國
立
交
通
大
學
資訊科學與工程研究所
碩
士
論
文
以 雙 分 群 方 法 分 析 基 因 微 矩 陣 資 料
Using biclustering algorithms to analyze microarray expression
data
研 究 生:陳貫中
指導教授:胡毓志 教授
國 立 交 通 大 學
資 訊 科 學 與 工 程 研 究 所
碩 士 論 文
A Thesis
Submitted to Institute of Computer Science and Engineering
College of Computer Science
National Chiao Tung University
in partial Fulfillment of the Requirements
for the Degree of
Master
in
Computer Science
June 2006
Hsinchu, Taiwan, Republic of China
7
以雙分群方法分析基因微矩陣資料
研究生:陳貫中 指導教授:胡毓志博士
國立交通大學資訊科學與工程研究所
摘要
從基因表現資料找出有意義的基因群組,長久以來都是分析微矩陣資料的一 個重要課題。由於傳統演算法在先天上的限制,許多雙分群演算法被發展出來, 用以解決此問題,並有著不同的目標和策略。我們基於分析頻繁項目集的架構下, 在此提出一個雙分群法。和以往較為不同的是,我們把微矩陣資料的雙分群問題, 轉換為挖掘頻繁項目集的問題。為了驗證我們演算法可行,我們首先和代表性的 數個傳統分群法進行比較 ,而實驗結果顯示我們的方法穩定度和精確度比傳統 方法好。接著,我們也和近年來的幾個雙分群系統 ,在已知且公開的資料下,進行 一連串比較。最後,將顯示我們演算法在多個測試項目下,確實超越近年來的知名 雙分群法。Using biclustering algorithms to analyze microarray expression
data
Student: Kuan-chung Chen Advisor:Dr.Yuh-Jyh Hu
Institute of Computer Science and Engineering College of Computer Science
National Chiao Tung University Hsinchu, Taiwan, Republic of China
Abstract
Finding meaningful clusters of gene expression data has always been one of the most important topics of microarray data analysis. Due to the limitations of conventional clustering algorithms, numerous biclustering methods, with different aims and strategies, have been developed to mitigate the problems. We propose a new biclustering algorithm under the framework of market basket analysis focused on frequent itemset analysis. Unlike previous works, we transform the biclustering problem into a frequent itemset finding task where significant biclusters are described as frequent itemsets. To verify its feasibility, we first compared it with several representative conventional clustering algorithms. The experiments show very promising results. We also conducted a comparative study of current biclustering systems based on the widely-used prior knowledge, Gene Ontology. The study demonstrates that our method
9
致謝
首先 ,在此感謝我的指導教授-胡毓志老師在這兩年來對我的指導與教誨。 老師平常以自由、開放的方式 ,讓我們在甚少壓力的環境下 ,主動去獲得新知。 平時 ,老師更花許多寶貴時間 ,和 LAB 每個學生 ,一對一的個別 meeting ,引領 我們研究方向 ,指導我們做研究的方法和態度 ,並給我們慣輸正確的觀念-不論 是在課業上還是為人處事上 ,這都有極大的助益。最後甚至花了老師許多寶貴時 間,在論文校稿和叮嚀上,在此相當感謝。 在這實驗室兩年, 感謝子緯學長、鈞木學長、世彥學長幫助我對於生物領域 的認識,大幅縮短了我入門這個領域的時間。也感謝音璇學姊、勁伍學長給予我 在作研究上面的建議。 當然 ,在程式的實作過程 ,最需要感謝程式經驗豐富的 昀君學長、繼養學弟 ,幫助我在程式實作上和除錯時的建議 ,讓我頭痛的程度得 以減緩。 最後 ,感謝家人們給予我在精神上的支持和幫助 ,讓我在困惑、迷惘之時 , 能獲得鼓勵和安慰,度過每個難關。 在此感謝大家目錄
摘要...7 Abstract...8 致謝...9 目錄...10 第一章--序論...12 1.1 問題源起...12 1.2 論文架構...12 第二章--文獻探討...13 2.1 中心法則...13 2.2 調控網路...13 2.2 調控網路...14 2.3 雙分群...15 2.4 FP-Tree 演算法...18 第三章--研究方法...20 3.1 實驗假設...20 3.2 實驗測試資料...22 3.2.1 資料格式...23 3.3 實驗前處理...25 3.3.1 正規化...25 3.3.2 正規化測試分佈圖...26 3.3.3 正規化結論...29 3.3.4 離散化...30 3.3.5 離散化結論...313.4 PIFP (Progress Iterative Frequent Pattern-tree)演算法...32
3.4.1 PIFP 系統流程圖...35 3.4.2 PIFP 輸出結果...37 3.4.3 PIFP 特色之處...39 第四章--結果與討論...40 4.1 評分準則...40 4.1.1 評分公式:超幾何分佈...41
11 4.4 結果討論 - Clustering Programs...55 4.5 測試程式 –Biclustering Programs...56 4.5.1 雙分群參數設定...57 4.6 測試結果 - Biclustering Programs...58 4.7 結果討論 - Biclustering Programs...63 4.8 FP vs PIFP...64 第五章--結論與展望...65 5.1 結論...65 5.2 未來展望...66 參考文獻 (依照字母順序排列)...67
第一章--序論
1.1 問題源起 為了瞭解複雜的生物系統和眾多基因間彼此的作用關係,學者從微矩陣 (Microarray)中取得實驗數據,進而產生各種基因表現(Gene Expression)的資 料庫 ,目標希望能夠建構出完整的基因網路,方便研究彼此之間的交互關係。 但因為資料過於龐大且複雜,要找出基因之間彼此的關係並不容易;其複雜度已 知為 NP-Hard 問題。在此,我們將採用分群演算法(Clustering Algorithm) 和資 料探勘(Data Mining)的方式,分割出有高度相關的調控模組(Transcription Module)。所謂的調控模組包含正相關(activate,synexpress)和負相關 (suppress ,negative)的調控關係 , 而找出真正的調控模組是我們演算法想找 出的主要目標 。最後希望能夠重建調控網路,提供正確的資訊給生物學家作研 究。 1.2 論文架構 首先將在第二章介紹我們的相關的名詞定義、理論來源,和目前學者們所提 出的一些相關研究方法 ; 以及在本篇論文中,我們所提出方法中的貢獻和可取 之處。接著在第三章將進行生物資料的前處理分析,包括數種正規化 (Normalization) 和離散化(Discretization)的前處理方式 ,並且顯示處理後 的資料分佈 ;第四章將我們會進行 PIFP 演算法的流程圖和實際操作情況, 並與 近幾年學者們所提出的數種分群法(Clustering Algorithm)和雙分群法 (Biclustering Algorithm),在相同資料與評分公式的條件下,進行完整且公平13
第二章--文獻探討
為了瞭解後續所提及的方法和研究假設 ,必須要先有一些背景和相關知 識。底下的章節將會介紹一些我們引用的相關理論和演算法。已經有基礎者可以 直接跳過。2.1 中心法則
