行政院國家科學委員會專題研究計畫 成果報告
應用近紅外光技術於幾丁類物質去乙醯率之檢測
計畫類別: 個別型計畫 計畫編號: NSC92-2313-B-002-106- 執行期間: 92 年 08 月 01 日至 93 年 07 月 31 日 執行單位: 國立臺灣大學生物產業機電工程學系暨研究所 計畫主持人: 陳世銘 共同主持人: 陳力騏 計畫參與人員: 蔡志成、楊宜璋、蕭賢義、陳加增、楊翕雯 報告類型: 精簡報告 處理方式: 本計畫可公開查詢中 華 民 國 94 年 5 月 10 日
應用近紅外光技術於幾丁類物質去乙醯率之檢測
陳世銘、蔡志成、陳力騏、楊宜璋、蕭賢義、陳加增、楊翕雯
國立臺灣大學生物產業機電工程學系
摘 要
幾丁類物質可用來生產高附加價值之生醫材料,但其產製過程中之純度控 制,常常是影響產品品質的重要環節。本研究以近紅外光檢測技術,針對幾丁質 (chitin)與幾丁聚醣(chitosan)之粉末、懸浮液與幾丁聚醣皮膜進行光譜掃描 分析,建立幾丁質與幾丁聚醣之近紅外光定量分析模式。定量分析之研究則以多 重線性迴歸(MLR)、修正部份最小平方迴歸(MPLSR)二種模式來探討並分析 幾丁類物質之去乙醯率檢測,並藉以改善幾丁類物質製程之品管監控。 以幾丁質混合粉末模擬不同去乙醯率之樣本,在 MLR 模式分析所挑選的重 要分析波長為 1528,1592,1662,1692,2022 及 2102 nm。去乙醯反應後之粉 末樣本的 MLR 檢量線分析結果,以二次微分的六波長組合(898,1402,2310, 428,1990 及 2016 nm)為最佳,rc=0.996,SEC=0.013,RSEC=1.0%,rp=0.996, SEP=0.015,RSEP=1.3%,RPD=13.8。在粉末幾丁類物質產品的近紅外光去乙醯 率測定,此為最佳之檢量線預測模式。 幾丁質在去乙醯化過程中,即時量測懸浮液樣本光譜的分析結果,以使用 部份特徵波段 600∼1000 nm 與 1400∼1500 nm 之二次微分光譜 MPLSR 分析, 可 得 最佳 光譜 檢量 線, 其 rc=0.991 , SEC=0.019 , RSEC=1.4% , rp=0.990 , SEP=0.022,RSEP=3.4%,RPD=9.4,此模式可作為幾丁質製備幾丁聚醣即時線 上檢測及品質監控之應用。 去乙醯和尚蟹皮膜的分析結果在濕膜的最佳檢量線為全波段(400∼2500 nm)光譜之二次微分的 MPLSR 分析結果,rc=0.885,SEC=0.065,rp=0.800, SEP=0.076 。去乙醯和尚蟹皮膜之乾膜分析結果以 MPLSR 全波段二次微分 rc=0.932,SEC=0.052,rp=0.921,SEP=0.056 為預測去乙醯率之最佳檢量線模式。 從廢棄蟹殼純化出幾丁質與幾丁聚醣,並應用近紅外光技術快速檢測其去乙醯 率,在水產資源的利用層面而言是非常有價值的。 本研究已成功的建立幾丁類物質之去乙醯率檢測模式,包括幾丁質與幾丁 聚醣粉末之去乙醯率近紅外光檢測模式、幾丁質在製備幾丁聚醣的去乙醯化過程 中,樣本即時檢測去乙醯率的近紅外光檢測模式,以及去乙醯和尚蟹皮膜濕膜與 乾膜去乙醯率檢量線的建立。 【關鍵詞】: 近紅外光、幾丁質、幾丁聚醣、去乙醯率The Study of Deacetylation in Chitinous Materials Using Near
Infrared Spectroscopy
Suming Chen, Chih-Cheng Tsai, Richie L. C. Chen, I-Chang Yang, Hsien-Yi Hsiao, Chia-Tseng Chen, Ci-Wen Yang
Dept. of Bio-industrial Mechartronics Engineering National Taiwan University.
ABSTRACT
Chitinous materials are the important sources for bio-medical applications, and the process monitoring is one of key factors for the quality control of products. In this study, chitin and chitosan in powder, suspension and membrane forms were analyzed using near infrared (NIR) spectroscopy. Quantitative models were developed for determining the content of chitin and chitosan. Two models including multiple linear regression, modified partial least square regression were adopted for studying the degree of deacetylation in chitinous materials in order to assure a better quality monitoring for chitosan production.
The spectrum analysis of the chitin and chitosan powder mixture exhibited absortion wavelengths at 1528, 1592, 1662, 1692, 2022 and 2102 nm by MLR (Multiple Leaner Regression)model. The best results of deacetylated chitinous powder from suspension liquid were given by MLR with six wavelengths combination ( 898, 1402, 2310, 428, 1990 and 2016 nm ) and the second derivative treatment, which were rc=0.993, SEC=0.013, RESC=1.0%, rp=0.996, SEP=0.015,
RSEP=1.3%, RPD=13.8. For the determination of the degree of deacetylation of
chitinous powder, the above model is the best prediction model.
During the process of the deacetylation, the real-time measurement of suspension was adopted. The MPLSR(Modified Partial Least Squares Regression) model with second derivative spectra in the range of 600~1000 and 1400~1500 nm yielded the best results, which were rc=0.991, SEC=0.019, RESC=1.4%, rp=0.990,
SEP=0.022, RSEP=3.4%, RPD=9.4. This model could be used for real-time and on-line DD determination in preparing chitosan from suspension.
The degree of deacetylation calibration model of the Mictyris brevidactylus membrane was MPLSR method with second derivative spectra in the range of 400~2500 nm, and its results are rc=0.885, SEC=0.065, rp=0.800, SEP=0.076 for wet
membrane, while rc=0.932, SEC=0.052, rp=0.921, SEP=0.056 for dried membrane.
materials from crab shell, which is regarded as waste.
This study had successfully developed the models for chitinous DD determination using NIR Spectroscopy, including DD determination model for chitin and chitosan powder, the real-time DD determination model for the chitinous suspension, and the DD determination model for Mictyris brevidactylus membrane in dried and wet state.
