To find new candidate loci predisposing individuals to Kawasaki disease, an acute vasculitis that affects children, we conducted a genome-wide association study in 622 individuals with Kawasaki disease (cases) and 1,107 controls in a Han Chinese population residing in Taiwan, with replication in an independent Han Chinese sample of 261 cases and 550 controls. We report two new loci, one at BLK (encoding B-lymphoid tyrosine kinase) and one at CD40, that are associated with Kawasaki disease at genome-wide significance (P < 5 × 10−8). Our findings may lead to a better understanding of the role of immune activation and inflammation in Kawasaki disease pathogenesis.
Kawasaki disease (MIM 611775) is an acute, self-limiting vasculitis that affects infants and young children. Symptoms include prolonged fever, polymorphous skin rash, swollen glands, red eyes, mouth inflammation and swollen hands and feet1. Coronary aneurysms
develop in 15–25% of untreated children with Kawasaki disease1,2,
and this disease is the leading cause of acquired heart disease among children in industrialized countries.
Genetic determinants have been suggested to contribute to disease susceptibility. Populations in Asian countries have higher incidence rates of Kawasaki disease than those in Western countries: Japan has the highest annual incidence rate3,4, followed by Korea5 and Taiwan6,7.
Although the cause of Kawasaki disease is unknown, clinical and epidemiological findings suggest that an infectious agent triggers an inflammatory response, leading to host immune dysregulation in genetically predisposed individuals8,9. Thus, in addition to loci
related to cardiovascular function, genes with a role in immune activ-ity have been a focus of candidate gene studies of Kawasaki disease susceptibility and disease outcome10. A genome-wide linkage
ana-lysis conducted in samples from Japanese sibling pairs with Kawasaki disease11,12 and four genome-wide association studies (GWAS) in
individuals of European ancestry and in Korean and Taiwanese popu-lations identified biologically plausible candidate loci for Kawasaki disease13–16. However, these loci do not fully explain the genetic
risk for Kawasaki disease, suggesting that additional genetic factors remain to be discovered.
We performed a case-control GWAS to search for loci associated with increased risk of Kawasaki disease using the Affymetrix 6.0 SNP chip. We initially analyzed 905,358 SNPs in 627 Kawasaki disease cases and 1,118 controls in a Han Chinese population residing in Taiwan. After strict quality control filtering (Supplementary Table 1), we analyzed 716,935 SNPs (79.19%) in 622 Kawasaki disease cases and 1,107 controls. Analysis of population structure by principal-component analysis (PCA) did not give any significant evidence of population stratification between Kawasaki disease cases and controls (Supplementary Fig. 1). The genomic inflation factor was 1.000.
The association results for Kawasaki disease susceptibility in the 622 Kawasaki disease cases and 1,107 controls are shown (Fig. 1). We found 101 SNPs associated with Kawasaki disease at P < 1 × 10−4
(Fig. 1 and Supplementary Table 2). We validated these SNPs by Sequenom MassARRAY and further genotyped the validated SNPs in an independent cohort of 261 Kawasaki disease cases and 564 controls (Supplementary Table 2). After kinship analysis, 261 Kawasaki disease cases and 550 controls remained in the replication cohort.
