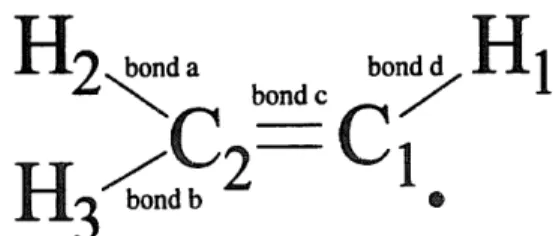ELSEVIER
Chemical Physics 206 (! 996) 43-56Chemical
Physics
Theoretical study of isomeric structures and low-lying electronic
states of the vinyl radical C2 H 3
Jeng-Han Wang
a,
Hung-Chang Chang
a,
Yit-Tsong Chen a,b,,
a Department o f Chemistry, National Taiwan University, Taipei 106, Taiwan, ROC
b Institute of Atomic and Molecular Sciences, Academia Sinica, P.O. Box 23-166, Taipei 106, Taiwan, ROC
Received 11 November 1995
Abstract
The molecular structure, intramolecular rearrangement and dissociation energy of C2H 3 have been studied with high-level ab initio calculations using ACES II and MOLCAS-2 programs. In the structural calculations of C2H 3, the optimized geometry and vibrational frequencies of X 2K, the vertical electronic transition energies (,~ 2h~' , - X 2h~ and 2K ~ X 2h~), the vertical ionization potential and the permanent dipole moment of X 2K have been computed. The harmonic vibrational frequencies and infrared intensities of C2H 3 X 2~ obtained from this calculation will help the spectroscopic observation for the vibrational modes, most of which are unobserved. The calculated vertical transition energy, 25529 c m - ~ for A 2h/' ~ X 2A~, and the vertical ionization potential, 8.33 eV from an MRCI method with atomic natural orbitals, are in excellent agreement with the experimental values of 24815 cm-~ and 8.25 eV, respectively. The vertical transition of B 2~/~ ,q 2~/, predicted to be 43910 cm-~ from this work, will facilitate the experimental search for the undiscovered B state of C 2 H 3 through spectroscopic observation. In calculating the intramolecular rearrangement in C 2 H 3 2K, using CCSD(T)/Dunning's triple zeta polarizations, the non-classical structure with a hydrogen atom bridged between the C = C bond has been found to lie at least 47 kcal/mol above the classical equilibrium structure. The calculation also indicates that the non-classical C2H 3 X 2.a/ is an unstable isomer, corresponding to a transition state. The computed barrier for the tunnelling of a-H in C2H 3 X 2A' is also in excellent agreement with the upper bound limit of < 1500 cm- determined from high-resolution infrared spectroscopy. The dissociation energy of C2H 3 ~ C2H 2 + H and the energy difference between the isomers of acetylene and vinylidene, calculated in the present study, are also consistent with experimental measurements.
1. Introduction
The vinyl radical, C 2 H 3 , is one of the most important transient hydrocarbons in flame chemistry [1-3]. The mechanisms for the decomposition of C 2 H 3, such as thermal decomposition, reactions with
* Corresponding author. Fax: + 886 2 362 0200; E-mail; [email protected].
hydrogen [4] and oxygen [5] etc., have demonstrated to be of fundamental importance in combustion pro- cesses. It is also a significant chemical inte~lediate in various chemical reactions, e.g. the addition to and the polymerization of an acetylenic bond, and the decomposition of ethenoid compounds. More- over, owing to its important role in stereochemistry, vinyl radicals have attracted the interest of organic chemists for a long time [6].
0301-0104/96/$15.00 © 1996 Elsevier Science B.V. All rights reserved
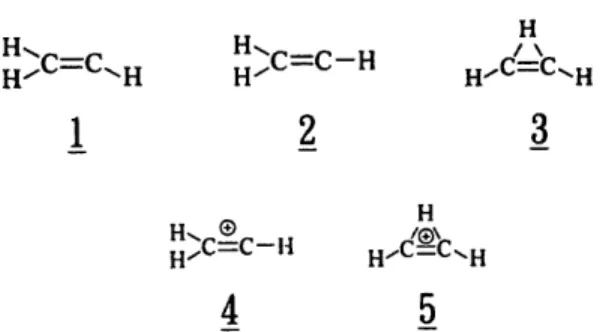
44 J.-H. Wang et a l . / Chemical Physics 206 ('1996) 43-56 H H~ HXC=C__ H / \ H / C = C - . H H" H / C - ' C ~ H
1
2
3
H H,, ® H c=c-H4
5
Fig. 1. The isomeric structures of vinyl radical and vinyl cation.
Geometry calculations for C 2 H 3 in the ground (X2A~) and the first excited (,~ 2~,) electronic states have also been the subject of many theoretical stud- ies [7-9]. The calculated planar structures with C s symmetry (1 in Fig. 1) in the X 2A~ and the ,~ 2g, states of C 2 H3, and their associated electronic ener- gies were used in the analysis of the low-resolution (A v ---- 45 cm- ! ) electronic ,~ 2~, ~_ ~[ 26 ~ absorption spectrum observed by Hunziker et al. [7]. In the observed vibrationally resolved electronic absorption spectrum of C2H 3, the attribution for the lack of rotational features as due to the low instrumental resolution or the nature of molecular predissociation remains uncertain [7]. The ionization potential of C2H 3 reported from several experiments [10-12], ranging from 8.95 to 8.25 eV, has also been the subject of some controversy.
Although the vibrational frequencies and dipole- moment derivatives of C2H 3 X 2AP were calculated from generalized valence bond wavefunctions many years ago [13], the v 7 vibrational band at 895 cm- reported by Kanamori et al. [14] in gas-phase spec- troscopy ( ~ 900 cm-l in matrix isolation by Shep- herd et al. [15]) has so far been the only vibrational observation. In the high-resolution infrared spec- troscopy of C 2 H 3, Kanamori et al. have reported the radical to be of C 2v effective symmetry (2 in Fig. 1), referring to a tunnelling motion in the double-mini- mum potential due to the rocking a-H in C2H 3 (as shown in Fig. 2a). The tunnelling was evidenced by (i) the spectroscopic splitting of the vibrational com- ponents in the excited v 7 = 1 and in the ground vibrational states, and (ii) the spectral intensity ratio of 3" 1 for the ortho- and para-nuclear-spin statistical weights, which can only be rationalized by a C2v molecular symmetry in C2H3 ~ 2A~. The energy
barrier for the rocking motion was estimated to be < 1500 cm -I from the observed spectrum [14], which is lower than the previous predictions from some relatively low-level calculations [8,13]. In this study, we have calculated the nine vibrational normal modes and infrared intensities of C2H 3 ~[ 2~, and have tried to accurately determine the barrier of the a-H rocking motion in C 2 H 3-
In contrast to the remarkable isomerization pro- cess in C2H ~ (as shown in Fig. 2b) extensively investigated both experimentally [16] and theoreti- cally [17], the study about the isomerization of C 2 H 3 (Fig. 2c) has been relatively little reported. While the energy difference between classical (4 in Fig. 1) and non-classical (5 in Fig. l) structures of CEH ~" is only ~ 4 kcal/mol, Harding predicted the barrier for the H-migration in C 2 H 3 a s high as 57 kcal/mol in an earlier study [ 18]. In this paper, we shall report our re-examination of the energy difference between the classical (1 in Fig. l) and non-classical (3 in Fig. 1) structures of C 2 Ha with high-level ab initio calcu- lations. The very recent calculation of the methylcar- byne-vinyl isomerization conducted by Nielsen et al. is worth noticing [19]. The calculated isomeriza- tion barrier and energy difference from methylcar- byne to vinyl radical are 9 and - 4 9 kcal/mol, respectively, in the doublet potential, and 57 and - 3 kcal/mol in the quartet. The calculations have signi- fied that the spectroscopic observation from vinyl to methylcarbyne is much more difficult than the re- versed isomerization process.