一、 前 言
幾 丁 質 是 由 葡 萄 糖 胺 ( glucosamin ) 及 N– 乙 醯 葡 萄 糖 胺 (N–acetlglucosamine),以 ß–1,4 鍵結而成的直鏈狀高分子多醣聚合物,其結構 近似於纖維素,從甲殼類的蝦蟹、昆蟲、植物的藻類、菌類細胞壁上都含有幾丁 質的構造與組織,資源蘊藏量為地球第二豐富的天然聚合物。幾丁質去乙醯率達 40%以上時,即可稱為幾丁聚醣。 幾丁質不溶於水、稀酸、鹼、酒精與其他有機溶劑中之特性,使應用性受到 限制。而為了提高幾丁質的應用性,可將蝦蟹甲殼純化所得的幾丁質經高溫熱鹼 的去乙醯處理而獲得幾丁聚醣,其多項優點(生物相容性、生物再製性、生物分 解性、生物活性)更勝於幾丁質,它是一種天然無毒性之高分子聚合物,在醫藥、 工業、農業、食品、生物技術研究上都有其應用的例子存在,如醫藥方面可應用 於傷口之繃帶、降低膽固醇;工業方面可當作楊離子交換樹脂、用於水質淨化系 統;農業方面可當殺蟲劑或農產品保鮮添加劑;食品應用上主要作為果汁澄清 劑、魚漿的凝結劑;生物技術可應用在酵素及細胞的固定化擔體、分離及純化蛋 白質。 隨著去乙醯率的提高,幾丁聚醣則可溶於稀酸溶液中,其應用性也比幾丁質 更為廣泛(Ravindra et al.,1998)。Nil(1996)提到幾丁聚醣可當作膳食纖維, 降低膽固醇及預防高血壓,增加腸內有益菌生長及重金屬排泄,提高人體免疫 力。Tomihata et.al.(1997)研究在老鼠的背部皮膚下層植入幾丁質/幾丁聚醣的 皮膜,觀察其生物降解性,發現與去乙醯率達 73.3 mol %的幾丁聚醣比較,幾丁 質和去乙醯率為 68.8 mol %的幾丁聚醣在老鼠體內的生物降解性很高。高去乙醯 率的幾丁聚醣與金屬離子的結合能力較好,可應用於污水處理方面(Chu, 2002 ); 在 薄 膜 製 程 上 , 高 去 乙 醯 化 程 度 的 幾 丁 質 所 製 備 的 薄 膜 較 為 堅 韌 (Nunthanid et al.,2001);另外,去乙醯率也關係著酵素結合能力(Ylitalo et al., 2002)。幾丁質的來源一般是以蝦蟹類的甲殼經過純化後得到,若欲得到不同去乙醯 率之幾丁聚醣可將幾丁質經高溫熱鹼的去乙醯化處理而得。Chang et al.(1997), Methacanon et al.(2003),以不同 NaOH 濃度、不同加熱時間、及不同溶液樣本 含量比例探討高溫熱鹼處理對去乙醯率的影響,結果顯示去乙醯率主要受溫度與 NaOH 濃度的影響,其中溫度可以提高去乙醯率的反應速度,NaOH 濃度則決定 了去乙醯率的高低。
Chang et al.(1997)研究異質幾丁質在熱鹼處理的去乙醯率過程中,各項反 應條件對異質幾丁質的去乙醯率影響,結果顯示幾丁類物質的去乙醯效率不受異
質幾丁質之影響,去乙醯率之計算如式 1。 DD= × + 203 161 161 Y X X 100 (mol %) (1)
X:葡萄胺糖質量(the weight of D-glucosamine residues)
Y: N-乙醯葡萄糖質量(the wight of N-acetyl-D-glucosamine residues in the N-deacetylated chitin) 其中 161 為葡萄胺糖單體分子量,203 為 N-乙醯葡萄糖單體分子量。 而幾丁質/幾丁聚醣產品去乙醯率的測定方法有很多,如紅外光光譜法 (IR)、核磁共振光譜法(NMR)、酸水解-高效能液相層析法(HPLC)、膠體滴 定法、苦味酸法、紫外光光譜法(UV)。這些測定方法各有其優缺點,如膠體滴 定法操作簡單但準確度欠佳。紅外光光譜法(IR)其優點為操作簡單、測定快速 與樣品用量少,但有基線飄移之缺點,容易影響其測定結果。Ottøy et al.(1996) 以核磁共振法測定固態樣本之去乙醯率,其優點為不須複雜的前處理步驟,且不 用擔心樣本無法溶解的問題,因此準確率最高,但 NMR 儀器設備昂貴非一般實 驗室方便使用之技術;酸水解-高效能液相層析法會有樣本無法溶解的問題,不 適合大範圍去乙醯率的量測;苦味酸法沒有樣本不溶解的問題,但是它測定費時 且過程繁複。(Shigemasa et al.,1996;Marvão et al.,1998;Vårum et al.,1991; Raymond et al.,1993;Heux et al.,2000;Martinou et al.,1995;Tan et al.,1998)。
13C CPMAS NMR 檢測技術已經證實是測定幾丁質/幾丁聚醣去乙醯率非常 重要且有力的一項工具,不論固態樣本能不能溶解都可以直接量測,且不須繁複 的樣本前處理過程。Harish et al.(2002)利用 13C NMR 檢測技術研究幾丁聚醣 在不同 N-deacetylation 方式的固態分子結構。Duarte et al.(2001)以 13C CPMAS NMR 做幾丁類物質去乙醯率的定量分析研究。Ottøy et al.(1996)用 13C CPMAS NMR 來測定幾丁聚醣中 acid-insoluble 部份的 FA 值(乙醯化程度),並且比較 13C NMR 與 1H NMR 測定 acid-soluble 部份的 FA 值,發現測定結果一致;NMR 測定幾丁聚醣的乙醯化程度由分子中的碳環(IC1-C6)與甲基的相對共振強度計 算得到,計算式(2)、(3)。 乙醯化程度 6 / ) ( 1 2 3 4 5 6 3 C C C C C C CH A I I I I I I I F + + + + + = (2) DD(%)=(1-FA)× 100 (3) ICH3:乙醯分子中之甲基吸收峰面積
IC1-6 : 碳環之吸收峰面積,將碳環上 6 個碳原子的總吸收峰面積除以 6 即 表示幾丁聚醣分子中胺基與乙醯基的總含量
本研究探討和尚蟹皮膜去乙醯率的檢測法,因此在標準測定方法的選擇以紫 外光法量測和尚蟹皮膜之去乙醯率,紫外線光譜法使用的原理與近紅外光理論相 同,皆是利用溶液中待測物的濃度 c 與吸收度A(或穿透率 T)呈線性關係,並 以比耳定律(Beer’s law)的數學式表示:A=-log T=e× b × c,其中 e 為莫耳 吸光係數,b 為光在透明溶液槽中行走的光路徑。紫外光光譜區一般波長範圍是 從 160∼380 nm,可定性與定量分析無機物及有機化合物(Tan et al.,1998)。