Two new susceptibility loci for Kawasaki disease
identified through genome-wide association analysis
Yi-Ching Lee
1,2,19, Ho-Chang Kuo
3,4,19, Jeng-Sheng Chang
5,19, Luan-Yin Chang
6,19, Li-Min Huang
6,
Ming-Ren Chen
7, Chi-Di Liang
3,4, Hsin Chi
7, Fu-Yuan Huang
7, Meng-Luen Lee
8, Yhu-Chering Huang
9,10,
Betau Hwang
11, Nan-Chang Chiu
7, Kao-Pin Hwang
3,12,13, Pi-Chang Lee
14, Li-Ching Chang
1, Yi-Min Liu
1,
Ying-Ju Chen
1, Chien-Hsiun Chen
1,13, Taiwan Pediatric ID Alliance
15, Yuan-Tsong Chen
1,16,
Fuu-Jen Tsai
13,17,18& Jer-Yuarn Wu
1,131Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan. 2Institute of Molecular Medicine, National Tsing Hua University, Hsinchu, Taiwan. 3Department of Pediatrics, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan. 4Graduate Institute of Clinical Medical Science, Chang Gung University College of Medicine, Kaohsiung, Taiwan. 5Department of Pediatrics, China Medical University Hospital, Taichung, Taiwan. 6Department of Pediatrics, National Taiwan University Hospital, Taipei, Taiwan. 7Department of Pediatrics, Mackay Memorial Hospital, Taipei, Taiwan. 8Department of Pediatrics and Divisions of Pediatric Cardiology, Changhua Christian Hospital, Changhua, Taiwan. 9Division of Pediatric Infectious Diseases, Chang Gung Memorial Hospital at Linkou, Taoyuan, Taiwan. 10College of Medicine, Chang Gung University, Taoyuan, Taiwan. 11Department of Pediatrics, Taipei City Hospital, Zhongxiao Branch, Taipei, Taiwan. 12Department of Pediatrics, Division of Pediatric Infectious Diseases, China Medical University Hospital, Taichung, Taiwan. 13School of Chinese Medicine, China Medical University, Taichung, Taiwan. 14Department of Pediatrics, Taipei Veterans General Hospital, Taipei, Taiwan. 15A complete list of members and affiliations appears in the Supplementary
Note. 16Department of Pediatrics, Duke University Medical Center, Durham, North Carolina, USA. 17Department of Medical Genetics, China Medical University Hospital, Taichung, Taiwan. 18Department of Health and Nutrition Biotechnology, Asia University, Taichung, Taiwan. 19These authors contributed equally to this work. Correspondence should be addressed to Y.-T.C. ([email protected]), F.-J.T. ([email protected]) or J.-Y.W. ([email protected]).
Received 30 August 2011; accepted 29 February 2012; published online 25 March 2012; doi:10.1038/ng.2227
npg
© 2012 Nature
America, Inc.
Nature GeNetics VOLUME 44 | NUMBER 5 | MAY 2012 523 A total of 23 SNPs showed nominal evidence of replication (P < 0.05)
(Supplementary Table 3). All these SNPs showed similar evidence for association with Kawasaki disease after PCA adjustment (for com-ponents 1 to 10) in the GWAS collection samples (Supplementary
Table 4). We observed no strong evidence of heterogeneity between
samples from the GWAS and the replication study for these SNPs (I2 = 0, P
het > 0.43) (Supplementary Table 5). A joint analysis of both
GWAS and replication samples for these 23 SNPs resulted in three SNPs located at two loci exceeding the threshold for genome-wide significance (P < 1 × 10−8; Table 1).
The rs2736340 (P = 9.01 × 10−10; odds ratio (OR) = 1.54) and
rs2618476 (P = 1.96 × 10−9; OR = 1.52) SNPs were found to be
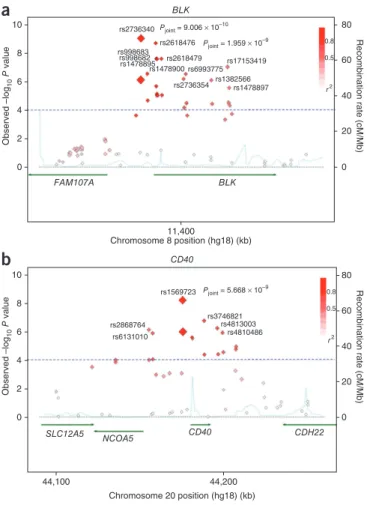
in strong linkage disequilibrium (LD) (D′ = 0.988 and r2 = 0.971; Fig. 2a and Supplementary Fig. 2a) and mapped to a 12.2-kb
LD block (position 11,378,539–11,390,744) at 8p23.1; the block comprises the promoter and the first intron of BLK (encoding B-lymphoid tyrosine kinase). Ten nearby SNPs clustered in the first intron of BLK did not reach genome-wide significance; however, their P values were significantly associated with Kawasaki disease (P = 2.68 × 10−6 to 2.44 × 10−8) in the joint analysis (Fig. 2a and Supplementary Table 3). Subsequent logistic regression analyses conditioned on
rs2736340 indicated that most of the observed associations resulted from strong LD with rs2736340 (Supplementary Fig. 2b). We per-formed haplotype analysis to investigate the effect of combinations of these Kawasaki disease–associated SNPs; however, no haplotype showed stronger association than the single-marker association of rs2736340 (strongest P = 9.35 × 10−7).
