable or insufficient descriptions of important interactions account for increased numbers of false positives [48].
iGEMDOCK, an integration of VS and post screening methods is based on the original evolutionary docking algorithm GEMDOCK, currently one of the pioneer methods used for combining VS with visualizing, organizing, analysing and data mining of lead compounds. It has an advantage over SIFt and VISCANA primarily due to the attempt of eliminating two key issues: 1) if a docking tool is used for VS, which post screening analysis can complement it best and 2) if a post screening analysis method is decided, which docking tool or VS method is most suitable. The difference in the post screening approach of iGEMDOCK and other methods (VISCANA and SIFt) is the use of a module which clusters compounds based on interaction profiles and atomic compositions. Selecting representative compounds from each cluster enables the maintaining of compound diversity and reduces the number of false positives. In addition, its pharmacological scoring function can reduce the ill-effect of energy-based scoring functions
23
which often favor high molecular weight or highly-polar compounds. This improves the screening accuracy when the molecular weights of the active compounds are less than 400 Daltons (Da) [52]. Most notably, GEMDOCK, the earlier version of iGEMDOCK was used successfully to screen and identify inhibitors for influenza virus neuraminidases and flaviviruses [6, 8].
We also emphasize on the use of VS and post screening analysis in the mining of novel compounds for various other applications (e.g. industry, agriculture, cosmetics and nutritional supplements). These areas have not been getting much attention in comparison to drug design whereas certain protein-ligand complexes constitute key compounds in developing various biochemical products [29-31]. VS and post screening analysis used in computer-aided drug design reveal great potential in such applications since prospect candidates used in cosmetics and other industries may be retrieved employing interaction profiles.
Although the methods investigated in this study, SIFt, VISCANA and iGEMDOCK employ different techniques (structural interaction fingerprint, ab initio FMO method and interaction energy modules) they have one common feature; the use of protein-ligand interaction profiles which can be further exploited in developing new and improved methods to retrieve and analyze potential candidates for drug design and other applications. Through the development of better techniques, measures and description of interaction energies can aid methods of novel compounds retrieval and analysis, improve in accuracy and selection of active compounds. In addition, these observations point to an important aspect in the computer-aided drug design and discovery, the necessity for more than one stage of clustering in post screening analysis. From this point we proceeded with developing our new method Two-Stage Combinative Clustering (TSCC) [48] which combines our specifically optimized docking tool (GEMDOCK) with two stages of clustering for an optimized post screening analysis.
24
CHAPTER 4
TSCC: Two-Stage Combinative Clustering for Virtual Screening Using Protein-ligand Interactions and Physical-Chemical Features
4.1 Introduction
Continuous advancements in high-throughput X-ray crystallography and genomics [2, 28] account for increased numbers of available crystal structures enabling a more rapid development of new therapeutic targets. However, prospect ligands and proteins need to be screened in order to downsize groups [22, 23, 53] and select suitable candidates for post-screening analysis. Clustering methods based on structural similarity which are employed in post-screening analysis generally improve the scoring function performance. In developing methods for 3D compound retrieval, a detailed understanding of intermolecular interactions between proteins and their ligands is critical to structure-based inhibitor design. Various post-screening analysis methods and clustering [23, 54-56] employ RMSD values, protein-ligand interactions and computation and comparison platforms for measuring distances. Since the above methods as well as TSCC encounter challenges of specific selectivity and false positives, we aim to provide advantages to our post screening analysis method by using two combined clustering stages to rank all compounds and select final representatives more efficiently and accurately. The final representatives can be confirmed through bioassays to verify their target and the proper activity and application.
Although similar methods (IBAC, SIFt and VISCANA) [21-23] have used visualization and clustering of compounds to enrich VS, they have not identified novel compounds for any practical applications (drug design or industrial purposes). In addition, with the use of such methods one main issue remains unsolved: which combination of VS and post screening analysis is the most efficient. Our goal is to provide a more efficient method for post screening analysis to identify novel compounds, their possible functions and practical applications. Thus, we employ the empirical energy function from GEMDOCK [26] and the basic premise of SIFt [23] to encode additional interaction-specific information into the real number strings, hydrogen bonds, van der Waal and electrostatic forces. By representing interactions at the atomic-level as opposed to the residue level and including measures of interactions strength, protein-ligand interactions can be described better and a more precise analysis of virtual screening can be obtained.
25
TSCC is accomplished by joining two clustering stages; one of protein-ligand interactions (e.g. hydrogen bonds, electrostatic interactions, and van der Waals forces) with another of physico-chemical features (e.g. atom composition). We employed our docking tool, GEMDOCK, to generate protein-ligand interactions and used the Accelrys CeriusQSAR module for obtaining physico-chemical features for the compounds. Based on normalized feature profiles, hierarchical and K-mean [57] clustering methods were used to cluster and select compound candidates. Since clustering based upon similarity requires a quantitative measure (descriptor) of the similarity between two molecules, 2D and 3D methods were used to generate a descriptor such as the atom pair descriptor (i.e. compound topological similarity) [58].