近紅外光檢測技術為非破壞性,測定時間快速且可作為線上製程分析之儀 器。在過去的研究中,NIR 技術成功的應用在檢測水分含量,蛋白質,脂肪,酸 洗纖維。陳等人(1998)在果汁糖度檢測模式一文中,應用多重線性迴歸(Multiple Linear Regression,MLR)、部份最小平方迴歸(Partial Least Square ,PLSR)與 類神經網路(Artificial Neural Network,ANN)三種分析模式探討不同的光譜處 理(原始光譜、一次微分光譜、二次微分光譜)對果汁糖度檢測的影響。該研究 中所探討的檢測模式是針對所得到的果汁光譜,以不同的光譜處理模式(MLR、 PLSR、ANN)做討論比較,找出最佳的檢測模式。不同的檢測模式所得到的最 佳光譜處理步驟並不相同,所以應該配合所建立的檢測模式,選用最適合的光譜 處理。 由於幾丁類物質去乙醯率的測定在大部分的方法中皆須經過溶解的步驟,但 是在低去乙醯率的樣本常常發生無法溶解的問題,故而限制了樣本的量測範圍。 因此本研究將採用近紅外光技術非破壞性的檢測優點,對幾丁類物質之去乙醯率 進行光譜分析,希望能建立快速,簡單的幾丁類物質產品去乙醯率檢測模式。 本研究以近紅外光檢測技術,針對幾丁質(chitin)與幾丁聚醣(chitosan) 之粉末、懸浮液與和尚蟹皮膜進行光譜掃描分析,建立幾丁質與幾丁聚醣之近紅 外光定量分析模式。定量分析之研究則以多重線性迴歸(MLR)、修正部份最小 平方迴歸(MPLSR)二種模式來探討並分析幾丁類物質之去乙醯率檢測,並藉 以改善幾丁類物質製程之品管監控。去乙醯率之標準值之建立,去乙醯過程之懸 浮液、粉末以 NMR 方法測定,而和尚蟹皮膜則以紫外光法測定。
二、 實驗材料與方法
本研究利用近紅外光檢測技術,針對幾丁類物質進行光譜分析,探討幾丁質 製備幾丁聚醣過程中,各階段產物(粉末、溶液、薄膜)之光譜吸收特性,建立 各型態幾丁類物質之去乙醯率檢測模式,包括粉末、溶液、薄膜三種不同形態,並建立光譜資料庫。本研究採用之樣本,按照幾丁質製備幾丁聚醣的過程分別量 測幾丁質與幾丁聚醣粉末原料吸收度光譜、去乙醯反應中之溶液吸收度光譜、去 乙醯後之幾丁聚醣粉末吸收度光譜、以及和尚蟹製備之天然幾丁聚醣薄膜,圖 1 為實驗流程圖,說明近紅外光檢測技術在幾丁質去乙醯過程及最終產物幾丁類物 質的檢測模式。圖 2 為幾丁質與幾丁聚醣轉換反應示意圖,幾丁質在高溫(反應 溫度須在 90℃以上)之鹼液(Base)中去乙醯化反應才會進行,且幾丁質與幾 丁聚醣不溶於鹼中;在酸液或酵素(Acid/Enzy)中則會使幾丁質長鏈分子結構 降解為小分子單體 N-Acetyl-D-glucosamine(N-乙醯葡萄糖胺)以及 D-glucosamine (葡萄糖胺);藉由乙醯化(N-Acetylation)處理的方式可將幾丁聚醣還原為幾 丁質。依照實驗流程圖 1 之架構,可將實驗分為四部份: 第一部份:購自 Wako 藥品公司之幾丁質與幾丁聚醣粉末,以混合的方式模 擬不同去乙醯率之粉末樣本。由粉末反射光量測可以得知幾丁質與幾丁聚醣之基 本基本光譜性質,並找出 N-乙醯基(N-CH3COOH)與胺基(NH2)之特徵波長。 第二部份:以去乙醯反應過程中之幾丁質懸浮溶液為樣本,建立幾丁質去 乙醯反應之即時檢測模式,並以此溶液之光譜資料庫作為將來線上檢測之依據。
圖 1 實驗操作流程圖
第三部份:以去乙醯反應後之幾丁聚醣粉末為樣本,建立不同去乙醯率粉末 樣本檢量線,以供幾丁聚醣產品一套快速檢測去乙醯率的模式。 第四部份:以實驗室製備之和尚蟹去乙醯皮膜為樣本,探討薄膜光譜之吸收 特性,並建立薄膜樣本去乙醯率檢測模式。 Chitin 粉末 熱鹼 處理 清洗 烘乾 和尚蟹 去乙醯皮膜 Chitin (去乙醯) 粉末量測NIR光譜 量測NIR光譜 量測NIR光譜
建立即時 檢測模式 建立粉末 檢測模式 建立薄膜 檢測模式
在建立近紅外線光譜檢測模式過程中,主要可分為兩部份。第一部份是建立 實驗數據,其中又可分為近紅外線光譜的取得以及欲檢測對象物之化學成份值, 化學成份值量測儀器為 NMR 核磁共振儀。光譜之取得依對象物的性質而採用不 同量測方式的近紅外光分光光度計,幾丁類物質的粉末與薄膜之樣本由於取樣容 易,故採用實驗室型分光光度計(NIRS 6500)量測光譜;幾丁質去乙醯過程溶 液則採用線上型分光光度計(on-line NIRS 6500)量測樣本光譜,因為去乙醯過 程反應係在高溫熱鹼下進行故危險性較高,須採用非接觸式的操作方式取得光譜 資料。第二部份則是建立校正方程式,校正方程式的種類來自於數學分析模式, 主要有 MLR 與 MPLSR 兩種檢量線分析模式,分析程式為 FOSS NIRSystems 公 司所出的 WinISI 軟體(Foss NIRSystems, 1999)。
在使用 NIRS 6500 時,因其不同的量測方法,需要搭配不同的配件。由於實 驗室型分光光度計在本研究中扮演的角色為幾丁質粉末反射光譜量取與薄膜透 射光譜的量取,因此所選擇之搭配配件為樣本自動傳送配件。自動傳送配件亦可 依夾具的不同選擇不同的樣本容器(mini-cup,small ring cup 等),在本研究所使 用的夾具為粉末槽夾具。其中粉末夾具所使用之容器為半徑 2.5 cm 的圓形容器, 粉末樣本可置於圓形容器中進行反射光譜量測;薄膜量測方式是將薄膜樣本固定 在圓形容器中間的小孔中,不同於粉末量測的地方是圓形容器不加背蓋,再放置 於自動傳送配件使其可做透射光譜之量測。去乙醯過程溶液光譜之量測則以培養 皿承裝溶液樣本,並置於線上型分光光度計中量測反射光譜。 CH2OH NH2 O O H OH O H D-glucosamine O O H CH2OH NH CO CH3 O O CH2OH NH CO CH3 O O H CH2OH NH CO CH3 O O H Chitin O O H CH2OH NH2 O O CH2OH NH2 O O H CH2OH NH2 O O H Chitosan CH2OH NH CO CH3 O O H OH O H N-Acetyl-D-glucosamine Bas Acid/E N-Acet Acid/E
圖 2 幾丁質與幾丁聚醣之轉換反應示意
樣本製備與光譜掃描
(1) 不同去乙醯率的混合粉末樣本 樣本製備:購自 Wako 藥品公司之幾丁質,038-13635 幾丁聚醣粉末 chitosan 10TM,100 kDa,039-16122 chitosan 100 TM,1000 kDa,032-16092 chitosan 500 TM,5000 kDa,032-14412 chitosan 1000 TM,10000 kDa,039-14422 實驗目的:配製不同去乙醯率的混合粉末樣本與光譜掃描 68 個樣本去乙醯率的分布範圍 27.6∼89.8 mol%。 (2) 去乙醯過程懸浮液樣本 樣本製備:購自 Wako 藥品公司之幾丁質粉末購自 Niho Shiyaku 藥品公司之 Sodium Hydroxide,SK371842 實驗目的:量測幾丁質去乙醯過程懸浮液之近紅外光光譜 配置濃度 40 %~60 %(w/v %)之 NaOH 溶液,稱取 Chitin 粉末 2 克 重,放入血清瓶中(可不攪拌),放回烘箱裡加熱,將去乙醯反應進行中之 NaOH 幾丁質去乙醯反應溶液,在每隔 15 分鐘的加熱時間間隔後(為了使 樣本去乙醯率涵蓋每個百分比程度,每次實驗取樣之間隔時間,依照實驗 的 NaOH 濃度而改變,取其懸浮溶液倒在直徑 9 cm 之培養皿裡。以 on-line NIRS 6500 量測去乙醯反應進行中之懸浮液樣本反射光譜,反射光譜為 16 次掃描後輸出之吸收度光譜,每個樣本取 5 次重複量測反射光譜。 (3) 以去乙醯反應後之幾丁聚醣粉末為樣本 樣本製備:去乙醯反應終止後之幾丁聚醣與 NaOH 混合液 Whatman 1 號濾紙,90 mmψ 抽氣過濾馬達 抽氣漏斗 實驗目的:量測去乙醯反應後之幾丁聚醣粉末近紅外光光譜 將量測完光譜之溶液樣本以去離子水稀釋冷卻,終止去乙醯化反應。以去 離子水反覆沖洗至中性,勿使 NaOH 殘留,影響去乙醯率之分析及近紅外光 譜量測。將清洗完之幾丁質粉末放入烘箱中以 50℃乾燥 24 小時,即可得到 自行製備之不同去乙醯率的粉末樣本。取 0.5 克之去乙醯率的粉末樣本,以 NIRS 6500 取得粉末樣本反射光譜,每個樣本取三重複量測反射光譜,在每 次量測完光譜後將樣本倒回容器中充分攪拌均勻,再取出做下一次的光譜量