The third SNP to reach genome-wide significance was rs1569723 (P = 5.67 × 10−9; OR = 1.42; Table 1), which mapped to a 17.2-kb LD
block (position 44,164,170–44,181,354) at 20q13.12 located upstream of the CD40 gene (Fig. 2b). Five nearby SNPs encompassing the region upstream of CD40 and CD40 itself showed suggestive asso-ciations (P = 1.46 × 10−6 to 1.93 × 10−7) in the joint analysis (Fig. 2b
and Supplementary Table 3). These SNPs are in LD with rs1569723 (0.75 < D′ < 0.89 and 0.51 < r2 < 0.67) (Fig. 2b and Supplementary Fig. 3a). In two-point logistic regression analyses conditioned on
rs1569723, the significant associations at the other SNPs disappeared (Supplementary Fig. 3b), indicating that the associations at the six SNPs were not independent of each other.
BLK is a Src family tyrosine kinase that transduces signals down-stream of the B-cell receptor. Expression of BLK is highly restricted to the B-cell lineage and is dependent on developmental stage17,18.
B-cell receptor signaling is important for establishing the B-cell rep-ertoire during development of these cells19 and has a critical role
in B-cell activation and antibody secretion. Genetic variants in the region upstream of the transcription initiation site of BLK have been associated with expression levels of BLK and increased risk of sys-temic lupus erythematosus (SLE)20 (rs13277113: OR = 1.39; P = 1 ×
10−10). The SLE-associated SNP rs13277113 was in strong LD with the
Kawasaki disease–associated SNP rs2736340 (D′ = 1 and r2 = 0.957
in the HapMap Japanese in Tokyo (JPT) and Han Chinese in Beijing (CHB) populations). Very recently, the rs2736340 SNP was shown to be associated with rheumatoid arthritis21 (P = 5.69 × 10−9; OR = 1.19).
Altered BLK protein levels could influence tolerance mechanisms during B-cell development and B-cell activation, predisposing indi-viduals to systemic autoimmunity. Our finding that BLK is associated with increased risk for Kawasaki disease suggests that autoimmu-nity and antibody-mediated immune responses may be involved in Kawasaki disease pathogenesis. We also observed that the distribution of rs2736340 in BLK alleles differs according to ancestry, in agreement with the prevalence of Kawasaki disease in Europeans and Asians. The frequency of the T allele is higher in Asians (0.68–0.77) and lower in Western Europeans (0.239) (based on 1000 Genomes Project data). Further elucidation of the role of BLK in Kawasaki disease susceptibility and its association with differences in Kawasaki disease prevalence among Europeans and Asians is required.
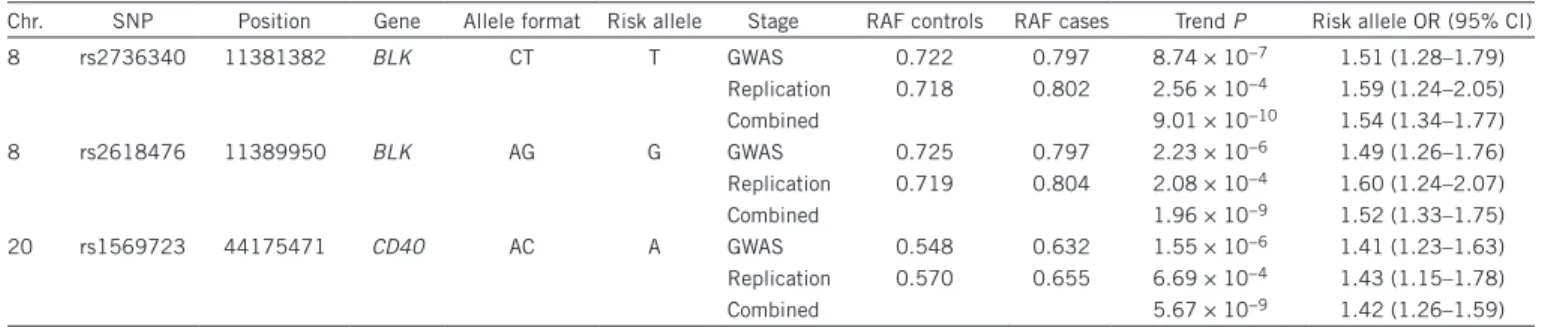
CD40 is a member of the tumor necrosis factor receptor (TNFR) superfamily. It is expressed on the surface of B cells and is inducibly table 1 Association analyses for the three SNPs reaching genome-wide significance in the joint analysis
Chr. SNP Position Gene Allele format Risk allele Stage RAF controls RAF cases Trend P Risk allele OR (95% CI)
8 rs2736340 11381382 BLK CT T GWAS 0.722 0.797 8.74 × 10−7 1.51 (1.28–1.79) Replication 0.718 0.802 2.56 × 10−4 1.59 (1.24–2.05) Combined 9.01 × 10−10 1.54 (1.34–1.77) 8 rs2618476 11389950 BLK AG G GWAS 0.725 0.797 2.23 × 10−6 1.49 (1.26–1.76) Replication 0.719 0.804 2.08 × 10−4 1.60 (1.24–2.07) Combined 1.96 × 10−9 1.52 (1.33–1.75) 20 rs1569723 44175471 CD40 AC A GWAS 0.548 0.632 1.55 × 10−6 1.41 (1.23–1.63) Replication 0.570 0.655 6.69 × 10−4 1.43 (1.15–1.78) Combined 5.67 × 10−9 1.42 (1.26–1.59)
Chr., chromosome; gene, genes containing the SNP or the closest gene up to 50 kb upstream or downstream of the SNP; RAF controls, risk allele frequency in controls; RAF cases, risk allele frequency in Kawasaki disease cases; OR, odds ratio; 95% CI, 95% confidence interval. Supplementary table 3 reports all SNPs with P < 1 × 10−4 in the
Kawasaki disease GWAS collection and with P < 0.05 in the Kawasaki disease replication collection and the results of the joint analysis.