細胞內各種活動的運作都需要蛋白質參與,其製造藍圖則記載於遺傳物質中 (DNA 或 RNA)。一個生命體中的所有遺傳物質稱為基因組(genome),其中每一段 可以合成一個具有功能的蛋白質或 RNA 分子之 DNA 序列則稱為基因(gene)。基因 可 以 分 為 轉 錄 區 (transcription region) 和 轉 錄 調 控 區 (transcription regulatory region),轉錄調控區是轉錄因子與 DNA 作用的區域。每一個轉錄因 子會與特定的 DNA 序列作用,此序列則稱為轉錄因子結合區。基因的轉錄,必須 靠轉錄因子的作用而啟動、增強、或抑制。當 DNA 被轉錄成 mRNA 之後,經過若 干步驟才會被轉譯成蛋白質。由 DNA 轉錄成 mRNA,再轉譯成蛋白質的步驟稱為 中心法則(central dogma),此為地球上大部份生命運作所遵從的法則。(徐英哲 2003) Central Dogma DNA RNA 細胞核 RNA Protein 轉錄 transcription 轉譯 translation 逆轉錄 reverse transcription2.2 調控網路
我們透過基因晶片(gene chip)或微矩陣(microarray)所產生的大量資料, 幫助我們一次觀察基因們數百個實驗狀態(condition)下的表現值。 並藉由各種 分析方法,希望找出基因們之間的調控和互動關係, 並建立所謂的調控網路。例 如:布林網路(boolean network , Shmulevich et al. 2002) 、機率網路 (probabilistic network)、分群法…等。有了調控網路之後,可以方便我們觀察 基因們彼此的關連性和影響。 下圖為基因網路的大致架構: 圖 1 :代表正向調控 , 例如 GET140、NODE1、HMG1、MEP3…等彼此在相同的一 些時間點上 , 有正向調控關係。 : 代表延遲調控, 例如 EX:ENO1、YKL177W 彼此之間有正向調控關係 ,但 不在同一時間點上; 此即所謂的『時間延遲』(Time Delay)。
15
2.3 雙分群
眾多的學者,根據中心法則提出假設:一群參與生化反應的基因 ,份扮演正 相調控(activate)的基因群應該要有
相似
的基因表現(gene expression);另一 部份扮演抑制(suppress)的基因群 ,其基因表現程度應該與正向調控的基因群組的基因表現
相反
。這種把基因們歸類成群組的演算法,我們稱之為基因的分群法 (cluster algorithm) (Dembele D et al.2003 ; Getz G et al.2000)
然而,透過生物學家的驗證 ,我們發現事實上一個基因可以同時參與多種生 化反應 ,也許甚至可以同時扮演抑制和調控的角色。 也就是說 ,若使用以往傳 統的分群方式將會有下面兩個缺點: 1. 一個基因只能出現在一個基因群組(gene cluster),且只能扮演正向或反向 的調控角色 ,無法正確詮釋真實的生化反應。 2. 分群法大多一次考慮全部的實驗狀態 ,這也是很大的一個缺點。因為,對一 個基因來說,並不是每個階段的數據都有意義,有些勢必要忽略 ; 部分參與 正向調控和部分負向調控的實驗數據 ,並無關連 ,應該要彼此獨立!。 根據上述兩個缺點,使用分群法(cluster algorithm)分析基因資料會產生極大 誤差。因此也才有雙分群的想法提出, 作為改良方式 ,彌補傳統方法的缺失和不 足之處。
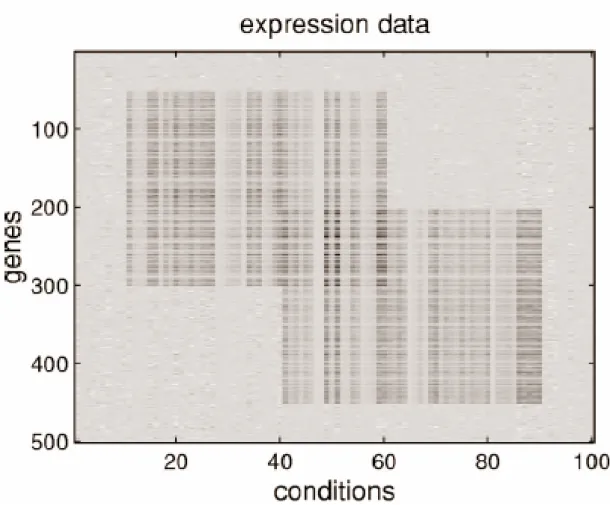
圖 2 為雙分群之後,基因群組(Gene cluster)的示意圖。
傳統方法的缺點 ,正是近幾年來雙分群方法(Bicluster algorithm)的改良 之處 (Creighton C et al.2003; Sheng Q et al. 2003 ;Sara C. et al. 2004 ; Kloster M, 2005)。下圖為雙分群的示意圖:
所謂的基因群組就是由基因(Genes)和實驗狀態(Conditions)所組成的群組 ,經 由演算法切割而成 ; 也可以稱之為調控模組(Transcription module ) (Bergmann S et al.2003)。藉由雙分群法所分割出的基因群組 ,具有重疊 (overlap)的特性,並允許不連續的實驗狀態參與反應。
17
雙分群特色在於:
1. 可以忽略某些實驗數據 , 避免無意義的資料參與分群 。 根據我們統 計,6325*300 的酵母菌微矩陣資料中 ,有多達『42594』筆在微矩陣中檢測 不到基因表現而呈現空白(missing values)! 使用雙分群法 ,在此即可忽 略而不影響分群結果。但若為傳統的分群法 ,則很容易受到這些數值影響, 產生誤差。 2. 允許基因群組之間 ,有重疊(overlap)的關係。換句話說 ,就是允許每個基 因可以出現在多個基因群組 ,並產生調控反應。由圖 2 可明顯看出此現 象。這樣一來比較符合生物上的實際情況也符合我們所提出的假設。 3. 只考慮部分的實驗數據 ,就可以決定分群。如圖 2 所示 ,圖中的兩個基因 群組, 都只包含部分的實驗數據 , 而且可以不用連續 ;彈性比以往的分群 演算法大很多 ,能夠挖掘出更多的基因群組。2.4 FP-Tree 演算法
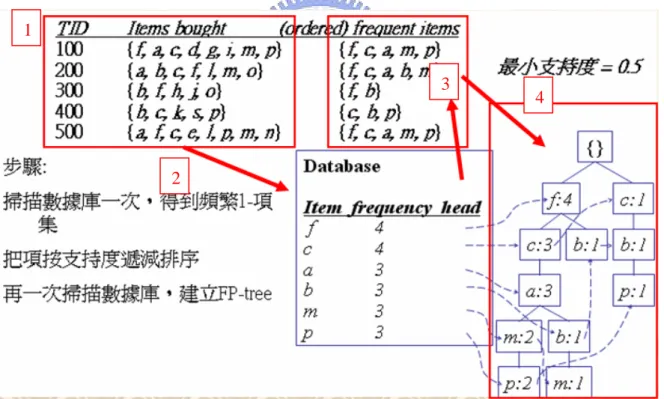
為了能夠連貫後面第三、第四章的內容,在此先介紹 Frequent–Pattern tree Algorithm(簡稱 FP-tree) (J.Han, J.Pei ,and Y.Yin 2000) ,此法是用 來幫助使用者找出資料中,彼此之間的關連性,而不需產生候選探勘頻繁項目集 合(mining frequent itemsets without candidate generation)。根據研究數 據顯示,該運算速度比 Apriori Algorithm (Creighton C, Hanash S 2003)快 很多 ,而且不用產生一大堆的資料。 相對來說,掃瞄資料的次數也少很多,只要 兩次即可,和 Apriori Algorithm 的暴力掃瞄方式不同。 底下為 FP-Tree 的運 作步驟和說明: 2 3 4 圖 3 FP-Tree 演算法的步驟 1
19 開始說明圖 3 的運算步驟: Step 1: 假設目前有五筆交易 , 內容為使用者購買的產品 。現在希望透過 FP-Tree 找出其商品之間的關連性。 Step 2: 開始累計出現的商品個數 ,並存入資料庫 中 ,並按照順序排列。出現 次數少於門檻值者于以刪除。如圖所示, item f 在五筆交易中出現四 次,次數最多所以排最前面 ; 而 item k、s、e、g…等出現次數少於 門檻值 3 者 ,則在此刪除。通過此階段的 items ,稱之為頻繁項目 (Frequent item)。其餘以此類推。 Step 3: 根據上一步找出的頻繁項目 , 來對原本的交易項目作排序。如圖所 示:順序為 f、c、a、b、m、p。而原本的第一筆交易,則改另存為:f、 c、a、m、p ;原本尚有其他的項目,因為通過 Step1 所設定的門檻值 3 ,所以不保留下來。其餘以此類推。
Step 4: 開始建樹 ,樹根(root)初始化設定為 NULL。依照 Step2 所過濾過的交 易資料 ,依序填入樹中。若遇到相同的項目資料, 則該節點(node)的 記次數目+1 ; 若遇到不同的項目資料 ,則另外產生新的子孫 (childern)。 Step 5: 有了 FP-Tree 之後 ,我們可以根據自己設定的門檻值, 找出所謂的共 同頻繁項目。例如:我們希望找出次數超過 3 次 ,且至少 3 個一起出 現的交易。則根據圖形顯示,答案為 f、c、a 這三個項目。此時我們 可以說,f、c、a 在這總共五筆的交易中 ,有很高的出現關連性。
第三章--研究方法
3.1 實驗假設
我們以 中心法則 為理論基礎 ,在此認為擁有相似基因表現的基因群,會一
起參與生化反應; 而共同參與反應的基因群組, 基因彼此之間可能存在正相關 (activate,synexpress)和負相關(suppress ,negative) 的調控關係(Sheng Q et al .2003 ; Sara C et al .2004 ; Creighton C et al .2003 ; Kloster M et al .2004)。 在不失一般性的情況下,我們所持的假設如下: 1. 會存在調控模組內的基因 ,應該是強烈表現(significant-expressed) 的基 因 , 而非無表現(none–expressed) 的基因。 所以會優先過濾不好的基因 表現資料,避免干擾, 影響準確度。 2. 在同一個調控模組內的基因 , 才是有可能實際參與生化反應 , 並互相調控 的基因! 即要在同一個基因群組內才算有效。我們將依照 正相關
(activate,synexpress)和負相關(suppress ,negative) relation ,來實作 系統。(Qian J, et .al 2001)