測。取得樣本近紅外光譜後,每個樣本稱取 1 克之量送至成功大學貴重儀器 中心,以13 C Solid-NMR 量測樣本頻譜圖。 (4) 和尚蟹皮膜去乙醯處理 樣本製備:實驗室製備之和尚蟹去乙醯皮膜 購自 NS 藥品公司之 NaOH,試藥特級,SK371842 抽氣過濾馬達 抽氣漏斗 實驗目的:和尚蟹皮膜去乙醯處理與近紅外光譜量測 取和尚蟹皮膜,以熱鹼處理(50 % NaOH,100℃)製備不同去乙醯率之 幾丁聚醣皮膜樣本。將去乙醯反應進行中之和尚蟹皮膜,在每隔 15 分鐘的加 熱時間間隔取出 3∼5 個皮膜樣本,置於去離子水中冷卻並終止去乙醯化反 應,實驗所得共 60 個去乙醯化和尚蟹皮膜樣本。 利用 NIRS 6500,量測和 尚蟹溼膜之透射光譜,每個樣本取五重複量測透射光譜。將和尚蟹溼膜放入 烘箱中乾燥(50℃,24 小時)。和尚蟹乾膜以 NIRS 6500 進行反射光譜量測, 每個樣本取三重複量測反射光譜。以紫外光法量測樣本之去乙醯率作為標準 值。
三、 模式分析
在分析模式的精準度方面,主要是比較實驗值與預測值,看二者之間的關係 來判定校正方程式的好壞,在本研究中用來判斷檢量線模式優劣的,除了相關係 數 r 值外,還有以下幾項統計指標,只要比較各模式的統計指標,就可清楚知道 模式的適用程度及優劣。 標準校正誤差(SEC)= 1 ) ( 1 − − −∑
= m n Yr Yc n i i i (4) 相對校正誤差(RSEC)= 1 ) ( 1 − − −∑
= m n Yr Yr Yc n i i i i (5) 標準預測誤差(SEP)= 1 ) ( 1 2 − −∑
= n Yr Yp n i i i (6) 相對預測誤差(RSEP)= 1 ) ( 1 − −∑
= n Yr Yr Yc n i i i i (7) RPD= SEP SD (8) 其中,n=樣本個數, m= MLR 模式之波長數目 Yr=實驗值, Yc=校正值, Yp=預測值, SD=預測組之標準差 在上述統計式中,RPD 值要越大越好,而 SEC、SEP 要越小越好。3-1 MLR 模式分析(Multiple Leaner Regression)
多重線性迴歸 (Multiple Leaner Regression),應用在光譜分析中,為一個至 數個選定波長之光譜吸收值所組成的線性迴歸分析模式,用來預測待測物之化學 成份值。由於其原理簡單易懂,因此在近紅外線光譜領域中是最常被用來當作檢 測之分析模式。一般 MLR 多重線性迴歸方程式如下, Y=K0+
∑
= n i i iX K 1 λ (9) Y=待測成份值。(本研究為去乙醯化程度值) Xλi=方程式變數。(本研究為波長λi 之吸收度) Ki=方程式常數。 在特徵波長選取方面,此研究所使用之分析軟體 WinISI 提供了 Step-up 與 Step-wise 二種不同之波長選擇方式來建立迴歸檢量線。其中 Step-up 是由使用者 來選擇相關性較高之波長,再與待測成份值進行迴歸,建立校正方程式。而 Step-wise 則是以淨偏相關係數及偏 F 統計量作為標準,以決定波長是否採用, 進而與待測成份值建立校正方程式。 當 MLR 模式為單波長組成,一般而言代表其波長吸收度為此光譜最具代表 性數值;而當 MLR 模式為數個波長所組成,這些波長的吸收度也都應該是此光 譜之重要訊息提供者。因此被選取來建立 MLR 模式之波長,最好是符合待測成 份之主要吸收波峰,而且在同一吸收波峰附近,最好避免選取太多波長,如此才 算較佳之近紅外光 MLR 分析模式。3-2 MPLSR 模式分析(Modified Partial Least Squares Regression)
修正部份最小平方迴歸(Modified Partial Least Squares Regression),其主要特 點在於光譜正規化,其餘原理與 PLSR(Partial Least Squares Regession)部份最 小平方迴歸雷同,也因此通常其預測效果比 PLSR 更佳。應用在光譜分析中,為 一段或數段波長範圍之光譜吸收值所組成的線性迴歸分析模式,用來預測待測物 之化學成份值。與 MLR 模式之最大不同處在於,MLR 模式是採用少數幾個特定 波長之資訊,而 MPLSR 模式則是選用整段波長範圍內之所有波長資訊。由於 MPLSR 模式以不失去整段光譜資訊為原則,而且一般比 MLR 有更好之分析結 果,因此在近紅外線光譜領域中已慢慢躍昇為常用之檢測分析模式,其使用頻率 也漸漸趕上 MLR 模式。
四、結果與討論
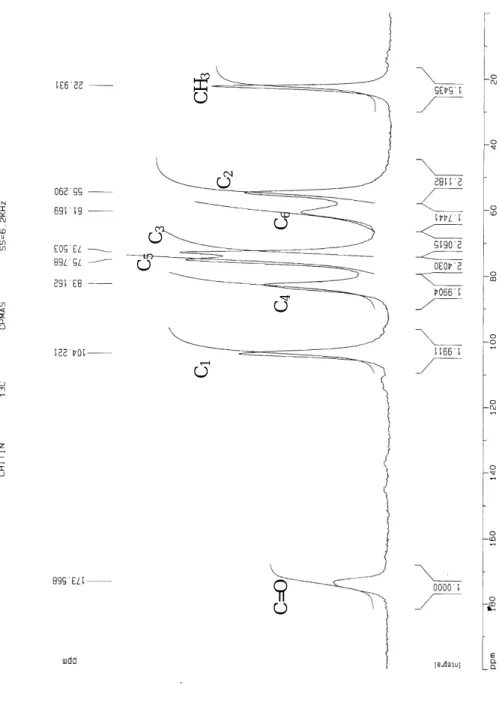
研究結果分為四個部份,首先對幾丁聚醣去乙醯率之 NMR 檢測結果做分析 討論,證明去乙醯率檢測的準確度。第二部份以混合粉末樣本的分析結果討論幾 丁類物質之近紅外光特徵波長範圍,以及去乙醯反應後之粉末樣本建立幾丁類物 質粉末去乙醯率檢測模式。第三部份則為分析幾丁質去乙醯過程懸浮液之光譜, 並建立即時的線上去乙醯率檢測模式。第四部份以實驗室製備之和尚蟹去乙醯皮 膜,分析濕膜與乾膜之近紅外光吸收光譜,以及建立薄膜去乙醯率檢測模式。 4-1 NMR 去乙醯率測定準確度分析 在許多研究幾丁質/幾丁聚醣的相關研究中,去乙醯率測定的方法皆是以 NMR 的測定值為最可靠的依據。本研究中之去乙醯率測定乃委託成功大學貴重 儀器中心量測。由於樣本的顆粒大小不均勻,在量測上產生的雜訊也不同,為了 使所有樣本在測量去乙醯率有相同的檢測條件,因此固定所有樣本的掃描次數 (number of scans)NS = 1000,磁場強度 6.2 KHz。NMR 準確度的分析以標準品(購自 Wako 公司,實驗級藥品) chitin, chitosan 10 做驗證,其標稱去乙醯率分別為 25%、85%(以 NMR 之方法測得);準確度 的驗證方法是以 NMR 測定的結果,如圖 3 之頻譜圖(橫軸為 ppm,縱軸為分子 共振強度),由圖中各 C 核的共振強度依式 3,以頻譜圖中各波峰底下的面積計 算去乙醯率,分別計算得到 chitin 與 chitosan 10 之去乙醯率為 24.76 %與 85.037 %,與 Wako 公司提供之標準值吻合。因此,本研究幾丁類物質之粉末及懸浮液 之 DD 值以 NMR 的檢測值作為標準值。
圖 3 幾丁質粉末以 NMR 測定之頻譜圖