Chr. 1 Chr. 9 Chr. 17 Chr. 2 Chr. 10 Chr. 18 Chr. 3 Chr. 11 Chr. 19 Chr. 4 Chr. 12 Chr. 20 Chr. 5 Chr. 13 Chr. 21 Chr. 6 Chr. 14 Chr. 22 Chr. 7 Chr. 15 Chr. 8 Chr. 16 –log 10 P value 7.25 BLK CD40 7.00 6.75 6.50 6.25 6.00 5.75 5.50 5.25 5.00 4.75 4.50 4.25 4.00 3.75 3.50 3.25 3.00 2.75 2.50 2.25 2.00 1.75 1.50 1.25 1.00 0.75 0.50 0.25 0
Figure 1 Results of genome-wide association analysis (−log10 P) shown in
chromosomal order for 716,935 SNPs tested for association in initial sample of 622 Kawasaki disease cases and 1,107 controls. The x axis represents each of the SNPs used in the primary scan. The y axis
represents the −log10 P value of the trend test. Horizontal lines indicate
−log10 P = 4 and 8. Signals in the BLK and CD40 regions are indicated.
l e t t e r S
npg
© 2012 Nature
America, Inc.
expressed on a variety of immune and nonimmune cell types22. CD40
potentially contributes to inflammation and autoimmune disease processes through the selection of autoreactive T cells in the thymus23
and the activation of B and T cells24. In addition, increased CD40
signaling leads to the production of proinflammatory cytokines and chemokines within targeted tissues, which contributes to tissue destruction and inflammatory cell influx. It has been proposed that aberrant expression of CD40 is a contributing factor for the initia-tion of autoimmunity in Graves’ disease25, type 1 diabetes26, multiple
sclerosis27, psoriasis28, Crohn’s disease29, rheumatoid arthritis30 and
SLE31. A functional polymorphism located −1 to the start codon of
CD40, rs1883832, was previously reported to alter the translation effi-ciency of CD40. This polymorphism was associated with increased risk of Graves’ disease32–34 and may have an effect on susceptibility
to rheumatoid arthritis35 and multiple sclerosis36. The significant
SNP (rs1569723) identified in the current study in the CD40 region was in strong LD with this functional polymorphism (D′ = 0.96 and
r2 = 0.93; in 1000 Genomes Project JPT and CHB data). Strategies for
alleviating these autoimmune diseases by inhibiting CD40 signaling have been explored37. There is increasing evidence that interactions
between CD40 and CD40 ligand on T lymphocytes and platelets have an important role in acute coronary syndrome38. Furthermore,
Kawasaki disease is characterized by overactivation of the immune system that specifically targets vascular endothelium, resulting in systemic vasculitis or even coronary artery aneurysm. Indeed, the expression of CD40 ligand on CD4+ T cells and platelets is associated
with coronary artery lesions and disease progression in Kawasaki disease39. These findings support our observation of an association
between CD40 and Kawasaki disease susceptibility and shed light on a possible mechanism for Kawasaki disease pathogenesis. We suggest that inhibiting CD40 signaling may be an effective strategy for treat-ing Kawasaki disease.