Of particular interest to us with the vinyl radical
a. HH~c=CxH -" "- H~C--CtHH / -- H H~C~=C__H ~ /OX -" H / C = C ~ H H H ..._ / \ ¢. ~.C=C-'H -- H / C = C ~ H Fig. 2. The intramolecular rearrangements in vinyl radical and vinyl cation.
J.-H. Wang et al. / Chemical Physics 206 (1996) 43 -56 45 is to experimentally investigate the isomerization
and the dissociation processes of C2H 3 X 23/ in our
laboratory by vibration-rotationally state-selective laser spectroscopy, such as stimulated emission pumping (SEP) [20] or two-color (IR-UV) laser-in- duced grating (LIG) [21]. To facilitate the experi- mental performance in the SEP and LIG spectro- scopies, we have calculated the vertical electronic transition frequencies. Owing to the likely predisso- ciation in the first excited electronic state (,~ 23/,) of C2H 3 as discussed by Hunziker et al. [7], we have sought an alternative and calculated the second ex- cited electronic state of the radical. We have also calculated the vertical ionization potential of C2H 3 to compare with the experimental result [12] to test the accuracy of the computational process. In this paper, we shall also report the calculations of the energy required for the C - H bond cleavage in the dissociation of C2H 3 ~ H + HCCH (acetylene) or C2H 3 ~ H + H2CC (vinylidene). The calculated re- suits will show that the isomerizations are energeti- cally above the dissociation threshold of C2H 3 ~ H
+ HCCH.
This paper is organized as follows. In Section 2, we will describe the computational details for the calculations of the molecular structure, intramolecu-
iar rearrangement and dissociation of C 2 H 3 using
ACES II [22] and MOLCAS-2 [23] programs. For
the molecular structure of C2H 3, we have calcu-
lated (i) the equilibrium geometry and vibrations of the ground electronic state (X: 2~/), (ii) the isomeric structures on the ground electronic potential, (iii) the vertical transitions from the ground (X: 23/) to the first (,~ 2,~,), and to the second (B 23/) excited elec- tronic states, respectively, (iv) the vertical ionization potential, and (v) the permanent dipole moment of ~: 23/. Calculations of the intramolecular rearrange-
ment in C2 H3 include isomerization and a - H rock-
ing motion as shown in Fig. 2. In the study of the
dissociation in C2H 3 X 2A', we have calculated the
dissociation energies of C2H 3 ~ H + HCCH, and of C 2 H 3 --' H + H 2CC- The energy difference between the isomers of acetylene and vinylidene has also been computed. In Section 3, we will discuss the calculated results obtained in the present study using high-level ab initio methods with various efficient basis sets. Concluding remarks will be addressed in Section 4.
2. Computational details
All of the calculations carried out in this study were executed on an IBM RISC6000/590 computer. The computational schemes for molecular structure, intramolecular rearrangement and dissociation of C2H 3 using the programs of ACES II and MOL- CAS-2 will be described in the following.
2.1. Molecular structure
2.1.1. Equilibrium geometry and vibrations o f C 2 H 3
g2a,
Using the ACES II program, we have optimized the ground electronic structure of C2H 3. The ab initio methods, including many body perturbation theory (MBPT) and coupled cluster (CC), have been employed in the full optimization for the nine inter-
nal co-ordinates in C2H 3. The basis sets for the
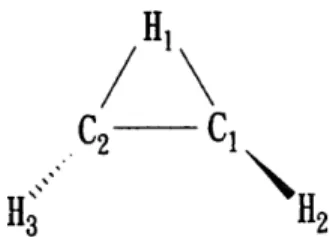
carbon and hydrogen atoms of C 2 H 3 were 6-31G * * and Dunning's DZP, TZP, TZ2P and PVTZ. In the optimization, UHF MOs were used as approximate starting wavefunctions, and the vinyl radical was set to be of C i symmetry. The labelling for each atom in the equilibrium structure of C 2 H 3 is shown in Fig. 3. The completion of the optimization was reached at an energy gradient of ~< 1 X 10 -4 hartree/bohr. The optimized geometry of the ground electronic state of C2H 3, using various methods and basis sets, together with the calculated rotational constants are listed in Table 1. Since the dihedral angles in the optimized geometry of the ground electronic state of C2H 3 are very small ( 4 0.02 ° in all of the calcula- tions) and are not shown in Table 1, the structure is essentially planar. The symmetry of the ground elec- tronic state of C 2 H 3 is therefore an 3/ representation belonging to the C s molecular symmetry group, i.e.
R23/.
H 2 " ~ d L bondc
°n H1
H3
C1 o
Fig. 3. The labelling for each atom in classical C2H 3. The core electrons ( i s 2) of the carbon are not shown.