CH 3 C6 C3 C4 C1 C=O C2 C54-2 幾丁質粉末光譜之去乙醯率分析結果 4-2-1 混合粉末樣本光譜之去乙醯率分析結果 配置混合粉末樣本共有 68 個並計算混合樣本之去乙醯率,在進行光譜有 效樣本的選擇時,將閥值 T 設定為 2.5,僅刪除 1 個無效樣本;樣本分組上, 從有效樣本數中選擇三分之二的樣本為校正樣本,三分之一為預測樣本,校正 樣本的範圍涵蓋整個預測樣本,以得到校正組與預測組間一致的分析結果。 表 1 為幾丁質/幾丁聚醣混合粉末以 MPLSR 模式分析去乙醯率之結果,表 1 中的分析主要為比較原始光譜、一次微分光譜、二次微分光譜的三種光譜前 處理方式分析結果,以及相同光譜前處理下的全波段(400-2500 nm)與特徵波 段的光譜分析結果。在 MPLSR 模式的分析結果下,除了原始光譜外,不論一 次微分或二次微分的光譜前處理在全波段與部份特徵波段之 rc 和 rp 皆高達 0.99,SEC 及 SEP 也只有 0.01 左右。MPLSR 模式分析之最佳結果為全波段(400 ∼2500 nm),二次微分光譜前處理,rc=0.999、SEC=0.006、rp=0.999、SEP=0.006、 RPD=28。混合粉末 MPLSR 分析結果就全波段與部份特徵波段的比較,以全波 段結果較佳,但全波段與部份特徵波段(1100-1200 nm,1500-1700 nm, rc=0.996、SEC=0.012, rp=0.991、SEP=0.016)的差異相當的小,rc的比較僅相 差 0.003,SEC 也僅差距 0.006;在預測組 rp相差 0.008,SEP 則相差 0.01。
在利用實驗室型分光光度計搭配自動樣本傳送配件,並以 small ring cup 量 測粉末反射光譜,從原始光譜所得之 MPLSR 分析結果已非常高,全波段原始 吸收光譜 rc=0.985,SEC=0.021,rp=0.979 、SEP=0.025;原始吸收光譜部份特 徵波段(1100∼1200 nm,1500∼1600 nm,2100∼2200 nm),所得結果 rc=0.99、 SEC=0.018,rp=0.989、SEP=0.019。微分分析結果的表現較佳,這是因為幾丁 質/幾丁聚醣混合粉末樣本具有顆粒大小不均的性質,會影響光源照射在樣品上 產生反射、散射、繞射、穿透等反應,因此藉由微分的處理可以消除此一干擾, 提升分析之效果。 混合粉末之去乙醯率以 MLR 分析結果如表 2 所示。原始光譜前處理之特 徵波長組合多集中於長波長 2000∼2500 nm 範圍,此一波段屬於纖維鍵結的吸 收區,在單一波長 2022 nm 之檢量線所得結果 rc=0.763、SEC=0.085 為文獻上 提到之特徵波段之一。而在雙波長之組合分析時,rc大幅提升至 0.984,此時之 特徵波段為 1528 nm 與 2318 nm 的雙波長組合,由原始吸收光譜圖中可以清楚 的看到幾丁聚醣在 1528 nm 有一明顯的吸收峰值,而幾丁質原始吸收光譜則沒 有,這是因為 1528 nm 屬於 N-H 的第一泛音吸收區,故幾丁質在胺基鍵結含量 遠少於幾丁聚糖的特性下在此波長沒有明顯的吸收波峰。將組合波長增加到三 個以上時,rc與 rp的分析結果都在 0.99 以上。因此作為幾丁質/幾丁聚糖特徵波 段的選擇,1528 nm、1592 nm、1662 nm、1692 nm、2022 nm、2102 nm,這些
MLR 分析在原始光譜、一次微分光譜及二次微分光譜所挑選之雙波長都是具有 很好的分析能力,rc與 rp皆高於 0.9。 表 1 混合粉末去乙醯率之 MPLSR 分析 表 2 混合粉末去乙醯率以 MLR 分析結果 400-2500 3 0.985 0.021 1.20% 0.979 0.025 3.00% 6.8 1100-1200 1500-1600 0.990 0.018 1.20% 0.989 0.019 2.05% 9 2100-2200 400-2500 5 0.996 0.010 0.60% 0.996 0.010 0.60% 17 700-800 1700-1800 400-2500 5 0.999 0.006 0.28% 0.999 0.006 0.50% 28 1100-1200 1500-1700 10 1.80% 11 0.996 0.012 0.60% 0.992 0.015 1.20% 0.012 0.90% 0.991 0.016 二次微分光譜 4 3 0.995 原始吸收光譜 一次微分光譜 3 MPLSR 光譜種類 Spectrum 波長範圍 校正樣本(45) 預測樣本(22) 因子數
Factor rc SEC RSEC% rp SEP RSEP% RPD
1 2022 0.763 0.085 7.9% 0.789 0.082 9.70% 2.1 2 2315+1528 0.984 0.022 1.7% 0.981 0.023 3.50% 7.4 3 2478+2368+1530 0.990 0.017 1.4% 0.985 0.021 2.90% 8.1 4 1532+1112+2488+2376 0.994 0.014 1.0% 0.992 0.015 1.80% 11.3 1 1662 0.985 0.021 1.4% 0.969 0.030 4.20% 5.7 2 1668+2102 0.993 0.014 0.8% 0.989 0.017 2.40% 10 3 1574+1670+2104 0.994 0.013 0.7% 0.991 0.016 2.30% 10.6 4 1660+1574+1668+2104 0.995 0.013 0.7% 0.992 0.015 2.10% 11.3 1 1592 0.990 0.018 1.0% 0.982 0.023 3.20% 7.3 2 1592+1692 0.991 0.016 0.8% 0.987 0.019 2.60% 8.9 3 1516+1660+1468 0.994 0.014 0.8% 0.992 0.015 1.90% 11.3 4 1468+1516+1662+2032 0.995 0.012 0.7% 0.993 0.015 1.70% 11.3 RSEP% RPD rp 波長個數 選定波長 預測樣本(22) SEC RSEC% rp SEP
rp SEP RSEP% RPD 選定波長 rc SEC RSEC% 原始吸收光譜 一次微分光譜 波長個數 選定波長 rc SEC RSEC% 校正樣本(45) rc 預測樣本(22) 校正樣本(45) 預測樣本(22) 校正樣本(45) SEP RSEP% RPD 二次微分光譜 波長個數