3. 一個實驗資料通常多達數百個實驗狀態(Time Points ,Conditions) ,在調控 模組中真正互相參與反應的基因 ,不一定會有連續相似的基因表現資料 (Expression Data)。所以在此依照雙分群(Bicluster)的基本想法假設 ,允 許不連續的實驗狀態參與反應 , 即允許時間間格(Gap)的加入。(Qian J, et .al 2001)
21 4. 實際參與反應的調控模組,所包含的基因個數都相當地多。 所以我們預期的 調控模組 , 應該要包含多個基因(Genes ,簡稱 G) 和多個實驗狀態 (Conditions 簡稱 C) 。換句話說 ,調控模組中 G * C 的值應該要越大越好 , 比較符合現實情況。G , C 參數值在程式中 ,雖可由使用者自行設定 ,但我 們給予的預設值為: G=10~25 , C=10~20 。 5. 由於目前缺乏連續的實驗數據 ,目前暫時不考慮考慮時間延誤(Time Delay) 的問題。包括:正相關延誤(Delay-Synexpress) ,負相關延誤(Delay-Invert) 等可能情況。 而文獻中也指出 ,目前延誤(Delay)和負相關(Invert)基因群 組所佔整體群組的比率很少 ,絕大多數依然為正相關 (Qian J,et al .2001)。
3.2 實驗測試資料
根據文獻所得,我們蒐集酵母菌的基因表現資料,其內容包含 6335 個基因和 121個實驗狀態 (參考錯誤! 找不到參照來源。) 。蒐集部分經驗法則和參數設 定之後,我們將經驗法則套用在較龐大的資料庫 (6325基因*300實驗狀態, Hughes TR et al.2000) 並用來測試章節4.2所包含的實驗 ,以及章節4.4所用的 BiCAT 預設酵母菌資料庫 (2997基因*173實驗狀態, Prelic A.et .al 2006) 。 以上均為多數學者所使用的實驗數據來源 ,我們將在下個章節展示我們測試前 處理的成果。
表 1 研究用的基因表現來源
Reference Dataset Description Type Number
DeRisi et al. 1997 Diauxic shift,
repressor TUP deletion, activator YAP1 overexpression
Time series, cDNA microarray
9
Eisen et al. 1998; Lashkari et al. 1997
Heat shock, DTT shock, cold shock
Time series, cDNA microarray
14
Chu et al. 1998 Sporulation,
sporulation ndt80 knockout
Time series, cDNA microarray
9
Holstege et al. 1998 Transcription factor mutant, SAGA chromatin modification complex mutant Multiple experiments, oligonucleotide chip 11
Spellman et al. 1998 Cell cycle α-facotr arrest, cell cycle elutriation, cdc15 arrest
Time series, cDNA microarray
60
Cho et al. 1998 cdc28 arrest Time series,
oligonucleotide chip
23
3.2.1 資料格式
一般而言 ,系統大多實作 .CDT 的資料格式。在此介紹.CDT file format , 範例如下(http://rana.lbl.gov/FuzzyK/cdt.html): 主要規格如下: 1. 第一行必須包含獨特的基因名稱, 標頭為『UID』; 主要用來說明基因名稱。 2. 第二行標頭為『NAME』, 包含某些程式想要顯示給使用者的註解 ,用來說明 該基因的特性或生物上的意義 ; 此行不一定要有 ,看程式需求。 3. 第三行標頭『GWEIGHT』, 包含該基因的權重。此行在某些程式會用到。 4. 其他行的標頭非上述者 , 即為該實驗的測試數據。 5. 在 .CDT 檔案格式中 , 不可以有空白的『行』。避免格式錯誤 圖 4 CDT 的實際格式。文字或數字之間, 彼此以 TAB 當作間隔 。按照此格式標準 ,為了之後的比對字串方便實作 ,我們將資料載入到 MySQL 資 料庫中,並加入了『Key』這個行(column),可以方便索引 ,資料格式如下:
行的內容依序為: KEY ,ORF ,Data …。因不需要 Weight 的加權 , 所以該行省 略 ; 也因為有另外的評分公式系統 , 所以在此不需要生物註解(Annotation)。 圖表 5 PIFP 系統中 , MySQL 內儲存的資料格式 。
25
3.3 實驗前處理
在此要介紹我們實驗的前處理實作方式包括 正規化(Normalization)和離 散化(Discretization) 。 此處主要根據學者們對於前處理各種方法的分析(Qiu X et al. 2005), 及論文著作中再三強調前處理的重要性,所以此步驟不能省 略。底下將會介紹我們所實作的各種方法; 根據數據顯示 ,所有的前處理動作 將會影響整個實驗的輸出結果和準確度!3.3.1 正規化
拿到基因的表現資料之後 , 一般來說,肯定要做一些前處理的動作 ,過濾 初步的雜訊! 底下會介紹相關論文中, 較常出現的幾種實作方法(Qiu X et al. 2005)。1. Geometric mean normalization (通稱 GEO): 將資料除以其 Row or Column 的幾何平均來當作前處理步驟 ; 此實作方式會影響資料最大 。
2. Rank normalization (通稱 RANK) :以每個單一數據(cell data)為單位 , 將所有數據依大小排序後 , 將原本的數值以排序後的名次取代 。
3. Z-Score (Spellman): 分別對『行』和『列』套用運算 ,使得平均為 0 (zero mean) , 變異數為 1(unit variance)
3.3.2 正規化測試分佈圖
正規化分佈圖表: 使用酵母菌微矩陣資料6325(orf) * 300(con) (Hughes TR et al.2000), 透過3.3.1的各種方法, 分析後的結果。
製作方式:跑完各種正規劃方法之後 ,依照相同間距平均切為20份,有效間格距 離為『1~20』 , 群組『0』代表的是在微矩陣實驗中,檢測不到數據的資料個數, 在此稱為 Null data。 『None』 代表未做任何步驟的原始資料。
間隔『0』代表的是在微矩陣實驗中的 Null data, 在此已經先做過濾動作 ; 間 圖 6 正規劃處理後 ,資料分佈。
27
在此 , 我們將 圖 6 的內容畫成各種正規化測試方法的長條圖,方便觀察:
由圖 8 可看出相對於左上角未經過處理的原始資料 ,各種方法對於基因表現資 料的影響。
在此 , 我們將 圖 6 的內容畫成圖形方便瀏覽。 底下為各種正規化測試方法的 整體比較折線圖 : 以上的分佈圖 ,只為顯示資料分佈曲線彼此間的差異, 並無所謂的方法好或 壞。只有適合與不適合目前要處理的資料。下個章節將會對以上的方法做出結論 和整理。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
累
計
數量
None Rank GEO Spellman Rank + Spellman GEO + Rank 圖 8 經過正規化程序之後 , 資料分佈曲線。29
3.3.3 正規化結論
1. 正規化之後, 影響原始資料(Raw data) 彼此的關係非常大 , 肯定會降低 原本基因們之間的關係! 2. 正規化可以消除一些來自生化實驗上雜訊(Noise) ,但真正關連性強的調控 關係, 應該不會造成嚴重破壞。所以這樣做並無不妥。 3. 根據文獻顯示, 最有效率的正規化方法也無法完全消除雜訊 ; 所以這也是 我們想盡力去改善的部分。 也許以後可以套用 2 個上述的方法一起當作前 處理,或許會有改善的空間。 4. 經過測試之後 ,目前我們追尋學者們大多數使用的方法,採用『Z-Score』的 正規化方式 ,當作系統實作的主要方法。 5. 正規化的各種方法 ,都有其存在的必要。彼此之間無法直接比較好或壞 , 每個方法都有適合使用之處 !3.3.4 離散化