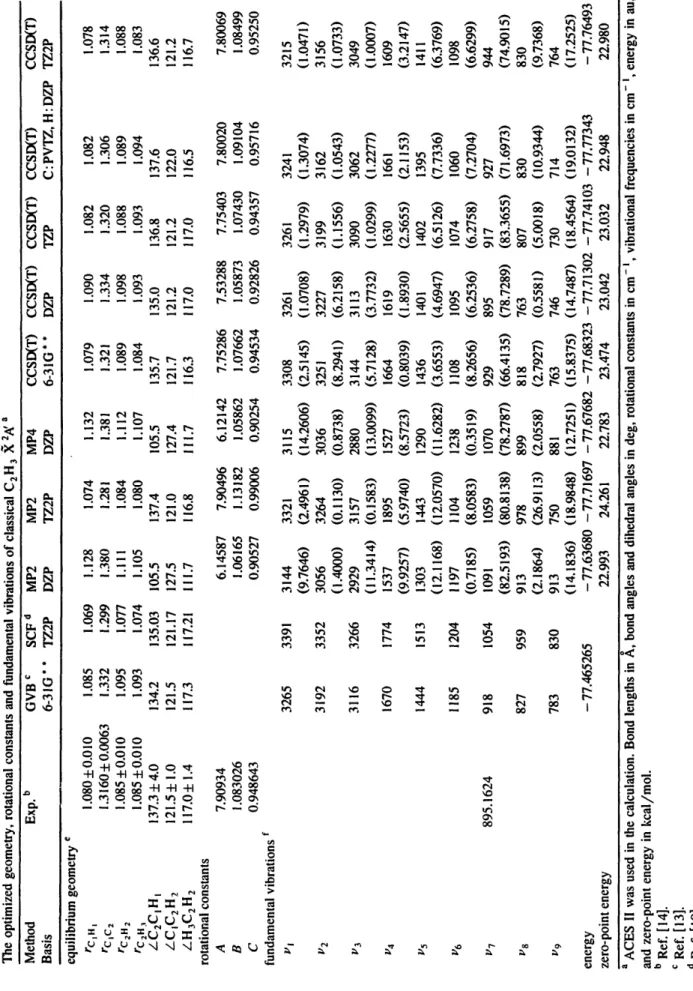
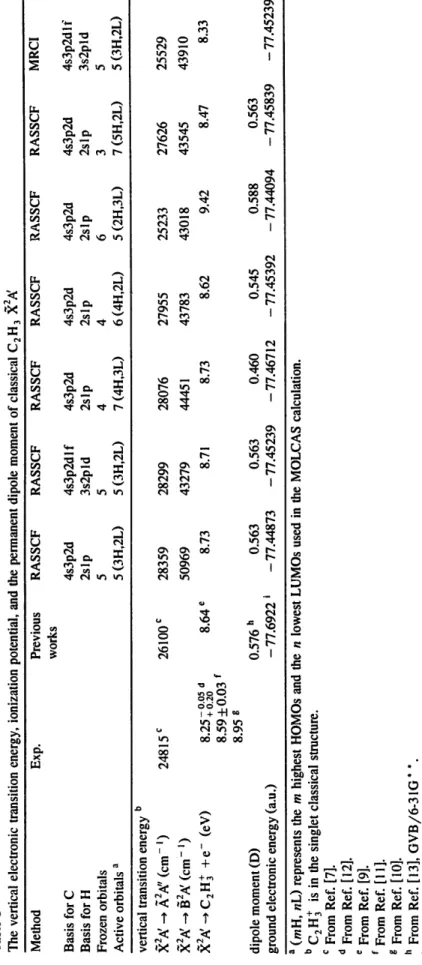
Table 1 The optimized geometry, rotational constants and fundamental vibrations of classical C2H 3 X 2~ a Method Exp. b GVB c SCF d MP2 MP2 MP4 Basis 6-3 IG * * TZ2P DZP TZ2P DZP CCSIXT) CCSIXT) CCSIXT) CCSIXT) CCSIXT) 6-31G * * DZP TZP C: PVTZ, H: DZP TZ2P equilibrium geometry e rCiH t rcic 2 rC zH 2 rC2H 3 /-C2CIH I /CIC2H 2 /-H3C2H 2 rotational constants A B C fundamental vibrations f /'1 /'2 /'3 /'5 /'6 /'7 /')8 i,, 9 energy zero-point energy 1.080±0.010 1.085 1.069 !.i28 1.074 i.132 !.079 1.090 1.082 !.082 1.078 1.3160~0.0063 1.332 1.299 1.380 1.281 1.381 1.321 1.334 !.320 1.306 1.314 !.085 ~0.010 1.095 1.077 i.lll 1.084 i.il2 1.089 !.098 1.088 1.089 !.088 1.085 ±0.010 1.093 1.074 1.105 1.080 1.107 !.084 !.093 1.093 i.094 !.083 137.3±4.0 134.2 135.03 105.5 137.4 105.5 135.7 135.0 136.8 137.6 136.6 121.5± 1.0 121.5 121.17 127.5 121.0 127.4 121.7 121.2 121.2 122.0 121.2 ll7.0~ !.4 117.3 il7.21 111.7 116.8 111.7 !16.3 117.0 ll7.0 !!6.5 116.7 7.90934 6.14587 1.083026 1.06165 0.948643 0.90527 895.1624 3265 3391 3144 (9.7646) 3 i 92 3352 3056 (!.4000) 31 ! 6 3266 2929 (11.3414) 1670 1774 1537 (9.9257) 1444 1513 1303 (12.1168) 1185 1204 1197 (0.7185) 918 1054 1091 (82.5193) 827 959 913 (2.1864) 783 830 913 (14.1836) - 77.465265 7.90496 6. ! 2142 7.75286 7.53288 7.75403 7.80020 7.80069 I. 13 ! 82 !.05862 !.07662 1.05873 1.07430 1.09104 1.08499 0.99006 0.90254 0.94534 0.92826 0.94357 0.95716 0.95250 3321 3115 3308 3261 3261 3241 3215 (2.496 !) (14.2606) (2.5145) (!.0708) (!.2979) (1.3074) (!.047 !) 3264 3036 3251 3227 3199 3162 3 ! 56 (0.1130) (0.8738) (8.2941) (6.2158) (I.1556) (!.0543) (! .0733) 3 ! 57 2880 3144 3 i 13 3090 3062 3049 (0.1583) ( ! 3.0099) (5.7128) (3.7732) (1.0299) (!.2277) (I .0007) 1895 1527 1664 1619 1630 1661 1609 (5.9740) (8.5723) (0.8039) (1.8930) (2.5655) (2. ! ! 53) (3.2147) 1443 1290 1436 1401 1402 1395 1411 (12.0570) (11.6282) (3.6553) (4.6947) (6.5126) (7.7336) (6.3769) i104 1238 i108 1095 1074 1060 1098 (8.0583) (0.35 ! 9) (8.2656) (6.2536) (6.2758) (7.2704) (6.6299) 1059 1070 929 895 917 927 944 (80.8138) (78.2787) (66.4135) (78.7289) (83.3655) (71.6973) (74.9015) 978 899 818 763 807 830 830 (26.9113) (2.0558) (2.7927) (0.5581) (5.0018) (10.9344) (9.7368) 750 881 763 746 730 714 764 (18.9848) (12.7251) (15.8375) (14.7487) (18.4564) (19.0132) (17.2525) - 77.63680 - 77.71697 - 77.67682 - 77.68323 - 77.71302 - 77.74103 - 77.77343 - 77.76493 22.993 24.261 22.783 23.474 23.042 23.032 22.948 22.980 r~ I a ACES II was used in the calculation. Bond lengths in ,~, bond angles and dihedral angles in deg, rotational constants in cm- ~, vibrational frequencies in cm- t, energy in au, and zero-point energy in kcal/mol. b Ref. [14]. c Ref. [ ! 3]. d Ref. [ 19]. e The dihedral angles in the optimized geometry are very small ( ~ 0.02" in all of the calculations) and are not listed. The structure of C2H 3 is essentially planar. f The numbers in parentheses are the infrared intensities in kin/tool.