The strongest Kawasaki disease susceptibility loci identified to date are on chromosome 19q13 and at FCGR2A. The 19q13 region was initially identified in Japanese sibling pairs11. Subsequent
finer-scale mapping and further in vitro functional analysis identified a functional SNP (rs28493229, P = 1.2 × 10−8) in ITPKC that affects
ITPKC expression and T-cell activation and thus may be involved in Kawasaki disease susceptibility12. More recently, a large-scale
interna-tional study identified two SNPs that were associated with Kawasaki disease susceptibility16. One SNP was located in FCGR2A (rs1801274,
P = 7.35 × 10−11), and the other SNP was located upstream of the MIA
and RAB4B genes at 19q13 (rs2233152, P = 2.51 × 10−9). The
previ-ously identified functional SNP in ITPKC was also verified in that study and showed the strongest association with Kawasaki disease susceptibility (rs28493229, P = 1.68 × 10−12). These three SNPs were
genotyped in a portion of our GWAS cohort comprising 438 cases and 446 controls and showed nominal association with Kawasaki dis-ease (rs2233152, P = 0.0036; rs1801274, P = 6.30 × 10−4; rs28493229,
P = 1.50 × 10−4)16. In addition, we observed that rs10401344
(NUMBL), rs17713068 (SNRPA), rs2233152 (MIA) and rs10403040 (RAB4B) at 19q13 were suggestively associated with Kawasaki disease (all with P < 1 × 10−4) in the current GWAS (Supplementary Fig. 4
and Supplementary Table 2). To establish independent evidence for association, the results for these four SNPs in our previously reported GWAS samples15 and the additional individuals genotyped in the
current study are listed (Supplementary Table 6). Although three SNPs (rs10401344, rs17713068 and rs2233152) were genotyped in our replication samples, they failed to replicate; however, the low minor allele frequency (MAF; 0.06 in controls) in the small replica-tion sample size may have affected our ability to observe an associa-tion (Supplementary Table 7). Because the ITPKC, MIA and RAB4B genes in this region are plausible biological candidates for Kawasaki disease susceptibility, detailed resequencing of this region and func-tional studies are required to provide further information and to identify disease-modifying variants. Although we did not observe any SNPs in FCGR2A that reached the threshold (P < 1 × 10−4)
for evaluation in the replication collection in our GWAS, six SNPs located in FCGR2A did show an association with Kawasaki disease at P < 0.005. The association of these six SNPs in the previous genotyped samples and the additional individuals genotyped in the current study are given (Supplementary Table 8). Our new results provide further support for an association between Kawasaki disease and the 19q13 region but do not add support for the FCGR2A association.
Early genetic studies of Kawasaki disease were focused on major histocompatibility complex (MHC) antigens10. We examined
the MHC region in our GWAS, but we did not identify any SNP associated with P < 1 × 10−4 in this region. However, some SNPs
a
b
10 80 60 40 20 0 rs2736340 rs2618476 rs998683 rs998682 rs1478895rs1478900 rs6993775rs2618479 rs2736354 rs1382566rs1478897 rs17153419 Pjoint = 9.006 × 10–10 rs1569723 rs3746821 rs4813003 rs4810486 rs2868764 rs6131010 Pjoint = 5.668 × 10–9 Pjoint = 1.959 × 10–9 BLK 8 6 Observed –log 10 P value Recombination rate (cM/Mb) 80 60 40 20 0 Recombination rate (cM/Mb) 4 FAM107A SLC12A5 NCOA5 CD40 CD40 CDH22 BLK 2 0 10 8 6 Observed –log 10 P value 4 2 44,100 44,200 0 11,400 Chromosome 8 position (hg18) (kb) Chromosome 20 position (hg18) (kb) 0.8 0.5 r2 0.8 0.5 r2Figure 2 Association plots for the BLK and CD40 regions. (a,b) Regional
association plot, recombination rate and LD for the BLK region on
chromosome 8 (a) and the CD40 region on chromosome 20 (b), with
gene annotations superimposed. Each SNP is plotted with respect to its
chromosomal location (x axis) and its −log10 P values (left y axis) for the
trend test from the primary GWAS scan and joint analysis at that region of the chromosome. The results from the joint analysis for key SNPs are indicated with their rs numbers. The estimated recombination rates (right y axis) based on the combined JPT, CHB and Chinese in Denver (CHD) samples from the HapMap Project are plotted in light blue. The color of each SNP symbol reflects its LD (using the D′ algorithm) with the top SNP (large red diamond) within the association locus. D′ values were calculated using data from the GWAS study.
npg
© 2012 Nature
America, Inc.
Nature GeNetics VOLUME 44 | NUMBER 5 | MAY 2012 525
l e t t e r S
located in the MHC region did show nominal association (P < 0.01) (Supplementary Fig. 5). Whether the MHC has a role in Kawasaki disease susceptibility needs to be further investigated by traditional human leukocyte antigen (HLA) genotyping methods.