我們將雙分群的搜尋問題,轉換為頻繁字串(Frequent Pattern)的搜尋問 題。其中,將基因表現行為程度轉換為一連串的字母,企圖在眾多字串中找尋較頻 繁字串的集合,以反應實際上雙分群所代表的意義。為了能夠方便底下的步驟--找出頻繁字串, 先做離散化(Discretize)的步驟是必須且重要的。在此介紹目前 已知的幾個方法及我們系統中採用的實作方式 (Becquet et al. 2002 , Creighton et al. 2003 ): 1. 取 log X >0.2 為正向基因表現(+1) , 取 log X <-0.2 為負向基因表現 (-1) , 其餘視為無基因表現(non-expressed) (0) ; 學者將此法套用在 Appriori Algorithm ,可成功找出已知的部分答案 2. 取 Max-25% 為門檻值(threshold) ; 大於此值者為正向基因表現(+1),小 餘者設定為負向基因表現(-1) 3. 取 MID 為門檻值, 即計算資料中的中位數; 所以前 50%者為正向基因表現 (+1), 後 50%者設定為負向基因表現(-1)。 4. 取 30% cut-off ; 前 30% 為正向基因表現(1) , 其餘 70%為負向基因表現 (-1)。5. 使用 Equal-interval partitioned data ; 按照相同間隔分 10 等分(1~10), 沒意義的數值設為無基因表現(non-expressed) (0) , 其餘有意義的數值¸
31
3.3.5 離散化結論
測試過以上數種方法之後。 3.3.4 的方法 2、3、4 並沒有考慮到一些無意 義的實驗數據, 將全部的實驗數據分為 『-1』 or 『1』 。以客觀的角度看來, 這樣的實驗步驟並不夠符合生物上的意義 ,因為實際上有很多實驗數據 ,確實 是無意義的。這樣的作法一定會產生較多的 Noise Data ,並影響實驗最後結果。 而在離散方法 1, 雖然會考慮無意義的資料 ,但微矩陣資料的表現程度 , 並非如此單純 ; 我們認為 ,即使同樣為正向或負向調控關係(Synexpress or Invert) ,在表現上也會有『相對高』或『相對低』的程度差異才對 ,不應該輕 易地忽略。 在經過我們測試此方法後 ,結果確實也不如我們所預期。 我們的系統目前採用方法 5: Equal interval 10 當作實作方式 ,因為實作 此法相當有效率, 且應用在我們的系統實作上,最能夠表達實際上基因表現的程 度 ,也能夠符合我們一開始的實驗假設。 對於要找出任一字串的互補字串 (complement pattern) ,藉由搜尋的技術, 也是能夠迅速的達成。 EX: Pattern : 1,1,2,3,4 其 Complement Pattern: 10,10,9,8,73.4 PIFP (Progress Iterative Frequent Pattern-tree)演算法
本研究的核心之處在於PIFP演算法。我們是以 FP-tree 演算法為基礎 ,加 入遞迴式的搜尋(Bergmann S et al. 2003)和刪除過於頻繁的項目(Kloster M, et al. 2005), 應用在生物資訊的探勘上面後 ,可以大幅提昇最後的輸出結果 ,加 強FP-Tree的探勘功能。 根據研究結果顯示 ,在相同測試條件下,可比FP-tree 演算法多增加100%~300%的輸出結果 , 並提高準確度。詳細評分過程和步驟 , 在第四章會介紹。因此,將PIFP應用在生物資訊的探勘上確實可以進一步找出潛 藏的基因模組,使輸出結果大幅提昇 ,並作為一個新的雙分群演算法。底下將介 紹我們異於原本的FP演算法之處 ,並配合一些人造資料, 作進一步說明。 PIFP異於原本的FP-tree演算法之處: z FP-tree的缺點: 根據FP-tree 建立頻繁集項目,並建立出完整的『樹』, 可以很容易地看出頻繁、出現次數多的字串,都集中在根節點(root)附近; 而越靠近葉節點(leaf node)的部分,則相對地出現次數越少。 也就是說, 當FP-tree要找出頻繁字串集合時 ,會選擇靠近樹根的字串,而非靠近樹葉 的字串! 然而,這樣的作法卻很有可能忽略由多組靠近樹葉的非頻繁項目, 所組成的頻繁項目。底下透過人造資料來說明FP 與 PIFP彼此間的差異。 此組資料包含 A~F共六個基因 , C1 ~ C6 共六個實驗狀態。而圖10中的數 值 , 為我們假設經過離散化處理的基因表現資料,『0』代表無基因表現。33
在此設定我們的 Minimum condition=2 、 Minimum gene=2 , 也就是說我們預 期找出的基因群組為至少包含兩個基因和兩個實驗狀態:
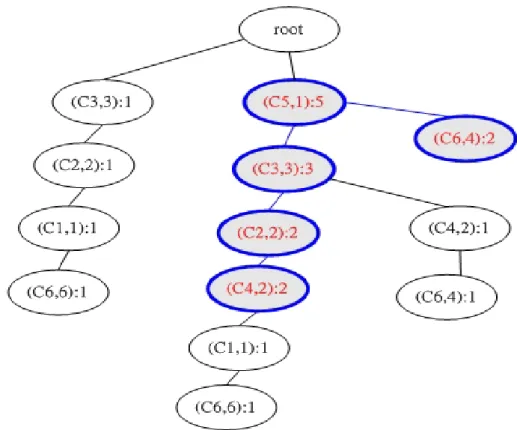
z 使用FP-tree所建立出的樹: ( (C5,1):5 代表在C5的狀態下,value=1共出
現5次)
z 使用FP-tree所找出的基因群組,以方匡表示:
z PIFP 使用『遞迴式的搜尋 & 刪除過於頻繁的項目』,所建立的樹:
35
以上範例說明, 藉由PIFP演算法 ,可以找出微弱的基因群組 (A ,B )。原因在於 其他出現次數過多的值 EX: (C5,1):5 , (C3,3):4, 會使得微弱的觀察值分散於 多處樹節點。藉由遮蓋掉這些過於頻繁的項目 ,並重建FP-tree , 可以讓微弱的 觀察值合併 ,且有可能符合 Minimum condition=2 、 Minimum gene=2 ,挖掘出 新的群組。
3.4.1 PIFP系統流程圖
在此介紹系統的詳細流程, 底下將會逐步解釋各步驟之間的運作細節。
流程圖步驟說明:
Expression Data: 從使用者指定的資料庫- DB_Yeast ,讀取資料 ,存入 PIFP 系統中。之後將在記憶體中進行底下運算步驟。 Normalization: 將輸入資料進行正規化動作。(參考 3.3.1) Discretization: 將正規化之後的資料 ,進行離散化動作(參考 3.3.4)。並將離 散化之後的資料 ,存入資料庫 DB_Token 中,方便之後的建造 FP 樹和字串查詢動 作。 FP-Growth: 從 FP-Tree 中 ,取出可能的頻繁字串集合。 將候選的頻繁字串,與 與我們設定的兩個門檻值: 出現次數和字串長度來作比較, 看是否滿足。 每個 符合參數的字串 , 在此將會形成會包含多個基因的基因群組 ,且同時包含正向 和負向調控關係 , 並記錄其參與基因表現的實驗狀態名稱。 Filtering: 用來過濾所找到的所有基因群組 。在此使用貪婪法(greedy method), 只挑選出雙分群大小(Gene_count * Condition_Count) 相對大者且雙 分群之間彼此覆蓋(overlap)程度小於 0.75 者; 如此可以避免過多重複的答案
Masking : 將這回合挑選出來的基因群組 ,執行 Mask(遮蓋)的動作。透過此步 驟可以凸顯較微弱的基因群組 ,方便下回合作搜尋。
37
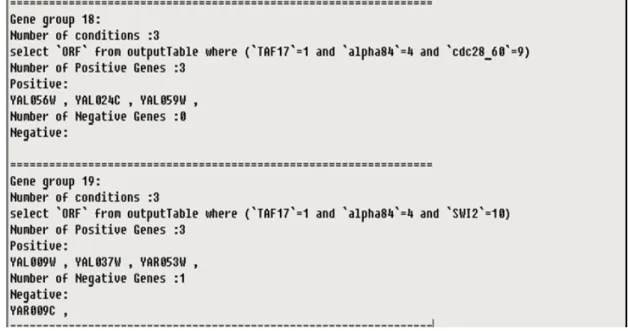
3.4.2 PIFP 輸出結果
為了與其他程式比較 ,PIFP 實作了兩種輸出格式 ,但其表達內容其實相同。 格式一:包含(1)群組編號 (2)正向的調控基因 (3)負向的調控基因(4)符合的實 驗狀態 (5) 符合的實驗狀態和基因個數 以酵母菌為測試資料的『基因群組 19』為例 , 可看出以下的資訊內容: 1. 調控模組彼此以線條分開 2. 該群組所符合的實驗狀態個數 = 3 ,及所屬的實驗狀態名稱為{TAF17 , alpha84 , SWI2} 3. 彼此為正向調控的基因個數 =3 ,分別是{YAL009W ,YAL037W,YAR053W} 4. 反向調控的基因個數 =1 ,為{YAR009C} 5. 可明顯看出此基因群組由 4 個基因所組成 , { YAL009W ,YAL037W,YAR053W , YAR009C } , 在 Condition {TAF17 , alpha84 , SWI2} 下 ,彼此可能有調 控關係格式二:與 Bimax Toolbox(Prelic A.et .al 2006)相同 ,包含:
(1)基因與狀態個數(第一行) (2)基因集合(第二行)(3)狀態集合(第三行)
為了與Bimax Toolbox的輸出格式相容,我們也遵從學者們的輸出格式,可以方便
日後與其他程式作結果分析。底下是實際包含內容:
採用此輸出格式之後 ,可與 OPSM (Ben-Dor et al. 2002)、 ISA (Ihmels et al.2004) 、CC (Cheng and Church ,2000)、xMotif(Murali and Kasif ,2003)、 Bimax(Prelic A et al. 2006) 等近年來發表的 Biclustering Algorithms ,在 相同的測試資料下,作合理且公平的分析、比較。在章節 4.2 、4.3 ,會提出與 多種方法的測試結果和比較。
39
3.4.3 PIFP 特色之處
接下來是本系統的主要核心— PIFP 的一些特色 : 1. 可根據使用者需求 ,找出龐大資料中的重要的頻繁字串。 只需調整 2 個參 數—出現次數和字串長度 , 即可找出符合的基因模組 ; 而此二參數可以 用%來做設定, 會根據資料庫大小作自動調整 ,也避免操作上的不便。 2. 如同雙分群的演算法一樣 , 可以搜尋間隔 (gap)的字串 ; 字串符合彈性 更大。也允許基因群組之間彼此有重疊 ; 即一個基因可以重複存在多個基 因群組中; 可以確實反映出實際的生化反應。 3. 演算法的實際運作相當快速 。如同前面所敘述 ,只要掃瞄兩次即可 ,複雜 程度相當低。使用 Pentium4 3.0 GHZ 和 512 Ram ,測試資料庫(6325*300) (Hughes TR et al.2000) ,約可在 15~30 分鐘內跑完,並輸出完整評分結果。 (時間差異為參數所造成) 4. 在相同條件下 ,使用 PIFP 和 FP 的差異 ,主要在於最後輸出的基因群組數 目 ; 使用 PIFP 演算法可找出數倍於傳統 FP 演算法的基因群組總數。可徹 底找出應該存在的調控關係。關於實驗的部分會在下個章節介紹。第四章--結果與討論