J.-H. Wang et al. / Chemical Physics 206 (1996) 43-56 47
/
/ /
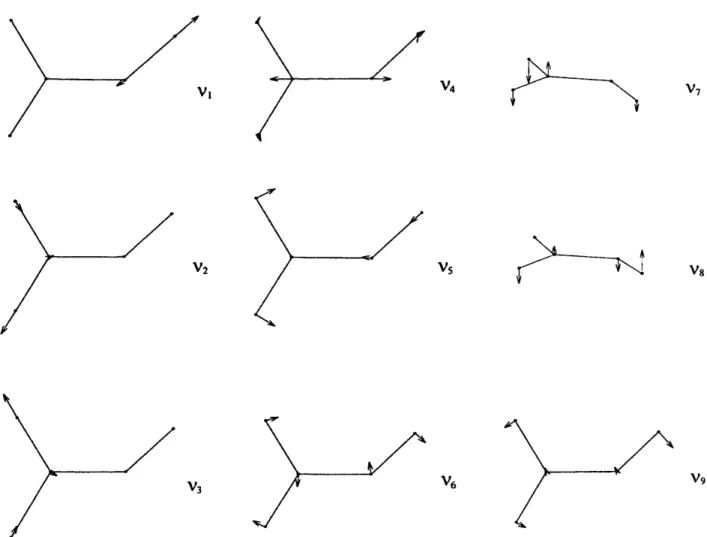
Vl v4 V7/
/
V2 Vs Vs/
v3./
v6Fig. 4. The vibrational normal modes of classical C 2 H 3 X 2~.
V9
After completing the optimization, we proceeded to calculate the fundamental vibrational frequencies of C2 H 3 ~ 2A~ by diagonalizing the force-constant matrix and transforming the dipole-moment gradient to normal co-ordinates. The calculated fundamental vibrational frequencies of C 2 H 3 ~ 2~6~ are also tabu- lated in Table I. The nine vibrational normal modes are shown in Fig. 4.
2.1.2. Electronic energies, ionizatit:n potential and dipole moment
In the MOLCAS calculations of C2H 3, the meth- ods from RASSCF to MRCI have been applied to compute the vertical transitions from the ground to the first, and to the second excited electronic states. The vertical ionization potential and permanent dipole moment of C2H 3 X 2A~ have also been included in the calculation. The generally contracted basis sets of
atomic natural orbital (ANO) [24] have been used in the present study. At the beginning, smaller basis sets of the ANO-type contractions were used, such as [4s3p2d] for the carbon and [2sip] for the hydrogen of C2H 3 composed of the primitive sets of (14s9p4d) and (8s4p), respectively. More complete basis sets of ANO contractions, [4s3p2dlf] from primitive
Table 2
The selected frozen orbitals in the M O L C A S calculation a
Number o f Frozen orbitals
frozen orbitals
cores a and b
cores a and b; bond c (or) cores a and b; bonds a and b cores a and b; bonds a, b and c (o') cores a and b; bonds a, b and c (ty + "rr) a Cores a and b are I s electrons o f C~ and C.,.
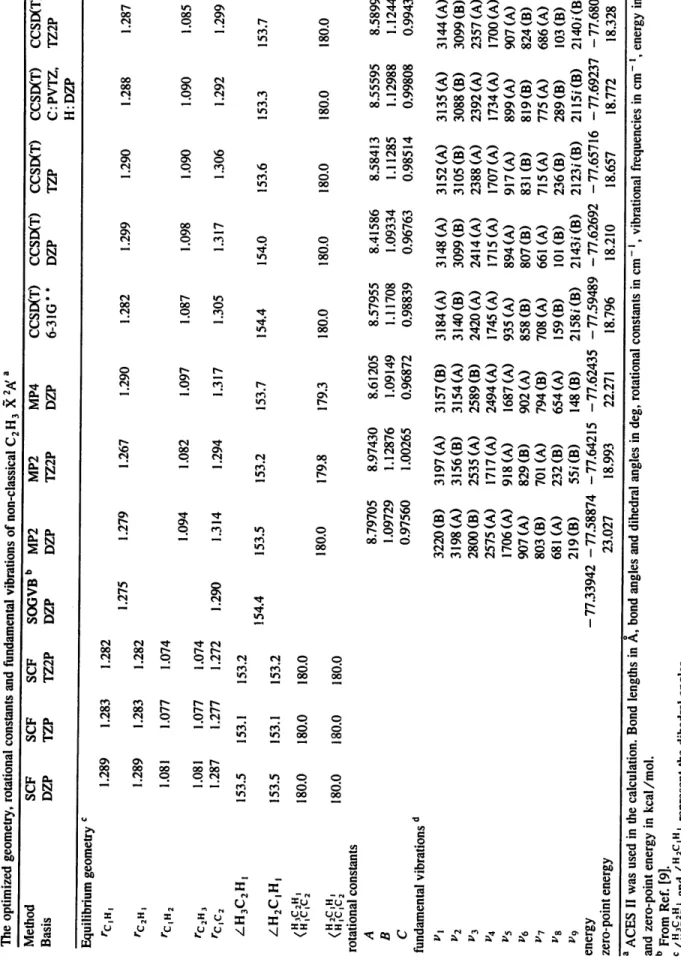
Table 3 The vertical electronic transition energy, ionization potential, and the permanent dipole moment of classical C 2 H 3 X2,a/ Method Exp. Previous RASSCF RASSCF RASSCF RASSCF works RASSCF RASSCF MRCI Basis for C 4s3p2d 4s3p2d ! f 4s3p2d 4s3p2d 4s3p2d 4s3p2d 4s3p2d I f .~ Basis for H 2s I p 3s2p ! d 2s ! p 2s 1 p 2s ! p 2s I p 3s2p I d .~ Frozen orbitals 5 5 4 4 6 3 5 Active orbitals a 5 (3H,2L) 5 (3H,2L) 7 (4H,3L) 6 (4H,2L) 5 (2H,3L) 7 (5H,2L) 5 (3H,2L) b vertical transition energy ~2 A, __, ~2g, (cm-') :X2A' --'C2H~" +e- (eV) [" ,,... 24815 c 26100 c - 0.05 d 8.25 + 0.20 8.59 + 0.03 f 8.95 ~ 8.64 e dipole moment (D) 0.576 h ground electronic energy (a.u.) - 77.6922 i 28359 28299 28076 27955 50969 43279 44451 43783 8.73 8.71 8.73 8.62 0.563 0.563 0.460 0.545 -77.44873 -77.45239 -77.46712 -77.45392 25233 43018 9.42 0.588 -77.44094 27626 43545 8.47 0.563 - 77.45839 25529 43910 8.33 -77.45239 a (mH, nL) represents the m highest HOMOs and the n lowest LUMOs used in the MOLCAS calculation. b C2H~- is in the singlet classical structure. c From Ref. [7]. d From Ref. [l 2]. e From Ref. [9]. f From Ref. [ i l ]. g From Ref. [10]. h From Ref. [13], GVB/6-31G * * i From Ref. [9], MP4/6-31 I G * * ~O I O~
J.-H. Wang et al./ Chemical Physics 206 (1996) 43-56 49 (14s9p4d3f) for the carbon and [3s2pld] from
(8s4p3d) for the hydrogen, have been employed later in the calculation. The sizes of the contracted basis sets for the smaller and for the more complete ones are 51 and 102, respectively.