In summary, we have identified and replicated the BLK and CD40 regions as two new loci associated with increased Kawasaki disease susceptibility and confirmed the previously identified ITPKC locus. All of these candidate loci are involved in immune and inflammatory responses and therefore broadly fit the current consensus regarding Kawasaki disease pathogenesis. Both BLK and CD40 signaling path-ways are potential targets for the treatment of Kawasaki disease.
MeTHOds
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Note: Supplementary information is available on the Nature Genetics website.
ACKNoWLeDgMeNTS
We thank all affected individuals and their families who devoted their time and effort to participate in this study. We gratefully acknowledge the members of the Translational Resource Center (TRC) (NSC100-2325-B-001-023) for Genomic Medicine and the National Center for Genome Medicine (NCGM) (NSC100-2319-B-001-001) at Academia Sinica for their support in subject recruitment, genotyping and statistical analysis. We especially thank H. Lue for his inspirational discussion. This study was supported by the Academia Sinica Genomic Medicine Multicenter Study, Taiwan (40-05-GMM). The funders had no role in study design, data collection or analysis, the decision to publish or preparation of the manuscript.
AUTHoR CoNTRIBUTIoNS
Y.-T.C., F.-J.T. and J.-Y.W. are the principal investigators who conceived and obtained funding for this project. Y.-C.L., C.-H.C. and J.-Y.W. organized and supervised the GWAS and replication genotyping pipeline and devised the overall analysis plan. Y.-C.L. wrote the first draft of the manuscript with input from C.-H.C. and J.-Y.W. Y.-C.L., L.-C.C. and C.-H.C. analyzed the data. C.-D.L., J.-S.C., L.-Y.C., L.-M.H., M.-R.C., H.-C.K., H.C., F.-Y.H., M.-L.L., Y.-C.H., B.H., N.-C.C., K.-P.H., P.-C.L., Y.-M.L., Y.-J.C. and the Taiwan Pediatric ID Alliance coordinated and contributed subject and database phenotype collections.
CoMPeTINg FINANCIAL INTeReSTS
The authors declare no competing financial interests.
Published online at http://www.nature.com/naturegenetics/.
Reprints and permissions information is available online at http://www.nature.com/ reprints/index.html.
1. Kato, H., Koike, S., Yamamoto, M., Ito, Y. & Yano, E. Coronary aneurysms in infants and young children with acute febrile mucocutaneous lymph node syndrome.
J. Pediatr. 86, 892–898 (1975).
2. Kato, H. et al. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation 94, 1379–1385 (1996).
3. Yanagawa, H. et al. Results of the nationwide epidemiologic survey of Kawasaki disease in 1995 and 1996 in Japan. Pediatrics 102, E65 (1998).
4. Yanagawa, H. et al. Incidence survey of Kawasaki disease in 1997 and 1998 in Japan. Pediatrics 107, E33 (2001).
5. Park, Y.W. et al. Epidemiological features of Kawasaki disease in Korea, 2006–2008.
Pediatr. Int. 53, 36–39 (2011).
6. Chang, L.Y. et al. Epidemiologic features of Kawasaki disease in Taiwan, 1996–2002. Pediatrics 114, e678–e682 (2004).
7. Huang, W.C. et al. Epidemiologic features of Kawasaki disease in Taiwan, 2003–2006. Pediatrics 123, e401–e405 (2009).
8. Matsubara, T., Furukawa, S. & Yabuta, K. Serum levels of tumor necrosis factor, interleukin 2 receptor, and interferon-γ in Kawasaki disease involved coronary-artery lesions. Clin. Immunol. Immunopathol. 56, 29–36 (1990).
9. Lin, C.Y., Lin, C.C., Hwang, B. & Chiang, B. Serial changes of serum interleukin-6, interleukin-8, and tumor necrosis factor α among patients with Kawasaki disease.
J. Pediatr. 121, 924–926 (1992).
10. Onouchi, Y. Molecular genetics of Kawasaki disease. Pediatr. Res. 65, 46R–54R (2009).
11. Onouchi, Y. et al. A genomewide linkage analysis of Kawasaki disease: evidence for linkage to chromosome 12. J. Hum. Genet. 52, 179–190 (2007).
12. Onouchi, Y. et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat. Genet. 40, 35–42 (2008).
13. Burgner, D. et al. A genome-wide association study identifies novel and functionally related susceptibility loci for Kawasaki disease. PLoS Genet. 5, e1000319 (2009).