本章將介紹我們實驗的成果。首先在 4.1 介紹評分準則,和學者們使用的兩
種評分公式 ,我們採用的是大家一致公認的資料來源--Saccharomyces Genome
Database。測試資料就如同前面所介紹 ,是酵母菌的微矩陣資料。我們將在同等
條件下,和之前學者們的系統 ,一起比較輸出結果。包含章節 4.3 的 Clustering Programs 和章節 4.5 的 Biclustering Programs。 最後並提供與傳統 FP 演算 法之間的差異, 以及敘述我們的貢獻。
4.1 評分準則
首先介紹評分公式中 ,最主要的資料訊息—生物註解(Annotation)。 如同 其他文獻中所說 ,我們也去 GO 找出相關的生物調控訊息 ,取得的註解包含許 多訊息, 如此便可成為我們輸出結果的參考答案,如下圖所示:
41
4.1.1 評分公式:超幾何分佈
為了評比各個系統所輸出的基因群組品質好或壞, 在此使用廣為學者們所 使用的超幾何分佈(Hypergeometric Distribution)當作『評分公式』來計算 P-value (Dembele D et al. 2003 ;Ihmels J et al. 2004), 定義如下:
經過此公式運算 ,我們將會期望此 P-value 越小越好 。 P-value 越小, 即代 表此基因群組和目前所知的生物註解相當符合!而為了方便使用者觀看輸出成 績 ,在此將會做進一步的運算 BFM (biological figure of merit) 的值 (Ihmels et al. 2004) 。BFM = -log10 (P-value) ; 此時我們將期望該值越大
P: p-value N: 基因群組中所包含的 M: 所有基因的總數基因個數 K: 生物註解中所包含的基因個數 Z: 基因群組和註解比較後,所包含的正 確基因個數 圖 16 生物註解所形成的關係樹狀圖(http://www.yeastgenome.org/)
M
N
N
Z
x
K
M
x
N
K
x
C
C
C
P
∑
=
−
−
⋅
=
越好 , 方便作最後的加權平均動作 , 最後以此平均值來衡量一個演算法的好 或壞。根據觀察 ,BFM 確實可以反映出一個演算法的準確度。我們以此當作仲裁 各演算法之優劣的方式之一。
4.1.2 評分公式:FuncAssociate(The Gene Set Functionator)
FuncAssociate (Berriz .et al. 2003), 此 web-based tool 亦為近年來學者們 評量雙分群演算法結果好壞的好壞之一 。該工具使用 Fisher's exact test 來 當作頻分公式, 可根據使用者輸入基因群組、基因來源、門檻值…等參數 , 找 出有意義的基因的分群資料, 並回傳多種重要資訊(Rank、p-value、
圖 17 為 『FuncAssociate』 所傳回的資料 , Rank 為該工具依據 p-value 來作排序的名次 ,N 為此群組命中的個數 ,X 為此群組對應之生物註解個數, p-value 為我們要參考的最重要數據 ,GO Attribute 為該生物註解的名稱。
圖 20 此工具為我們開發的『ClusterView』, 用來讀取各種雙分群程式的輸出檔案 ,並進行 4.1.1 和 4.1.2 兩種評分公式的計算。可以觀察每個基因群 組所包含的基因 、BFM 平均值 、生物註解、p-value (Func)、p-value(Hyper) 、p-valu 分佈情形…等多種資訊。
4.2 測試程式 –Clustering Programs
1. 首先 ,我們使用 Esien Lab 所製作的分群程式 『Cluster』。當初 ISA (Ihmels et al.2004) 等相關論文也曾經使用過此工具來作檢測 ,所以我們 認定該工具有一定的可信度。『Cluster』功能包含 :Hierarchical Cluster , K-Means Cluster , SOM , PCA 等多種傳統分群功能 ,其外觀如下: (下載 位置:http://rana.lbl.gov/EisenSoftware.htm)
45
2. 使用 Fuzzy C-Means 程式 ,在 Linux 底下的測試情形 (Dembele D et al. 2003) :
0
5
10
15
20
25
30
35
Fr
eq
ue
nc
y
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30-log(p-value)
Hierarchical Cluster
4.3 測試結果 - Clustering Programs
底下將會顯示各個程式所跑出來的結果 ,都是以內建的參數來做測試 , 並 在下個段落分析與比較。因顯示空間有限 , 只顯示 1~30 分 。 高於 30 分者 , 暫時以 30 分顯示 ,但不會影響各種演算法之間的優劣程度和比較結果。 參與測試的方法均為傳統的分群演算法, 包括: Hierarchical cluster、SOM 、 PCA 、K-Means 、Fuzzy C-Means 。 最後將我們的 PIFP 演算法以基因個數 G=21 狀態個數 =17 參與比較。4.3.1 BFM 分佈圖
關於資料繪圖部分 , 為了凸顯分數好壞的差異 ,在此分為 BFM 此三的分數 三個部分 : 1~10 、11~20 、21~30 , 每個部分以相異的材質來作繪製動作。
47 0 1 2 3 4 5 6 7 Fr eque nc y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 -log(p-value) SOM 0 20 40 60 80 100 120 140 160 Fr eq ue nc y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 -log(p-value) PCA 圖 24 PCA 之 BFM 分佈圖 , 紅色的低分較多,BFM 平均 1.7 圖 23 SOM 之 BFM 分佈圖 ,BFM 平均為 5.5
0 5 10 15 20 25 30 35 40 45 Fr eque nc y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 -log(p-value) K-Means 0 5 10 15 20 25 30 Fr eq ue nc y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 -log(p-value) Fuzzy C-Means 圖 18 Fuzzy C-Means 之 BFM 分佈圖 , BFM 平均 2.23 圖 19K-Means 之 BFM 分佈圖 , 平均 2.84
49 0 2 4 6 8 10 12 14 16 18 Fre qu en cy 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 -log(p-value) PIFP 演算法 基因群組數目 BFM 平均分數 BFM 標準差 Percentage of BFM >5 Hierarchical 68 3.22 3.72 16.18% SOM 27 5.44 5.83 37.04% PCA 300 1.70 0.85 0.33% K-Means 100 2.84 3.33 11.00% Fuzzy C-Means 64 2.63 3.60 7.81% PIFP (21,17) 98 10.20 5.97 88.78% 表 2 為上述各演算法分群結果 , 實際的測量數據 。 圖 20 PIFP 之 BFM 分佈圖 ,平均 10.06
0.00% 10.00% 20.00% 30.00% 40.00% 50.00% 60.00% 70.00% 80.00% 90.00% 1 00.00%
PI FP Hier ar cical SOM PC A K- Mean s Fu zzy C - Mean s
1 <= -log(p-value) <=5 6<= -log(p-value) <=10 11<= -log(p-value) 根據表 2 顯示, 我們所提出的雙分群演算法 ,就如同我們在前面章節假設的一 樣,可以彌補傳統分群演算法的不足 ,並提高預測能力、準確度。如圖 28 , 在 此將分數分為三個區間 分別是 1~5 , 6~10 , 11 以上 。 由此可見, 傳統方法 的結果大多集中在前半部的低分區域 ,而我們有超過 80%以上是集中在中高分 區域。因此我們認為,在相同的資料、相同的評分公式下 ,我們的輸出結果精準 度比之前的傳統演算法要好。 圖 21 顯示各種分群演算法之-log(p-value)分佈情況 。