Since the geometry optimization can not be car- ded out in the MOLCAS-2 program, the optimized equilibrium geometry of C2 H 3 X 2A', obtained from the ACES II calculation using CCSD(T) and P V T Z / D Z P , has been transferred into the MOLCAS computation to calculate the vertical transitions from the ground (~:) to the first excited (,~), and to the second excited (B) electronic states of C2H 3. We have tested various sets of frozen and active orbitals in the MOLCAS calculation. The selected frozen orbitals in different approaches are listed in Table 2 and the corresponding labelling for the chemical bonds in C2H 3 is shown in Fig. 3. We have taken the high occupied and the low virtual molecular orbitals of C2H 3, shown in Table 3, as a restricted active space (RAS), From the occupied molecular orbitals, the electronic configurations for the ,X, ,~, and B states of C2H 3 are (la')2(2a')2(3a') 2- (4a')E(5a~)2(6a')2(1 d')2(7a')i, (1 a')2(2a')2(3a')2(4a')2- (5at)2(6~)2(ld')l(7a') 2, and (la')2(2a')2(3a')2(4a') 2- (Sa')2(6a')2(l~')l(7a')i(2d') I, respectively, where the l d', 7a' and 2d' molecular orbitals have, in turn, been characterized by "rr-bonding, tr-bonding (oc- cupied by the single electron on C~ in Fig. 3), and "tr *-bonding. From the electronic configurations, we have been able to identify the symmetry of the ground, the first and the second excited electronic states of C2H 3 (in terms of a C s molecular symme-
try group) as X 2~, ,~ 2/~, and B 2/~, respectively. The reference configurations for the X 2/,,,, ,~, 2A? ' 2,6~ electronic states of C2H3, and the vinyl cation used in the calculations are listed in Table 4. The resulted vertical transition energies, ionization poten- tial and permanent dipole moment from the RASSCF calculations are tabulated in Table 3. After using the GUGA selection reference, we have tried an MRCI method with the basis sets of ANO contractions, [4s3p2dlf] for the carbon and [3s2pld] for the hydro- gen, to calculate the vertical electronic transitions of ~k 2~6~ ' ~ X 2~ and B 2Ap ~ X 2/~ and the vertical ionization potential of C2H 3. In the GUGA selec- tion, five frozen and five active orbitals have been chosen. For the active orbitals, three HOMO, and two LUMO, were involved in the calculation. The MRCI results are listed in Table 3. In comparison with the experimental values for the vertical transi- tion in ~ 2 a / , ~ ,~ 2A~ and the vertical ionization potential of C2H 3, the accuracy of the computational procedures in the present calculation has been tested, and will be discussed in Section 3.
2.2. lntramolecular rearrangement 2.2.1. Isomerization
In the study of the hydrogen-migration in C2H 3 2,a/, the energy difference between the classical (1 in Fig. l) and the non-classical (3 in Fig. l) isomers has been calculated exclusively with the ACES II program. At the beginning of the optimization for the non-classical C2H 3, we tried a full optimization
Table 4
The reference configurations for the X 2~, ,~ 2 K, and B 2a/electronic states of C2H 3 and the vinyl cation a Electronic state Reference configuration
ground (X 2.K) 22100 20120 2 ! I 10 02 ! 02
(0.9604) (0.035 ! ) (0.0062) (0.0033)
the first excited (.~ 2~,) 21200 21110 21020 20210 12200
(0.8848) (0.0567) (0.0483) (0.0058) (0.0039)
the second excited (B 2A') 21110 12200 i 1210 22100 2101 !
(0.9754) (0.0977) (0.0074) (0.0073) (0.0040)
vinyl cation (C2H ~" ) t, 22000 20020 1 i 110 02200 02002
(0.9456) (0.03 i 0) (0.0 ! 01 ) (0.0040) (0.0032) a The number in parentheses represents a weight for the configuration used in the MRCI calculation.
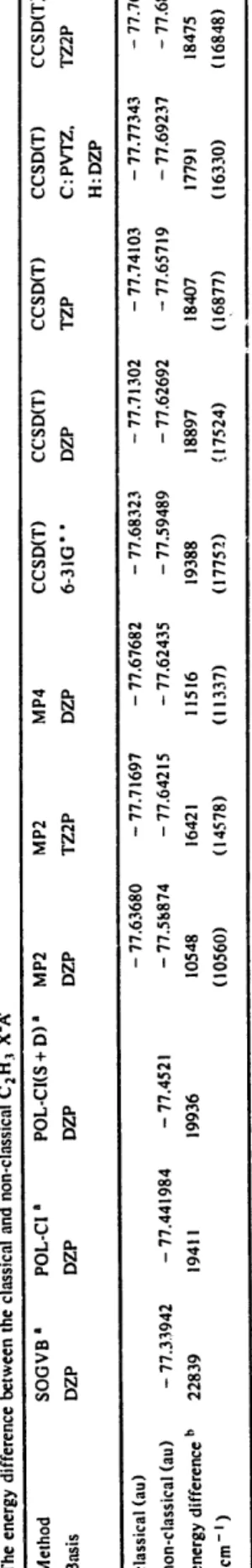
Table 5 The optimized geometry, rotational constants and fundamental vibrations of non-classical C2H 3 X 2~ a Method SCF SCF SCF SOGVB b MP2 MP2 MP4 Basis DZP TZP TZ2P DZP DZP TZ2P DZP Equilibrium geometry c rC,H, rCzH I FCtH 1 rc2tt 3 rcic 2 /-HaC2H ! /-H2C,H, H3C2H < I HiCIC2 <H2CIHI ICLC2 rotational constants A B C fundamental vibrations a Pi /"2 /23 /24 /25 /26 /27 /28 /)9 energy zero-point energy 1.289 1.283 1.282 1.275 !.279 1.267 1.290 1.289 1.283 !.282 1.081 1.077 1.074 1.094 i.082 1.097 1.081 i.077 1.074 1.287 i.277 1.272 !.290 !.314 1.294 1.317 153.5 153.1 153.2 153.5 153.1 153.2 180.0 180.0 180.0 180.0 180.0 180.0 CCSD(T) CCSD(T) CCSD(T) CCSD(T) CCSD(T) 6-3 ! G * * DZP TZP C" PVTZ, TZ2P H: DZP 1.282 1.299 1.290 1.288 1.287 i.087 i.098 i.090 1.090 1.085 1.305 1.317 1.306 i.292 1.299 154.4 153.5 153.2 153.7 154.4 154.0 153.6 153.3 153.7 180.0 179.8 179.3 180.0 180.0 180.0 180.0 180.0 8.79705 8.97430 8.61205 8.57955 8.41586 8.58413 8.55595 8.58991 1.09729 !.12876 1.09149 1.11708 1.09334 I.!1285 i.12988 i.12446 0.97560 1.00265 0.96872 0.98839 0.96763 0.98514 0.99808 0.99430 3220 (B) 3 ! 