14. Kim, J.J. et al. A genome-wide association analysis reveals 1p31 and 2p13.3 as susceptibility loci for Kawasaki disease. Hum. Genet. 129, 487–495 (2011). 15. Tsai, F.J. et al. Identification of novel susceptibility loci for Kawasaki disease in a
Han Chinese population by a genome-wide association study. PLoS ONE 6, e16853 (2011).
16. Khor, C.C. et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat. Genet. 43, 1241–1246 (2011).
17. Dymecki, S.M., Zwollo, P., Zeller, K., Kuhajda, F.P. & Desiderio, S.V. Structure and developmental regulation of the B-lymphoid tyrosine kinase gene blk. J. Biol.
Chem. 267, 4815–4823 (1992).
18. Wasserman, R., Li, Y.S. & Hardy, R.R. Differential expression of the blk and ret tyrosine kinases during B lineage development is dependent on Ig rearrangement.
J. Immunol. 155, 644–651 (1995).
19. Nemazee, D. & Weigert, M. Revising B cell receptors. J. Exp. Med. 191, 1813–1817 (2000).
20. Hom, G. et al. Association of systemic lupus erythematosus with C8orf13-BLK and
ITGAM-ITGAX. N. Engl. J. Med. 358, 900–909 (2008).
21. Gregersen, P.K. et al. REL, encoding a member of the NF-κB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat. Genet. 41, 820–823 (2009).
22. Peters, A.L., Stunz, L.L. & Bishop, G.A. CD40 and autoimmunity: the dark side of a great activator. Semin. Immunol. 21, 293–300 (2009).
23. Akiyama, T. et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance.
Immunity 29, 423–437 (2008).
24. Iezzi, G. et al. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17–producing CD4+ T cells. Proc. Natl. Acad. Sci. USA 106, 876–881 (2009).
25. Jacobson, E.M. et al. A CD40 Kozak sequence polymorphism and susceptibility to antibody-mediated autoimmune conditions: the role of CD40 tissue-specific expression. Genes Immun. 8, 205–214 (2007).
26. Wagner, D.H. Jr. et al. Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc. Natl. Acad. Sci. USA 99, 3782–3787 (2002).
27. Gerritse, K. et al. CD40–CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA 93, 2499–2504 (1996).
28. Ohta, Y. & Hamada, Y. In situ expression of CD40 and CD40 ligand in psoriasis.
Dermatology 209, 21–28 (2004).
29. Danese, S. et al. TNF-α blockade down-regulates the CD40/CD40L pathway in the mucosal microcirculation: a novel anti-inflammatory mechanism of infliximab in Crohn’s disease. J. Immunol. 176, 2617–2624 (2006).
30. Brennan, F.M. & McInnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Invest. 118, 3537–3545 (2008).
31. Pyrovolaki, K. et al. Increased expression of CD40 on bone marrow CD34+
hematopoietic progenitor cells in patients with systemic lupus erythematosus: contribution to Fas-mediated apoptosis. Arthritis Rheum. 60, 543–552 (2009). 32. Tomer, Y., Concepcion, E. & Greenberg, D.A. A C/T single-nucleotide polymorphism
in the region of the CD40 gene is associated with Graves’ disease. Thyroid 12, 1129–1135 (2002).
33. Ban, Y., Tozaki, T., Taniyama, M. & Tomita, M. Association of a C/T single-nucleotide polymorphism in the 5′ untranslated region of the CD40 gene with Graves’ disease in Japanese. Thyroid 16, 443–446 (2006).
34. Kurylowicz, A. et al. Association of CD40 gene polymorphism (C-1T) with susceptibility and phenotype of Graves’ disease. Thyroid 15, 1119–1124 (2005).
35. Raychaudhuri, S. et al. Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat. Genet. 40, 1216–1223 (2008).
36. Blanco-Kelly, F. et al. CD40: novel association with Crohn’s disease and replication in multiple sclerosis susceptibility. PLoS ONE 5, e11520 (2010).
37. Law, C.L. & Grewal, I.S. Therapeutic interventions targeting CD40L (CD154) and CD40: the opportunities and challenges. Adv. Exp. Med. Biol. 647, 8–36 (2009). 38. Hassan, G.S., Merhi, Y. & Mourad, W.M. CD154 and its receptors in inflammatory
vascular pathologies. Trends Immunol. 30, 165–172 (2009).
39. Wang, C.L. et al. Expression of CD40 ligand on CD4+ T-cells and platelets
correlated to the coronary artery lesion and disease progress in Kawasaki disease.