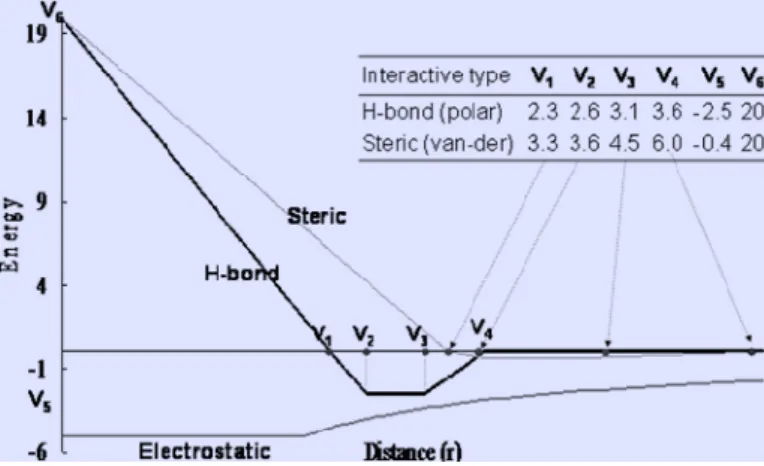
To handle the vast results from virtual screening and use more specific information for protein-ligand binding, we utilize the empirical energy function from GEMDOCK [26] specifically optimized for virtual screening of ligands. GEMDOCK uses piecewise linear potential (PLP), a simple scoring function (Fig. 9), comparable to similar scoring functions for estimating binding affinities [60, 61]. Our previous works showed a comparison of GEMDOCK and other docking methods for 100 protein-ligand complexes and two virtual screening targets [49-50]. In addition, GEMDOCK has been successfully applied to identify inhibitors and binding sites for some targets [6, 8]. Here, we utilize the PLP of GEMDOCK to generate the protein-ligand interaction profiles.
Figure 9. The linear energy functions of the pairwise atoms for the steric interactions and Hydrogen bonds in GEMDOCK (bold line) with a standard Lennard-Jones potential (thin line) Yang et al. [26].
26
To demonstrate the efficiency of our method we successfully applied its combinative two-stage concept in two separate post screening analysis studies. In the first study (sections 4.2, 4.3) two compound sets (testing and verifying) were designed to determine if the protein-ligand interaction descriptor is suitable for identifying compounds with similar binding modes. The two sets were also used to determine if the compound structure descriptor is suitable to identify similar structure compounds and to evaluate the database enrichment potential and the property of compounds in the same cluster by docking a diverse set of compounds spiked with known active compounds into the same target protein.
4.2 Materials and Methods
The Two-Stage Combinative Clustering (TSCC) Methodology
The overview of TSCC concept in our first study is shown in Figure 10. We first calculated the atom-based protein-ligand interactions by converting every docked pose into a one dimensional real number string in order to visualize and analyze large data obtained from virtual screening using Yang et al [26].
Figure 10. Overall process of TSCC in our first study (a) First stage clustering using P-L interactions generated via GEMDOCK. (b) Second stage clustering of first stage results using physico-chemical features. (Figure obtained from our published study [48]).
27
Due to protein-ligand interactions representation, we were able to evaluate the distance of binding modes between two docked poses and to carry out hierarchical clustering analysis. Compounds with a similar binding mode were visualized and grouped into clusters [59]. In our structure based clustering section, each structure was represented by a one dimension atom-pair descriptor, an approach proposed by Carhart et al [58]. After analyzing the distance between active and non-active compounds, a reference threshold was decided for demarcating similar compounds (Fig. 11).
Figure 11. Designing the reference thresholds for protein-ligand interaction and atom-pair descriptor (Figure obtained from our published study [48]). The complementation between atom-pair descriptor and the protein-ligand interaction descriptor is also shown in this figure. The distance threshold of atom-pair descriptor obtained was 0.55 (tanimoto coefficient) and the threshold of distance of protein-ligand interaction descriptor was 0.39 (correlation coefficient).
28
We generated two sets of structure-based virtual screening results: 1) to verify if the protein-ligand interaction descriptor is suitable for identifying compounds with similar binding mode and 2) to evaluate the database enrichment potential and the property of compounds in the same cluster by docking a diverse set of compounds spiked with known inhibitors into the same target protein.
4.2.1 Preparation of Target Protein and Compound Databases
The Ligand binding site was defined as a collection of amino acids using a cutoff radius of 10Å from each atom on the bound ligand, since most studies in lead discovery use a cutoff radius between 8 to 12 Å. Structure files were stored as a PDB format for GEMDOCK analysis.
Compound databases
We constructed two compound sets for screening against each target protein: thymidine kinase (TK) PDB id: 1kim, estrogen receptor alpha-agonist (ERα) PDB id: 3ert, estrogen receptor alpha-antagonist (ERα) PDB id: 1gwr, human dihydrofolate reductase (hDHFR) PDB id: 1hfr, tern n9 influenza virus neuraminidase (NA) PDB id: 1mwe. The structures used were obtained from the database Comprehensive Medicinal Chemistry (CMC) and American
Chemical Directory (ACD) and compounds with molecular weights between 200 and 800 D were chosen only based on the similar size of our active compounds. The active compounds (61 compounds, Appendix 1 a) were listed as the following: 1) TK: 10, 2) ER α antagonists: 11, 3) ER α agonists: 10, 4) hDHFR: 10, and 5) NA: 20. The two crystal structures of human estrogen receptors alpha have been intensively studied for their different functions (agonist 1GWR promotes coactivator binding while antagonist 3ERT blocks it) and ability to bind on the same site of the protein. The agonists play an important role in regulation of gene expression and prevention of osteoporosis while the antagonists have been used as treatment of hormone-dependent breast cancer [60, 61].