51 0 100 200 300 400 500 600 700 800 900 1000 0 20 40 60 80 100 120 Cluster Si ze PIFP Hierarchical SOM PCA K-Means Fuzzy C-means
4.3.2 基因個數分佈圖
上面的圖形都是根據評分公式所整理出來的統計圖, 但除了分數之外 ,每 個演算法所找出的基因群組大小 也是我們所關注的。如同我們的假設一樣 , 我們所期望的基因群組 ,應該是要包含多個基因 ,和多個實驗狀態 ,而且該群 組中(基因的個數 * 實驗狀態個數) 應該要越大越好。 但因為傳統方法是以全 部的實驗狀態作分群考量 ,所以在此僅呈現各演算法依照程式輸出之群組順序 內, 所包含的基因群組分佈圖。圖 29 上面幾個分佈圖的整體繪製 ,只顯示前 100 個 cluster 分佈。 因為只有 PCA 的分群個數較多 ,PCA 從 100~300 ,且都只包含一個基因 ,數值太小無法顯示, 在此省略其後半部的輸出結果。
PIFP 0 200 400 600 800 1000 1200 1400 1600 1 11 21 31 41 51 61 71 81 91 Cluster Si ze Hierarchical Cluster 0 50 100 150 200 250 300 1 11 21 31 41 51 61 Cluster Si ze
圖 23 Hierarchical Cluster 的 cluster size 分佈圖 圖 22 PIFP 的 cluster size 分佈圖
53 SOM 0 100 200 300 400 500 1 11 21 Cluster Si ze PCA 0 50 100 150 1 51 101 151 201 251 Cluster Si ze 圖 33 , genes 主要內容集中在前 100 clusters,100~300 的基因群組, 都只包含 一個基因。這是這個方法主要錯誤的原因。
圖 24 SOM 中, cluster size 的分佈圖
K-Means(100) 0 100 200 300 400 500 1 11 21 31 41 51 61 71 81 91 Cluster Si ze Fuzzy C-Means(100) 0 500 1000 1500 2000 2500 3000 1 11 21 31 41 51 61 Cluster Si ze
圖 26 K-Means 分群後的 Cluster size 分佈圖
4.4 結果討論 - Clustering Programs
由以上的圖表顯示 , PIFP 最高分的成績高於之前的方法數倍 ,包括 Hirarchical cluster 、SOM 、PCA 、Fuzzy C-Means ; 且我們的基因群組數量 和加權總分都相當高 , 主要分佈都高於 10 分 ,這和傳統方法普遍低分有明顯 不同 。 如此便可說明: 在相同的生物註解和評分公式底下 ,我們的方法可以找 出更多且品質更好的基因群組。 除此之外, 所有程式參數的設定 ,都盡量以預設值來作為測試標準 。由於 Fuzzy C-Means 會從 C/3 個群組開始找起 , 所以在此測試幾個不同的參數來 做參考。而我們的 PIFP , 只需要設定兩個參數 『出現次數』和『字串長度』; 在此也做了大範圍的測試 : 將記次個數設定 15~80 , 字串長度設為 30~70 。 結果顯示 ,雖然參數會影響我們的輸出結果 ,但分數下降幅度並不會很快 ;測 試了將近 2000 組不同的參數值, 其 BFM 的平均值幾乎都在 20 分以上。 這也代 表我們的系統穩定性高 , 擁有容錯的功能 , 以後即使遇到新的資料 ,也不會 因為參數的設定而完全找不到答案。
4.5 測試程式 –Biclustering Programs
之前的測試著重於傳統的分群程式,此處將著重於與近年來的演算法比較。 在2006年, Prelic與其伙伴提出系統化的雙分群法測試平台。 底下我們將拿 PIFP演算法 和 BicAT程式工具中所包含的5個雙分群程式進行測試, ,包括: OPSM (Ben-Dor et al. 2002)、 ISA (Ihmels et al.2004) 、CC (Cheng and Church ,2000)、xMotif(Murali and Kasif ,2003)、 Bimax(Prelic A et .al 2006)。我們將在相同評分公式和測試資料下, 所進行一連串的比較。由於此篇 論文具有相當的重要性 ,因此我們使用其內建測試資料和論文中所使用的評分 方式 ,和之前4.2、4.3章節的略有不同。
4.5.1 雙分群參數設定
表 3 為五個雙分群程式根據Prelic A et .al 2006所使用的參數設定 , 之後的輸出測試也是以此為標準。 而為了配合
其他程式的輸出 ,我們調整 PIFP 參數 G=12 ,C=11 ,使得輸出的群組個數大約和其他演算法相同 ,個數約等於 100 個 , 比較客觀和公平。
0
25
50
75
100
125
1 6 11 16 21 26 31 36 41 46 51 56 61 66 71 76 81 86 91 96 Cluster Number-l
og(
p-val
ue
)
PIFP4.6 測試結果 - Biclustering Programs
我們直接由 Bimax 的官方網站,抓取測試之後的實驗結果來和我們的程式 作比較。http://www.tik.ee.ethz.ch/sop/bimax/SupplementMaterials,Biclustering.html 評分公式使用 4.1.2 評分公式:FuncAssociate ,所計算的 p-value 當作標準。 底下會介紹我們的輸出結果-包含雙分群的相關資訊 ,以及和各個雙分群程式的 比較 ,最後是在多種門檻值下 ,所繪出的直條圖。我們希望在一切條件公平的情 況下 ,進行一連串的比較。圖 29 PIFP 根據 Bimax 內建的酵母菌資料, 測試之後的 –log (p-value) 分佈圖 , X 軸依序為每個基因 群組依照順序所找出的編號 , Y 軸為該基因群組根據 FuncAssociate 所得到的評分。PIFP 參數設定 為 G=12, C=11
1 10 100 1000 10000 1 11 21 31 41 51 61 71 81 91 Cluster Nu m be r of G en es PIFP Bimax CC ISA OPSM Samba xMotif 1 10 100 1 11 21 31 41 51 61 71 81 91 Cluster N um be r of c ondi tions PIFP Bimax CC ISA OPSM Samba xMotif 圖 38 雙分群演算法在酵母菌資料上 , 所得到的基因群組內之基因個數分佈圖。 圖 39 分群演算法在酵母菌資料上 , 所得到的基因群組內之實驗狀態個數分佈圖。
100
10000
1000000
Cluster size
PIFP Bimax CC ISA OPSM Samba xMotif 圖 30 雙分群演算法在酵母菌資料上 , 所得到的基因群組大小分佈圖 。 X 軸 為群組編號 ,Y 軸為該群組大小( 基因 個數* 實驗狀態個數)。透過分佈圖, 可 以看到我們的 PIFP, 透過遞迴和遮蓋 的方式 ,可以找出由大到小的群組,並 持續數個回合。表 4 PIFP 測試 Bimax 之酵母菌資料庫 ,經過 統計之後 ,所得到的部 分結果。我們的 p-value 值範圍: 9.70E-05~1.80E-119 , 在統計上來說 ,是 highly significance。 至於其他演算法的輸出 結果 ,可至 http://www.tik.ee.et hz.ch/sop/bimax/Supp lementMaterials,Bicl ustering.html 下載 BiCluster_Num Size p-value(Func) Anno(Func)
BiCluster1 228 1.80E-119 cytosolic ribosome (sensu Eukaryota)/80S ribosome BiCluster85 166 3.00E-39 ribonucleoprotein complex/RNP
BiCluster58 58 5.30E-32 ribosome
BiCluster41 101 3.90E-26 ribosome biogenesis BiCluster7 135 5.80E-24 protein complex BiCluster11 120 2.60E-19 RNA metabolism
BiCluster69 64 5.40E-19 non-membrane-bound organelle BiCluster13 128 4.00E-18 physiological process
BiCluster14 108 2.60E-16 cellular process