97 (A) 3157 (B) 3184 (A) 3148 (A) 3152 (A) 3135 (A) 3 i 44 (A) 3198 (A) 3156 (B) 3154 (A) 3140 (B) 3099 (B) 3105 (B) 3088 (B) 3099 (B) 2800 (B) 2535 (A) 2589 (B) 2420 (A) 2414 (A) 2388 (A) 2392 (A) 2357 (A) 2575 (A) 1717 (A) 2494 (A) 1745 (A) 1715 (A) 1707 (A) 1734 (A) 1700 (A) 1706 (A) 918 (A) 1687 (A) 935 (A) 894 (A) 917 (A) 899 (A) 907 (A) 907 (A) 829 (B) 902 (A) 858 (B) 807 (B) 831 (B) 819 (B) 824 (B) 803 (B) 701 (A) 794 (B) 708 (A) 661 (A) 715 (A) 775 (A) 686 (k) 681 (A) 232 (B) 654 (A) 159 (B) 101 (B) 236 (B) 289 (B) 103 (B) 219 (B) 55i (B) 148 (B) 2158i (B) 2143i (B) 2123i (B) 2115i (B) 2140i (B) - 77.33942 - 77.58874 - 77.64215 - 77.62435 - 77.59489 - 77.62692 - 77.65716 - 77.69237 - 77.68075 23.027 ! 8.993 22.27 ! 18.796 ! 8.210 18.657 ! 8.772 18.328 a ACES II was used in the calculation. Bond lengths in ,g,, bond angles and dihedral angles in deg, rotational constants in cm- ~, vibrational frequencies in cm- ', energy in au, and zero-point energy in kcal/mol. b From Ref. [9]. C /H3C2H ! /H2CIH t \. !c 1¢2 and \. ,c Ic2 represent the dihedral angles. d The A/B in parentheses is the symmetry representation of a C 2 molecular symmetry group. L~ 4~ I Lh
Table 6 The energy difference between the classical and non-classical C 2 H.~ X2A' Method SOGVB a POL-C! a POL-CI(S + D) a Basis DZP DZP DZP MP2 MP2 MP4 CCSD(T) CCSD(T) CCSD(T) DZP TZ2P DZP 6-3 IG" " DZP TZP classical (au) CCSD(T) C: Pv'rz, H: DZP CCSD(T) TZ2P - 77.63680 - 77.71697 - 77.67682 - 77.68323 - 77.71302 - 77.74103 - 77.77343 - 77.76493 ,.,.. ,...,. non-classical (au) - 77.33942 - 77.441984 - 77.4521 energy difference b 22839 19411 19936 (cm- i ) - 77.58874 - 77.64215 - 77.62435 - 77.59489 - 77.62692 - 77.65719 - 77.69237 - 77.68075 10548 16421 11516 19388 18897 18407 17791 18475 (10560) (14578) (11337) (17752) (17524) (16877) (16330) (16848) r~ ...,. t,o a From Ref. [18]. b The number in parentheses represents the zero-point energy corrections for both classical and non-classical C 2 H 3. 4~ I
52 J.-H. Wang et a l . / Chemical Physics 206 (1996) 43-56 H 1
/ \
C z ~
C1
~"'"
N
H
a
tt2Fig. 5. The atomic labelling of non-classical C, H 3 X 2,~.
process (including nine internal co-ordinates) using an SCF method and the Dunning basis sets of DZP, TZP and TZ2P. The optimized results are listed in Table 5 and the corresponding atomic labelling is shown in Fig. 5. The optimized non-classical C2H 3 is essentially planar and possessed of C2v symmetry. In the later calculations with higher-level methods,
Table 7
The optimized geometry, rotational constants, and fundamental vibrations of the transition state of C2H 3 ~ 2 ~ a
Method UHF b GVB c CCSD(T) CCSD(T) CCSD(T) Basis 6-31G * 6-31G * 6-31G * * DZP TZP CCSD(T) CCSD(T) C: PVTZ, H : DZP TZ2P equilibrium geometry d rc l u i 1.059 rC~C , 1.317 rc 2n 2 1.081
rC2H3
!.081 /-H2C2C I 122.1 / H 2 C 2 H 3 115.8 rotational constants A B C fundamental vibrations u I 3583 v 2 3303P3
3240
v 4 1621 v 5 1434 1'6 1060 v7 960 us 536 u9 917i energy - 77.37773 zero-point energy 22.50 1.070 1.065 1.322 1.310 1.096 1.090 1.096 1.090 121.8 122.5 116.4 115.0 1.076 1.068 1.067 i.064 !.324 1.310 1.296 1.304 !.099 1.094 1.095 1.089 1.099 1.094 1.095 1.089 122.1 122.3 122.4 122.2 115.9 115.3 i15.2 !15.5 9.89064 9.63495 9.78676 9.78403 9.86067 1.03016 1.01095 1.03022 1.04743 1.03942 0.93298 0.91495 0.93210 0.94614 0.94030 3485 3426 3432 3416 3426 3160 3149 3119 3075 3122 3103 3080 3058 3025 3060 1657 1611 1628 1661 1626 1466 1431 1432 1419 1435 989 968 963 951 976 932 895 926 932 930 619 592 657 713 644831i 806i 770i 744i 780i
- 7 7 . 6 7 3 2 6 - 7 7 . 7 0 3 0 2 - 7 7 . 7 3 2 6 3 -77.76581 -77.75625
22.030 21.663 21.750 21.717 21.756
a ACES II was used in the calculation. Bond lengths in ,~,, bond angles and diheral angles in deg, rotational constants in c m - ~, vibrational frequencies in c m - ', energy in au and zero-point energy in kcal/mol.
b Ref. [8]. c Ref. [ 13].
a The atomic labeling is shown in Fig. 3 except that the H t is along the line with C t = C 2.
Table 8
The calculated barrier height for the rocking motion of ot-H in C2H 3 X2A'
Method Exp. a GVB b MP4 c CCSD(T)
Basis 6-31G* * 6-31G* * 6-31G* *
classical (au)
transition state (au)
energy difference d ( c m - ~) < i 500 31 ! 3 - 7 7 . 6 8 3 2 3 - 7 7 . 6 7 3 2 6 2168 2188 (1539) (1683) CCSD(T) CCSD(T) CCSD(T) CCSD(T) DZP TZP C: PVTZ, H : DZP TZ2P - 77.71302 - 77.74103 - 77.77343 - 77.76493 - 77.70302 - 77.73263 - 77.76581 - 77.75625 2195 1844 1672 1905 (2029) (1396) (1241) (I 477) Ref. [ 14]. b Ref. [13]. c Ref. [8].