4-2-2 去乙醯反應後之粉末樣本 樣本全波段與部份特徵波段 MPLSR 分析結果如表 3 所示。全波段 MPLSR 分析結果中,以原始光譜之結果最好,校正組 rc=0.99、SEC=0.022,預測組 rp=0.989、SEP=0.022。全波段 MPLSR 分析之一次微分與二次微分結果 rc、rp、 SEC、SEP 皆相同,兩者使用的因子數也一樣,此部份之去乙醯反應後粉末樣 本為均質的情況下,微分的數學處理對提高樣本反射吸收光譜的分析結果並沒 有顯著的效益。MPLSR 部份特徵波段分析結果,仍然以原始吸收光譜結果最 好,部份特徵波段 1500-1800 nm、2000-2300 nm,rc=0.995,SEC=0.014,rp=0.99, SEP=0.021,RPD=9.8,為測定幾丁聚醣粉末樣本去乙醯率最佳檢量線方程式。 表 4 為去乙醯反應後之粉末樣本經由散射校正處理的 MLR 分析結果。原 始光譜的單波段選擇與混合粉末的分析結果有相同的特徵波長 2024 nm,在雙 波長組合以上 rc皆高於 0.98,表示 2148 nm 與 2254 nm 亦是重要之特徵波長。 在一次微分光譜與二次微分光譜的單波長 MLR 分析中,校正組 rc與預測組 rp 即可達到 0.976 以上的相關性,而 MLR 一次微分光譜單波長選擇為 2166 nm, 此波長屬於 N-H 鍵結的 combination 與 CONH2(R)之結構關係密切,二次微分 光譜單波長選擇為 1264 nm 包含在 C-H 鍵結的吸收區域內,都是與幾丁質/幾 丁聚醣分子結構中的代表性官能基乙醯胺基、胺基分子相符合。MLR 部份特徵 波段的選擇在一次微分與二次微分光譜光譜處理,三波長的組合以上即可有 0.99 的分析與預測能力,其中一次微分的多波長組合主要出現在 1000∼1200 nm 之間,此波段在近紅外光的鍵結為 N-H 以及 C-H 鍵結的 2nd Overtone。二 次微分的多波段組合則多出現在 2000∼2300 nm 的波長範圍,屬於 N-H 鍵結的 Combinations,此兩特徵波段都是分析建立幾丁質/幾丁聚醣去乙醯率之重要特 徵波段。 表 3 去乙醯反應後之粉末樣本 MPLSR 分析 400-2500 5 0.990 0.022 1.4% 0.989 0.022 2.3% 9.3 1500-1800 2000-2300 400-2500 2 0.988 0.024 1.5% 0.983 0.030 3.5% 5.7 400-700 1400-1800 1 0.987 0.024 1.6% 0.984 0.029 2.9% 7.1 2000-2300 400-2500 2 0.988 0.023 1.5% 0.984 0.028 3.4% 7.3 400-600 1400-1700 1 0.988 0.023 1.4% 0.984 0.028 3.5% 7.3 2000-2200 MPLSR 光譜種類 Spectrum 波長範圍 校正樣本(39) 預測樣本(19) 因子數
Factor rc SEC RSEC% rp
二次微分光譜 一次微分光譜 RSEP% RPD SEP 0.021 2.3% 9.8 原始吸收光譜 7 0.995 0.014 1.1% 0.990
表 4 去乙醯反應後之粉末樣本 MLR 分析 1 2024 0.235 0.181 10.3% 0.200 0.183 12.6% 1.1 2 2148+2254 0.985 0.026 2.0% 0.983 0.027 2.4% 7.6 3 706+2150+2256 0.987 0.024 1.9% 0.985 0.025 2.1% 8.2 4 866+2352+2246+1552 0.990 0.021 1.6% 0.987 0.023 2.0% 8.9 5 968+2348+2248+1552+1522 0.992 0.018 1.4% 0.991 0.020 1.8% 10.3 1 2166 0.982 0.028 2.4% 0.981 0.029 2.30% 5.8 2 1190+2166 0.989 0.022 1.8% 0.983 0.029 3.10% 5.8 3 2168+1184+1190 0.990 0.021 1.8% 0.991 0.020 2.70% 8.5 4 1002+2168+1184+1190 0.993 0.018 1.5% 0.991 0.021 2.30% 8.1 5 2170+1184+1190+914+1002 0.994 0.016 1.2% 0.993 0.018 2.20% 9.4 1 1264 0.976 0.033 2.9% 0.976 0.036 4.40% 5.7 2 1266+2296 0.985 0.025 2.2% 0.986 0.026 3.00% 7.9 3 2434+2016+2084 0.991 0.020 1.1% 0.988 0.024 2.80% 8.6 4 428+2434+2016+2084 0.992 0.019 1.1% 0.992 0.021 2.40% 9.8 5 2298+2016+2084+1404+428 0.994 0.016 1.1% 0.992 0.021 2.40% 9.8 6 898+1402+2310+428+1990+20160.996 0.013 1.0% 0.996 0.015 1.30% 13.8 校正樣本(39) 預測樣本(19) 校正樣本(39) 預測樣本(19) 二次微分光譜 波長個數 選定波長 rc SEC RSEC% 波長個數 選定波長 校正樣本(39) 預測樣本(19) RSEP% RPD rc SEC RSEC% rp SEP RSEP% RPD
rp
rp SEP 原始吸收光譜
一次微分光譜
波長個數 選定波長
4-3 幾丁質去乙醯過程懸浮液之光譜分析 因採用線上型分光光度計,其儀器搭配之光纖設備在 1900 nm 波長以後之光 譜雜訊非常大,雖然在 2000∼2400 nm 的近紅外光波長範圍也是幾丁質/幾丁聚 醣的重要分析波段,但為避免雜訊影響分析結果,故在 MPLSR 全波段分析中僅 選擇 400∼1900 nm 的波長範圍進行分析。總樣本數為 51 個,經由刪除樣本的光 譜處理後保留了 49 個分析樣本數,去乙醯率的分布範圍在 0.322 mol %∼0.960 mol %。 表 5 則為 MPLSR 400∼1900 nm 波段分析結果,整體來說 MPLSR 的分析預 測能力隨著微分的次數增加而提升,原始光譜與一次微分光譜校正組的 rc值雖然 有達到 0.9、0.97 的分析能力,但是預測組的 rp值只有 0.86、0.89 的預測能力, rc與 rp明顯的相差過多,表示其中仍有不利於分析的干擾雜訊存在,在幾丁質去 乙醯化的過程中光譜量測受到外界的干擾最大,由於樣本以 on-line NIRS 6500 擷取懸浮液樣本反射光譜,懸浮液置於培養皿中的容量(液面高度),去乙醯過 程所使用的鹼液濃度都是影響光譜吸收度差異的干擾,必須以數學處理的方式消 除雜訊,並進一步提升樣本特徵(如代表樣本特徵的乙醯胺基、胺基)在光譜吸 收度的表現。在 MPLSR 二次微分光譜的部份特徵波段,600∼1000 nm,1400∼ 1500 nm 皆為 C-H 鍵結的第一與第三泛音吸收區,600∼1000 nm 則是包含了部 份的 RNH2的吸收鍵結(800 nm),其分析結果 rc=0.991、SEC=0.019; rp=0.99、 SEP=0.022,為最佳之校正方程式,在即時的幾丁質去乙醯過程線上檢測應用此 為最佳之檢量線模式。 表 6 所列為幾丁質去乙醯過程中懸浮液光譜之 MLR 分析結果,原始光譜所 選擇之特徵波長主要介於 1200∼1300 nm ,單一波長時之檢量線結果 rc=0.564, 而在雙波長組合時,rc大幅提升至 0.824。 比較幾丁質去乙醯反應後之粉末樣本與去乙醯過程懸浮液之分析結果,在 MLR 的分析結果,幾丁質去乙醯反應後之粉末樣本所挑選之特徵波長為 1264 nm (C-H 吸收鍵結)、2166 與 2024 nm(N-H 吸收鍵結);幾丁質去乙醯過程懸浮 液所挑選之特徵波長為 976 nm(N-H 吸收鍵結),1226 與 1294 nm(C-H 吸收鍵 結),不論樣本之形態為粉末或是液態所得近紅外光分析結果皆為 N-H 以及 C-H 鍵結的泛音吸收區。