BiCluster76 70 8.00E-14 cytoplasm organization and biogenesis
BiCluster24 76 5.80E-12 cellular physiological process/cell growth and/or maintenance/cell physiology
BiCluster64 70 4.40E-11 translation factor activity, nucleic acid binding BiCluster77 62 2.40E-10 mitochondrial ribosome
BiCluster83 33 7.10E-09 generation of precursor metabolites and energy/energy pathways BiCluster67 42 1.20E-06 translation initiation factor activity
BiCluster82 19 7.70E-06 peroxisomal matrix BiCluster35 14 9.60E-06 cellular biosynthesis
BiCluster34 23 3.60E-05 fatty acid elongase activity BiCluster95 23 3.80E-05 molecular function unknown BiCluster94 32 9.70E-05 binding/ligand
表 5各個雙分群演算法的輸出結果,在『GO Process category』下(Saccharomyces cerevisiae),使用 FuncAssociate 輸出的 p-value ,
在不同的門檻值 α (0.001%,0.1%,0.5%,1%,5%), 進行統計上的檢測和分佈情況。(Prelic A.et .al 2006)
測試項目 / 演算法 PIFP OPSM Bimax ISA Samba CC xMotif
significant α <0.001% 95% 87.5 % 74 % 80 % 68 % 24 % 1 % significant α <0.1% 100% 87.5 % 81 % 80 % 68 % 24 % 1 % significant α <0.5% 100% 87.5 % 87 % 80 % 68 % 24 % 1 % significant α <1% 100% 93.75 % 92 % 85 % 74 % 29 % 2 % significant α <5% 100% 100 % 99 % 94 % 83 % 34 % 7 % 雙分群之總個數 100 12 100 66 100 100 306
0 10 20 30 40 50 60 70 80 90 100 Pr op or tio n of b ic lu ste rs p er s ig ni f. le ve l α %
PIFP OPSM Bimax ISA Samba CC xMotif
significant α <0.001% significant α <0.1% significant α <0.5% significant α <1% significant α <5%
4.7 結果討論 - Biclustering Programs
根據我們觀察 ,我們 PIFP 的演算法輸出結果, 整體來說比其他雙分群演算 法好, 而且雙分群的數量也相當多。單純以 FuncAssociate 這個評分工具來看, 我們的輸出結果在統計上是有不錯的表現,絕非以隨機的方式產生答案,這對我 們來說 , 是一個很有力的支持。 就生物學家的角度來看 ,我們能夠在真實的生 物實驗數據下,提供部分與已知的生物註解相符的答案 ,幫助學者們預測、分析 生物功能 ; 而非只是測試人造資料 。而統計圖中 PIFP 所表現的 highly significant, 並非表示我們系統完美無缺 ,而是顯示那五個門檻值的設定太過 於接近。不同的門檻值 α (0.001%,0.1%,0.5%,1%,5%),對於近年來雙分群法來 說 ,這樣的設定並不符合我們所需, 應該再小一點才對 ,否則不太容易區分出 分群結果彼此間的好壞。 圖 31 根據表格 2 所繪製的長條圖,可以明顯表示出各種演算法的分群結果,符合門檻值的比例。我們的 p-value 最大值恰巧在 1E10-6 附近 , 所以全部的值都小於 0.001%。(Prelic A.et .al 2006)4.8 FP vs PIFP
我們針對 FP 與 PIFP 在相同的條件、參數設定下 , 進行以上兩個資料庫 ( Hughes and bimax) 的測試,發現 FP 與 PIFP 彼此間的差異, 完全在於基因 群組的個數! 換句話說 ,FP 演算法的輸出, 屬於 PIFP 輸出結果中的子集合。 因 為群組個數差距過大, 且兩者重疊部份的輸出完全相同,我們不再強調 p-value 的比較 。 底下將會進行這兩者間差異較大之處進行比較。
表 6 FP vs PIFP
資料來源 Bimax Dataset Hughes Dataset
演算法 FP(12,11) PIFP(12,11) FP(21,17) PIFP(21,17) 群組個數 36 100 20 98 生物註解種類 9 20 3 9 所需時間(sec) 48 70 443 665 經由以上表可以得知 , 透過 PIFP 的遞迴式搜尋 , 在相同條件下 , 確實可以 搜尋出更多的基因群組 ,並提供更廣泛、更接近真實的生物註解。 至於運算的 時間方面 ,也在合理的範圍之內 ,只要 10 多分鐘即可獲得答案。
65
第五章--結論與展望
5.1 結論
本研究根據生物學家們所觀察到生物上的實際情況 ,提出一連串的假設 , 並根據假設實作出一個新的雙分群演算法-PIFP 。PIFP 可以在同時考慮基因、 實驗狀態下, 快速進行微矩陣資料的分群 ,並徹底找出可能的基因群組。相較於 其他演算法, 我們的基因群組 ,允許不連續的實驗狀態、允許群組間部分的重 疊、會過濾不好的基因表現資料,僅分析有意義的資料。 而與傳統方法比較起來 , 我們的演算法精確度超過 K-means 、Hirarchical 、SOM 、PCA…等多年前學者 們提出的方法。這也映證我們一開始假設正確無誤,應用在尋找基因群組上, 雙 分群法確實會優於傳統分群法。此外 ,我們採用了兩種評分公式Hypergeometric distribution 和 FuncAssociate 來和已知生物註解作驗證,所 測試出來的 p-value 甚至可以到 1.0E-200。這代表在統計上, 我們的輸出結果 有意義。 在運作計算上, 我們使用 Pentuim 4 的 cpu 搭配 400M 的可用記憶體來執行 測試。在實驗過程中 ,學者們所開放出來的工具 ,大多要運作 30 分鐘。然而我 們的 PIFP,即使是龐大的資料庫 ,一樣可以準確地在 10 分鐘內找出答案 ; 若套 用 Bimax 的內建資料庫 , 則可在 2 分鐘內給予預測的結果。 以上實驗結果顯示,本研究藉由分析生物的微矩陣資料,提高了基因模組預 測能力,其結果也與前人文獻記載相呼應。因此,本研究所預測的基因群組和 PIFP 演算法 , 可以供生物學家作為研究參考。
5.2 未來展望
我們在上面第四章所進行的測試 , 實驗的設計和步驟 ,都是跟隨之前學者 們的檢測方式。然而,不論是計算 p-value 或 significant , 都是以統計的角度 來衡量預測出的基因群組的穩定性和準確度。然而 ,統計和現實的生物反應比較 起來 ,還是有一段差距 ,只有統計資料 ,不足以讓人負予重任。 因此 ,未來可 以進行的部分 ,應該是著重於基因網路的重建 、真實生物路徑(pathway)的分析 上面 , 並提供更多的資料給生物學家分析、檢測、驗證。 最後 ,本研究目前所測試的數據 ,是目前最為大家所熟悉且易取得資訊的 酵母菌資料。未來希望能夠在我們測試酵母菌、果蠅…等微矩陣資料後 ,更進一 步將實驗套用在人類的資料上 , 透過基因層次上面的分析,用以預測、分析白血 病、癌症等重大疾。67
參考文獻 (依照字母順序排列)
徐英哲(2003)以基因表現相關性及轉錄因子結合區重建調控網路
Alter O, Brown PO, Botstein D (2000). Singular value decomposition for genome-wide expression data processing and modeling.Proc Natl Acad Sci.;97(18):10101-6.
Becquet C, Blachon S, Jeudy B, Boulicaut JF, Gandrillon O. (2002)
Strong-association-rule mining for large-scale gene-expression data analysis: a case study on human SAGE data. Genome Biol.;3(12)