J.-H. Wang et al. / Chemical Physics 206 (1996) 4 3 - 5 6
Table 9
The optimized geometry and energy difference of the isomeric C 2 H 2 a
53
Acetylene (H 2-C2 ~ C I - H t)
exp. b calc.
Ref. [26] c this work a
.Hl.~ Vinylidene (H 2 " C 2 = C ! )
exp. e calc.
Ref. [26] c this work a
r c i c 2 rc zn t rc, FI j 1.0622 rc 2n 2 1.0622 / - C I C 2 H ~ / H I C 2 H 2 / - C 2 C I H I /--CIC2H 2 energy (au) energy difference ( k c a i / m o l )
a Bond lengths in ,~,, bond angles in deg. b From Ref. [25].
c C C S D / T Z 2 P was used in the calculation. a C C S D ( T ) / T Z 2 P is used in the calculation. e From Ref. [27]. 1.2026 !.2017 1.2081 !.0616 1.0634 1.0616 1.0634 180 180 180 180 - 7 7 . 1 8 5 3 7 - 7 7 . 2 0 0 6 5 0 0
MBPT and CCSD(T), we have restricted the non- classical C2H3 to be of C2 symmetry with five internal co-ordinates in the optimization. The opti- mized structures from various methods and basis sets are tabulated in Table 5, in which the computed fundamental vibrational frequencies of the non- classical C2H 3 are also listed. In the CCSD(T) calculations for the vibrations of the non-classical C2H3, an imaginary vibrational frequency was al- ways found, thus indicating the non-classical struc- ture as an unstable isomer corresponding to a transi- tion state on the molecular potential energy hyper- surface. Comparisons for the calculated energy dif- ference between the non-classical and the classical isomers of C2H 3 are listed in Table 6.
2.2.2. a-H rocking motion
In a recent observation of the v 7 vibrational band in C2H 3 X 2A~, Kanamori et al. [14] have reported that the a-hydrogen (H~ in Fig. 3) of C2H 3 is rocking back and forth around the two equivalent sp 2 positions of the C~ atom, corresponding to a tun- nelling motion in a double-minimum potential. In this study, we have tried to accurately calculate the barrier of the rocking motion by using the ACES II program. We have optimized the planar transition state (2 in Fig. 1) and calculated its fundamental vibrational frequencies. The results are listed in Table
1.3008 1.3050 ! .08 ! 8 1.0840 1.0818 1.0840 120.13 120.22 119.74 ! 19.56 - 77. I 1765 - 77.13087 46.4 __+ 5.5 42.39 43.76
7 with the similar atomic labelling as in Fig. 3 except that H~ is along C ~ = C 2. The energy comparison between the transition state and the classical struc- ture of C2H 3 (1 in Fig. 1), which is located in the global minimum of the potential energy hypersur- face, is shown in Table 8.
2.3. Dissociation of C 2 Hj X 2A'
In calculating the dissociation of C2 H 3 ~ 2K, the decomposition channels to acetylene and to vinyli- dene have been considered. The CCSD(T) method and TZ2P basis set were used in the calculation with the ACES II program. The dissociation energies for C2H 3 --* HCCH + H and C2H 3 ~ H2CC + H are calculated to be 40.70 and 84.46 kcal/mol, respec- tively. The optimized geometries of acetylene and vinylidene and the energy difference between these two isomers are listed in Table 9.
3. Discussion
3.1. Molecular structure
3.1.1. Equilibrium geometry and vibrations of C 2 H 3 )(2 a'
Comparing the experimental values, the optimized geometry for the classical structure of C2 H 3 :~ 2~
54 J.-H. Wang et al. / Chemiccd Physics 206 (1996) 43-56
(in Table 1) has shown a better accuracy for the CCSD(T) method than for MP2 and MP4. The use of 6-31G * * in the CCSD(T) calculation seems to have a fairly good result in this case. While the structure calculated from Dunning's DZP basis set turned out to be not quite satisfactory, the triple zeta polariza- tions have much improved the results, especially for /C2CIH~. The calculated rotational constants (A, B, and C) using PVTZ/DZP or TZ2P in CCSD(T) are very close to the experimental values. Neverthe- less, one should note that the observed geometry is for the vibrational ground state and may differ slightly from the calculated equilibrium structure. Moreover, the observed rotational constants are responsible for a geometry of C2v effective symmetry in C2H 3, while the calculated one is of C s. The slightly larger rotational constant A in the observation, in compari- son with the calculated one, is in line with reasoning
the linearity along H IC iC2 due to c~-H tunnelling. The fundamental vibrational frequencies of the classical C2H 3 X 2~ obtained from the calculation are also tabulated in Table 1. As mentioned before, u 7 has the strongest infrared intensity, and is the only vibrational mode observed so far. In view of this ~,~ vibration, poor vibrational frequencies re- sulted from the calculations at levels of SCF and MP. The CCSD(T) calculations, on the other hand, have given quite satisfactory results for the ~'7 vibra- tional frequency, indicative of the substantial im- provement for the potential energy hypersufface due to electron correlation.
3.1.2. Electronic energies, ionization potential and dipole moment
In comparison with the observed vertical transi- tion energies of C2H 3 ,g, 2~, ~ ~ 2~ (24815 cm-1) and of the ionization potential (8.25 eV), the calcu- lated values (in Table 3) have gradually approached the experimental results from the uses of RASSCF to MRCI. The calculated 25529 c m - l for ,~, 2g, ,.. ~ 2?¢ and 8.33 eV for the ionization potential from the MRCI method are in excellent agreement with the observations. The small deviations between the cal- culated and the experimental values, i.e. only 714 cm-i for ,~ 2g, ~ ~ 2 g and 80 meV for the ioniza- tion potential, have demonstrated a very good quality in the present computation. The ve~iical transition energy of 43910 cm -I (,~ 228 nm) for B 2~
2~, obtained from the MRCI calculation, will be very helpful for us to experimentally search for the undiscovered B 2~ state of C2H 3 by laser spec- troscopy. The transition moment of B 2~ ~._ ~ 2A ~
should be substantial for spectroscopic observation due to its "rr * ~ ~ character. The bond orders be- tween the C - C of C2H3 for the ~:, ,~ and § elec- tronic states are 2, 1~ and 1, respectively. The bond-order calculation is indeed reconciled with the long vibrational progression, due to the C - C stretch mode, observed in C2H 3 ~. 2A~' ~--X 2~ [7]. The permanent dipole moment in C EH 3 ~ 2"A~ obtained from this work is also in good agreement with the previous generalized valence bond wavefunction cal- culation [ 13].