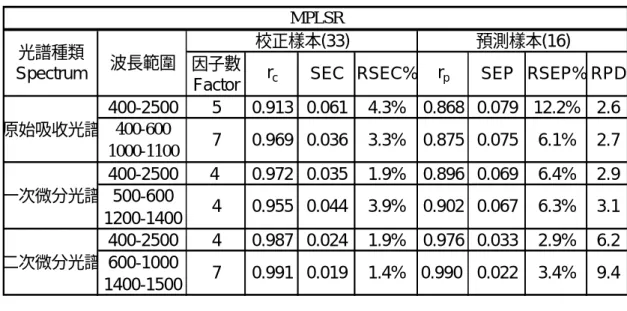
表 5 幾丁質去乙醯過程懸浮液樣本 MPLSR 分析 表 6 幾丁質去乙醯過程懸浮液樣本 MLR 分析 400-2500 5 0.913 0.061 4.3% 0.868 0.079 12.2% 2.6 400-600 1000-1100 400-2500 4 0.972 0.035 1.9% 0.896 0.069 6.4% 2.9 500-600 1200-1400 400-2500 4 0.987 0.024 1.9% 0.976 0.033 2.9% 6.2 600-1000 1400-1500 RPD 一次微分光譜 二次微分光譜 SEC RSEC% 3.4% 0.019 1.4% 0.990 0.022 MPLSR 光譜種類 Spectrum 波長範圍 校正樣本(33) 預測樣本(16) 因子數
Factor rc rp SEP RSEP%
0.075 6.1% 0.991 7 0.044 3.9% 0.902 0.067 4 0.955 2.7 原始吸收光譜 9.4 6.3% 3.1 7 0.969 0.036 3.3% 0.875 1 1294 0.564 0.136 7.8% 0.423 0.162 25.5% 1.3 2 1238+1250 0.824 0.086 5.3% 0.807 0.094 8.5% 2.2 3 1226+1234+444 0.878 0.072 4.6% 0.853 0.083 10.1% 2.5 4 440+1226+1234+444 0.926 0.056 4.3% 0.867 0.080 9.4% 2.6 1 1226 0.821 0.087 5.4% 0.735 0.110 8.7% 1.9 2 648+1226 0.898 0.066 4.8% 0.847 0.087 9.5% 2.4 3 608+1226+616 0.953 0.045 3.3% 0.922 0.059 7.1% 3.5 4 1246+608+1226+616 0.961 0.041 3.0% 0.943 0.051 5.7% 4.0 1 976 0.889 0.069 4.1% 0.748 0.107 11.4% 1.9 2 1050+976 0.941 0.05 4.4% 0.795 0.097 5.0% 2.1 3 1046+976+1680 0.949 0.047 3.6% 0.879 0.076 5.3% 2.7 4 1306+1046+976+1680 0.951 0.046 3.0% 0.920 0.062 4.5% 3.3 5 1388+1306+1046+976+1680 0.964 0.039 2.2% 0.921 0.061 5.1% 3.4 SEP RSEP% 校正樣本(33) 預測樣本(16) rc SEC RSEC% rp rp 波長個數 選定波長 校正樣本(33) 預測樣本(16) RPD RSEP% RPD RSEP% RPD 原始吸收光譜 一次微分光譜 波長個數 選定波長
rc SEC RSEC% rp SEP
SEP 二次微分光譜
波長個數 選定波長
校正樣本(33) 預測樣本(16)
4-4 去乙醯和尚蟹皮膜分析結果 去乙醯和尚蟹皮膜分析結果分為濕膜與乾膜,在濕膜的量測過程以每一個樣 本量測五次光譜(不做光譜平均),MPLSR 與 MLR 模式的分析則是取每一樣本 第一次量測之光譜值做迴歸分析,依此類推分析每一樣本第二次量測之光譜值, 藉由近紅外光的能量照射在去乙醯和尚蟹皮膜之濕膜上帶走水分,比較去乙醯和 尚蟹皮膜之濕膜光譜不同含水情況下分析結果,五次不同含水情況之分析結果以 第五次所量得之光譜分析結果為最佳,表 7 為第五次光譜 MPLSR 分析結果,表 7 中原始吸收光譜之 rc值雖然僅 0.530,但已是優於前四次所量得之光譜分析結 果,因為和尚蟹皮膜每隻重量約為 2∼6 mg(乾重),而其本身在含水情況下可 重達 30∼50 mg,兩者相差 8∼15 倍,故濕膜受到水之影響,使近紅外光大部分 被水吸收,相對之下去乙醯和尚蟹皮膜本身之吸收度變得很小,樣本本身的光譜 吸收特性便無法凸顯出來。表 7 為去乙醯和尚蟹皮膜之濕膜的全波段分析結果, 其中 MPLSR 模式在二次微分可得最佳之分析結果,rc=0.885,SEC=0.065, rp=0.800,SEP=0.076。部份特徵波段的分析結果無法得到理想的分析預測能力, 可能是因為受到水份的影響使得樣本在有限光譜資訊下迴歸分析結果無法得到 很好的相關性。 表 8 為去乙醯和尚蟹皮膜之濕膜在 MLR 模式的分析結果,表中所列為各光 譜在不同數學前處理前處理下所得之最佳組合波段,MLR 模式原始吸收光譜之 三波長組合 1402、1566、1926 屬於 N-H 鍵之吸收波長。而 MLR 模式二次微分 光譜所得結果為最佳 rc=0.817,SEC=0.065,rp=0.800,SEP=0.076,其所使用之 三波長組合 1168、2022、2096 則為 N-H 鍵與纖維素之吸收鍵結,2022 nm 的波 長再一次由去乙醯和尚蟹皮膜之濕膜的分析結果中被挑選出來,顯示 2022 nm 為 幾丁類物質之重要波長。在去乙醯和尚蟹皮膜之濕膜的去乙醯率檢測,建議以 MPLSR 的二微分全波段分析模式做為檢量線模式。 去乙醯和尚蟹皮膜之乾膜的 MPLSR 與 MLR 分析結果為表 9 與 10 所示。乾 膜不同於濕膜的分析之處在於吸收度光譜在三次重複量測後取平均值做後續之 分析處理。因為乾膜沒有含水量的干擾,故在量測光譜上只有樣本不平整以及乾 膜面積形狀無法填滿容器量測面積之操作問題,因此以光譜重複量測,取吸收度 平均值之方法減少誤差。表 9 為全波段 MPLSR 的分析結果,其中微分處理的效 果消除了樣本不平整造成的光譜差異性,使原始吸收光譜之 rc 值 0.53 提升至 0.867。乾膜最佳之檢量線模式為全波段二次微分的 MPLSR 分析結果,其 rc=0.932,SEC=0.052,rp=0.921,SEP=0.056。 表 10 為去乙醯和尚蟹皮膜之乾膜的 MLR 分析結果,在原始光譜中所挑選 之組合波段 2024 與 2154 nm 皆為 N-H 吸收鍵結,而 2024 nm 不論在何幾丁類物 質的分析下皆是重複出現的特徵波長。MLR 最佳之分析結果為二次微分四波長 組合(1188、1410、1438、1526),rc=0.904,SEC=0.060,rp=0.857,SEP=0.065。