Pediatrics 111, E140–E147 (2003).
npg
© 2012 Nature
America, Inc.
Board and the Ethics Committee of the Institutional Review Board of China Medical University Hospital, National Taiwan University Hospital, Changhua Christian Hospital, Taipei Veterans General Hospital, Kaohsiung and Linkou Chang Gung Memorial Hospital, Mackay Memorial Hospital and Academia Sinica, Taiwan. Written informed consents were obtained from the subjects’ parents in accordance with institutional requirements and Declaration of Helsinki principles.
Study subjects and phenotype definition. Individuals with Kawasaki disease
(n = 627) (including the 250 Kawasaki disease cases in the GWAS and the 208 Kawasaki disease cases in the replication study in our previous study15,
which were also used in the international replication study16) were
con-secutively recruited in Taiwan from the China Medical University Hospital in Taichung, the National Taiwan University Hospital in Taipei, Changhua Christian Hospital in Changhua, Taipei Veterans General Hospital in Taipei and Chang Gung Memorial Hospital in Kaohsiung and Linkou in collabora-tion with the Translacollabora-tional Resource Center (TRC) for Genomic Medicine of Taiwan. The 261 Kawasaki disease cases in the replication study were also recruited from these hospitals. All of the cases were diagnosed according to criteria for Kawasaki disease40,41 and were recruited as in our previous report.
The 1,118 control subjects in the GWAS and the 564 control subjects in the replication study were randomly selected from the Taiwan Han Chinese Cell and Genome Bank in Taiwan, as reported previously42. The prevalence of
Kawasaki disease in the Taiwanese population is less than 0.01%; hence, the controls were presumably disease free. The demographic and clinical char-acteristics of participants in the GWAS and replication study after kinship filtering are listed (Supplementary Table 9).
Genotyping and quality control. Genomic DNA was extracted from blood
using the Puregene DNA Isolation Kit (Gentra Systems). Each individual was genotyped using the Affymetrix Genome-Wide Human SNP Array 6.0 (with a total of 906,600 SNPs) according to the manufacturer’s protocols by the National Center for Genome Medicine (NCGM) at Academia Sinica. All of the sample call rates were >98%, and the mean individual sample call rate was 98.4 ± 0.7%. First-degree relatives (parent-offspring and full sibling pairs) in Kawasaki disease cases and in the control samples were identified by kinship analysis and were excluded from further analysis. Genotyping quality control
analysis if only one allele appeared in cases and controls, the total call rate was <0.95 or the total MAF was <0.05 and the total call rate was <0.99. In addi-tion, SNPs that departed significantly from Hardy-Weinberg equilibrium were excluded (P < 1 × 10−4).
Statistical analysis. Detection of possible population stratification that could
influence association analysis was carried out using EIGENSTRAT 2.0 to con-duct PCA43. We also estimated the variance inflation factor for genomic
con-trol. Genome-wide association analysis was carried out to compare allele and genotype frequencies between cases and controls using the Cochran-Armitage trend test. A quantile-quantile plot was used to examine the P value distribu-tion (Supplementary Fig. 1f). Two-point analyses were performed using a logistic regression model, regressing the affected status of two SNPs and their interaction. SNPs were coded as 0, 1 and 2 for the number of minor alleles and were treated as continuous variables. Heterogeneity tests (I2 and P values
of the Q statistics) between GWAS and replication groups were performed using described methods44.
Validation and replication. The top SNPs (P < 1 × 10−4) from the
genome-wide association analysis of the 622 Kawasaki disease cases and 1,107 controls were further validated in 94 controls and 188 Kawasaki disease cases using MALDI-TOF mass spectrometry (MassARRAY, Sequenom), and the SNP genotypes with over 99% successful rate and over 99% concordance between two platforms were then genotyped in an additional 261 Kawasaki disease cases and 564 controls for replication.
40. Newburger, J.W. et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics 114, 1708–1733 (2004). 41. Kim, S. & Dedeoglu, F. Update on pediatric vasculitis. Curr. Opin. Pediatr. 17,
695–702 (2005).
42. Pan, W.H. et al. Han Chinese cell and genome bank in Taiwan: purpose, design and ethical considerations. Hum. Hered. 61, 27–30 (2006).
43. Price, A.L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
44. Higgins, J.P. & Thompson, S.G. Quantifying heterogeneity in a meta-analysis.
Stat. Med. 21, 1539–1558 (2002).
npg
© 2012 Nature
America, Inc.