3.2. Intramolecular rearrangement
3.2.1. Isomerization
The optimized geometry, rotational constants and vibrational frequencies of the non-classical C2H 3
2A~ are listed in Table 5. The shortening of the C - C bond-length in the non-classical C2H 3 X 2~, in comparison with that of the classical structure, is caused by the bridging hydrogen, H 1, the presence of which results in a doubly occupied orbital which resembles a C - C "tr-bonding orbital. In all of the CCSD(T) calculations, the imaginary vibrational fre- quency around 2100 cm-~ has indicated the very unstable nature of the non-classical isomeric struc- ture. In Table 6, the energy difference between the classical and non-classical structures of C2H 3 is represented. Including zero-point energy, the non- classical isomer has been found to lie at least 47 kcal/mol above the classical, and energetically far above the dissociation threshold (40.70 kcal/mol) of C2H 3 ~ H C C H + H . The large energy difference between the two isomers and the unstable nature of the non-classical structure have made the isomeriza- tion process in CEH 3 quite a challenge for spectro- scopic observation.
3.2.2. a-H rocking motion
In the determination of the barrier height for a-H tunnelling in C2H 3 X 2~, we have optimized the transition state (2 in Fig. l) as listed in Table 7. The vibrational frequencies associated with the transition state have accordingly shown an imaginary value
J.-H. Wang et al. / Chemical Physics 206 (1996) 43-56 55
responsible for this saddle-point on the potential energy hypersurface. The calculated barrier height for the rocking motion of a-H in C2H 3 ]~ 2~ is tabulated in Table 8. The barriers with zero-point energy correction, computed from CCSD(T)/Dun- ning's triple zeta polarizations, are in excellent agreement with the upper bound limit of < 1500 cm-~ determined by high-resolution infrared spec- troscopy [ 14].
3.3. Dissociation o f C 2 H 3 3~ 2A'
In comparison with the experimental values of 80.0 _ 5.0 kcal/mol for the dissociation of C 2 H 3 --~
nECC -I- H reported by Ervin et al. [27], tb_e calcu-
lated 84.46 kcal/mol is in agreement with the mea- surement. The calculated energy difference of 43.76 kcal/mol between the isomers of acetylene and vinylidene is also consistent with the spectroscopic determination of 46.4 _ 5.5 kcal/mol [27].
4. Conclusion
We have carded out a systematic study of the molecular structure (including equilibrium geometry, vibrational frequencies and intensities, electronic en- ergies, ionization potential and dipole moment), the intramolecular rearrangement (isomerization and a-H rocking motion) and the dissociation of C2H 3 with high-level ab initio calculations using ACES II and MOLCAS-2 programs. In view of the calculated and the experimental values, such as the equilibrium geometry of C EH 3 ~: 2A~, the transition energy of ,A 2Ay ~ X 2A~, the ionization potential, the barrier height for the a-H rocking motion and the C - H dissociation energy, the computational accuracy in the present study has demonstrated to be of very good quality.
The vertical electronic transition B 2~ ~_ ,~ 2~ of C2 H 3 and the energy difference between the classi- cal and non-classical isomers in C2H 3 X 2~, re- sulted from the calculations in this work, have pro- vided valuable information which will facilitate the forthcoming experimental studies in our laboratory. The calculated vibrational frequencies and infrared intensities of C 2 H 3 X 2,A~ will assist the spectro- scopic measurement for the unobserved vibrational
modes. The knowledge from the calculated isomer- ization and dissociation processes in C2Iq 3 X 2 ~
will help us in the experimental investigations to be carried out with high-resolution IR-UV laser-induced grating spectroscopy.
Acknowledgement
We thank Professor S.H. Lin and Dr. J.-W. Yu for their comments on this manuscript. Mr. Chung- Jen Wu is gratefully acknowledged for his computer drawing of Fig. 4. This work is supported, in part, by the National Science Council of Republic of China under grant No. NSC-84-2113-M-001-037 CT.
References
[1] H. Okabe, Photochemistry of small molecules (Wiley, New York, 1978).
[2] W.C. Gardiner, Jr., ed., Combustion chemistry (Springer, New York, 1984).
[3] I.K. Purl, ed., Environmental implications of combustion processes (CRC Press, Boca Raton, 1993).
[4] A. Fahr, A. Laufer, R. Klein and W. Braun, J. Phys. Chem. 95 (1991) 3218.
[5] DJ. Donaldson, I.V. Okuda and J.J. SIoan, Chem. Phys. 193 (1995) 37.
[6] O. Simamura, in: Topics in stereochemistry, Vol. 4, eds., E.L. Eliel and N.L. Allinger (Wiley, New York, 1969) p. 1. [7] H.E. Hunziker, H. Kneppe, A.D. McLean, P. Siegbatm and
H.R. Wendt, Can. J. Chem. 61 (1983) 993.
[8] M.N. Paddon-Row aad J.A. Pople, J. Phys. Chem. 89 (1985) 2768.
[9] L.A. Curtiss and J.A. Pople, J. Chem. Phys. 88 (1988) 7405. [10] F.P. Lossing, Can. J. Chem. 49 (1971) 357.
[1 i] J. Berkowiz, C.A. Mayhew and B. Ruscic, J. Chem. Phys. 88 (1988) 7396.
[12] J.A. Blush and P. Chert, J. Phys. Chem. 96 (1992) 4138. [13] M. Dupuis and J.J. Wendoloski, J. Chem. Phys. 80 (1984)
5696.
[14] H. Kanamori, Y. Endo and E. Hirota, J. Chem. Phys. 92
(1990) 197.
[15] R.A. Shepherd, T.J. Doyle and W.R.M. Graham, J. Chem. Phys. 89 (1988) 2738.
[16~ M.W. Crofton, M.F. Jagod, B.D. Rehfuss and T. Oka, J. Chem. Phys. 91 (1989) 5139, and references therein. [17] C. Liang, T.P. Hamilton and H.F. Schaefer Ill, J. Chem.
Phys. 92 (1990) 3653, and references therein. [18] L.B. Harding, J. Am. Chem. Soc. 103 (1981) 7469. [19] I.M.B. Nielsen, C.L. Janssen, N.A. Burton and H.F. Schaefer
56 J.-H. Wang et ai./ Chemical Physics 206 (1996) 43-56
[20] C.E. Hamilton, J.L. Kinsey and R.W. Field, Annu. Rev. Phys. Chem. 37 (1986) 493.
[21] M.A. Buntine, D.W. Chandler and C.C. Hayden, J. Chem. Phys. 97 (1992) 707.
[22] ACES II is authored by J.F. Stanton, J. Gauss, J.D. Watts, W.J. Lauderdale and R.J. Bartlett., University of Florida, USA.
[23] MOLCAS is authored by K. Andersson, M.P. F'filscher, G.
Karlstrom, R. Lindh, P.-A. Malmqvist, J. Olsen B.O. Roos and A.J. Sadlej, University of Lund, Sweden.
[24] J. AlmlSf and P.R. Taylor, J. Chem. Phys. 86 (1987) 4070. [25] G. Strey and I.M. Mills, J. Mol. Spectry. 59 (1976) 103. [26] J.F. Stanton, C.-M. Huang and P.G. Szalay, J. Chem. Phys.
101 (1994) 356.
[27] K.M. Ervin, J. Ho and W.C. Lineberger, J. Chem. Phys. 91 (1989) 5974.