Ben-Dor A, Chor B, Karp R, Yakhini Z. (2003)Discovering local structure in gene expression data: the order-preserving submatrix problem. J Comput Biol. 10(3-4):373-84.
Bergmann S, Ihmels J, Barkai N (2003). Iterative signature algorithm for the analysis of large-scale gene expression data. Phys Rev E Stat Nonlin Soft Matter Phys. 2003 Mar;67(3 Pt 1)
Berriz GF, King OD, Bryant B, Sander C, Roth FP.(2003)Characterizing gene sets with FuncAssociate.Bioinformatics. 2003 Dec 12;19(18):2502-4.
Cheng Y, Church GM. (2000)Biclustering of expression data. Proc Int Conf Intell Syst Mol Biol. 2000;8:93-103.
Creighton C, Hanash S (2003).Mining gene expression databases for association rules. Bioinformatics. Jan;19(1):79-86.
Dembele D, Kastner P. (2003) ,Fuzzy C-means method for clustering microarray data. Bioinformatics. ;19(8):973-80.
Getz G, Levine E, Domany E. (2000) Coupled two-way clustering analysis of gene microarray data.Proc Natl Acad Sci ;97(22):
Hashimoto RF, Kim S, Shmulevich I, Zhang W, Bittner ML, Dougherty ER.(2004) Growing genetic regulatory networks from seed genes.
Bioinformatics ;20(8):1241-7.
Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J, Bard M, Friend SH.(2000) Functional discovery via a compendium of expression profiles .Cell. Jul 7;102(1):109-26.
Ihmels JH, Bergmann S. (2004), Challenges and prospects in the analysis of large-scale gene expression data.Brief Bioinform.;5(4):313-27.
Ihmels J, Friedlander G, Bergmann S, Sarig O, Ziv Y, Barkai N (2002). Revealing modular organization in the yeast transcriptional network. Nature
69
Ihmels J, Bergmann S, Barkai N.(2004) Defining transcription modules using large-scale gene expression data. Bioinformatics;20(13):1993-2003.
J.Han, J.Pei ,and Y.Yin (2000) .Mining frequent patterns without candidate generation.SIGMOD'00 ,pages 1-12 ,
Ji L, Tan KL (2004). Mining gene expression data for positive and negative co-regulated gene clusters. Bioinformatics.;20(16):2711-2718.
Kloster M, Tang C, Wingreen NS.(2005) Finding regulatory modules through large-scale gene-expression data analysis. Bioinformatics.(7) 1172-1179.
Michael B. Eisen, Paul T. Spellman, Patrick O. Brown, and David Botstein,(1998) Cluster analysis and display of genome-wide expression
patterns ,PNAS ,Vol. 95, Issue 25, 14863-14868,
Murali TM, Kasif S.(2003) Extracting conserved gene expression motifs from gene expression data. Pac Symp Biocomput. 2003;77-88.
Prelic A, Bleuler S, Zimmermann P, Wille A, Buhlmann P, Gruissem W, Hennig L, Thiele L, Zitzler E.A (2006) systematic comparison and evaluation of biclustering methods for gene expression data.Bioinformatics. 2006 May 1;22(9):1122-1129.
Qian J, Dolled-Filhart M, Lin J, Yu H, Gerstein M. (2001) Beyond synexpression relationships: local clustering of time-shifted and inverted gene expression profiles identifies new, biologically relevant interactions.J Mol Biol ;314(5):1053-1066.
Qiu X, Brooks AI, Klebanov L, Yakovlev N.(2005) The effects of normalization on the correlation structure of microarray data. BMC
Bioinformatics.;6(1):120.
Rice JJ, Tu Y, Stolovitzky G.(2005) Reconstructing biological networks using conditional correlation analysis.Bioinformatics.;21(6):765-773.
Sara C. Madeira ,Arlindo L. Oliveira,(2004) Biclustering Algorithms for Biological Data Analysis: A Survey , IEEE / TCBB Volume 1 , Issue 1 ) Pages: 24 - 45
Sheng Q, Moreau Y, De Moor B. (2003) Biclustering microarray data by Gibbs sampling. Bioinformatics. ;19 Suppl 2:II196-II205.
Shmulevich I, Dougherty ER, Kim S, Zhang W. (2002) Probabilistic Boolean Networks: a rule-based uncertainty model for gene regulatory networks Bioinformatics;18(2) : 261-74.
Tanay A, Sharan R, Shamir R.(2002) Discovering statistically significant biclusters in gene expression data.Bioinformatics.;18 Suppl 1:S136-44.
71
Torrente A, Kapushesky M, Brazma A. (2005) A new algorithm for comparing and visualizing relationships between hierarchical and flat gene expression data clusterings. Bioinformatics.;21(21):3993-3999.