表 7 去乙醯和尚蟹皮膜之濕膜 MPLSR 分析結果 表 8 去乙醯和尚蟹皮膜之濕膜 MLR 分析結果 表 9 去乙醯和尚蟹皮膜之乾膜 MPLSR 分析結果 表 10 去乙醯和尚蟹皮膜之乾膜 MLR 分析結果 原始吸收光譜 400-2500 4 0.530 0.117 8.3% 0.465 0.121 11.2% 1.1 一次微分光譜 400-2500 6 0.867 0.103 5.7% 0.658 0.103 9.4% 1.3 二次微分光譜 400-2500 4 0.885 0.065 5.5% 0.800 0.076 6.1% 1.8
SEC RSEC% rp SEP RSEP% RPD
光譜種類 Spectrum 波長範圍 校正樣本(41) 預測樣本(14) 因子數 Factor rc 原始吸收光譜 1566+1402+1926 0.625 0.108 7.0% 0.583 0.111 7.4% 1.2 一次微分光譜 1302+2218 0.658 0.100 6.8% 0.595 0.110 7.1% 1.3 二次微分光譜 1168+2022+2096 0.817 0.080 5.1% 0.716 0.099 6.2% 1.4 SEP RSEP% RPD 選定波長 光譜種類 Spectrum 預測樣本(14) 校正樣本(41) rc SEC RSEC% rp 原始吸收光譜 400-2500 6 0.756 0.090 8.1% 0.665 0.102 10.3% 1.4 一次微分光譜 400-2500 2 0.789 0.084 7.8% 0.733 0.093 9.0% 1.5 二次微分光譜 400-2500 3 0.932 0.052 5.5% 0.921 0.056 6.7% 2.5 RSEC% rp SEP RSEP% RPD
光譜種類 Spectrum 波長範圍 校正樣本(41) 預測樣本(14) 因子數 Factor rc SEC 原始吸收光譜 2154+2024 0.770 0.088 9.5% 0.752 0.090 12.0% 1.5 一次微分光譜 592+1214+1224+2032 0.861 0.070 7.9% 0.844 0.073 8.4% 1.9 二次微分光譜 1188+1410+1438+1526 0.904 0.060 4.9% 0.857 0.065 6.0% 2.1 RPD 選定波長 光譜種類 Spectrum 預測樣本(14) 校正樣本(41)
五、結 論
本研究針對幾丁類物質在去乙醯化的過程中不同形態下粉末、去乙醯過程 懸浮液,以 MPLSR 與 MLR 迴歸分析建立幾丁類物質的近紅外線光譜檢測模式, 並探討適用於各形態下幾丁類物質去乙醯率的檢測模式。 首先以幾丁質與幾丁聚醣標準品混合,所得的粉末樣本可模擬幾丁質在製 備幾丁聚醣後之不同去乙醯率粉末樣本,其光譜分析結果在採用 MPLSR 部份特 徵波段的分析結果,在原始光譜、一次微分光譜、二次微分光譜中均找到 1500-1600 nm 之特徵光譜波段,顯示此一波段為幾丁質/幾丁聚醣重要之特徵波 段。而 MLR 的分析結果,在原始光譜中挑選的特徵波長都集中在 2000∼2300 nm 之間,其中單一特徵波長為 2022 nm 與 Vårum(1995)的研究相同。另外,混合 粉末樣本在 MLR 模式分析所挑選的重要分析波長為 1528 nm、1592 nm、1662 nm、1692 nm、2022 nm、2102 nm。去乙醯反應後之粉末樣本 MLR 的分析結果 在原始光譜的單波段選擇中,與混合粉末 MLR 原始光譜的單波長選擇有相同的 特徵波長 2024 nm,表示 2024 nm 為幾丁質/幾丁聚醣相當重要之特徵波長。而 MLR 以二次微分的六波長組合(428、898、1402、1990、2016、2310 nm)為最 佳 之 檢量 模式 ,其 校正 組 rc=0.996 、 SEC=0.013 、 RSEC=1.0 % 、 rp=0.996 、 SEP=0.015、RSEP=1.3 %、RPD=13.8。在自行製備的幾丁聚醣粉末樣本建議可採 用 MLR 的二次微分光譜六波長組合為檢量線,作為預測粉末樣本之去乙醯率。 本研究將近紅外光之技術應用在幾丁質去乙醯過程懸浮液的去乙醯率檢 測,以去乙醯率變化下即時量測所得之光譜進行分析,不同於 Vårum et al.(1995) 將乙醯化程度在 0.01∼0.51 範圍的水溶性幾丁類物質做乙醯化處理的方式製備 樣本,且本研究實驗樣本的去乙醯率範圍為 32 %∼96 %,包含了整個低去乙醯 程度的範圍。另外,在建立近紅外光之去乙醯率檢量線模式,Vårum et al.(1995) 是以間接測定黏度的方法來預測樣本去乙醯率;而本研究則是以樣本去乙醯率直 接進行迴歸分析,因此本研究方法所建立之近紅外光檢測模式更具實用價值,並 可作為將來線上即時檢測之依據。最佳光譜檢量線為部份特徵波段 600∼1000 nm 與 1400∼1500 nm 之二次微分光譜的 MPLSR 分析結果,其 rc=0.991、 SEC=0.019、RSEC=1.4 %,rp=0.990、SEP=0.022、RSEP=3.4 %、RPD=9.4。 去乙醯和尚蟹皮膜之分析結果以乾膜之 MPLSR 全波段二次微分結果最 好,rc=0.932,SEC=0.052,rp=0.921,SEP=0.056,為預測去乙醯和尚蟹皮膜乾膜 狀態去乙醯率的最佳檢量線。若考慮和尚蟹皮膜乾燥後活性可能降低之問題,則 預測去乙醯和尚蟹皮膜濕膜之檢量線,則建議以 MPLSR 的二微分全波段模式 rc=0.885,SEC=0.065,rp=0.800,SEP=0.076 為最佳。。 本研究利用近紅外光快速且非破壞性的檢測優點,成功建立了幾丁質與幾丁聚醣粉末之去乙醯率近紅外光檢測模式。在樣本即時檢測去乙醯率的近紅外光 檢測模式研究中,更提供了幾丁質在製備幾丁聚醣的去乙醯化過程中品管的即時 監控方法。而去乙醯和尚蟹皮膜之濕膜與乾膜去乙醯率檢量線的建立,可以使蝦 蟹加工中的廢棄甲殼變成有價值之再生資源。
六、參考文獻
陳加增。2001。近紅外光應用於水果糖酸度線上檢測之研究。碩士論文。台北: 國立台灣大學生物產業機電工程學研究所。 陳世銘、張文宏、謝廣文。1998。果汁糖度檢測模式之研究。農業機械學刊 7(3): 41-60。 蔡志成。2004。以近紅外光檢測幾丁類物質去乙醯率之研究。碩士論文。台北: 國立台灣大學生物產業機電工程學研究所。 蔡志成、陳世銘、楊宜璋、黃怡靜。2004。以近紅外光檢測幾丁類物質去乙醯率 之研究。2004 生機與農機論文發表會論文摘要集。123-124。台北:台灣生物 機電學會。 國立成功大學貴重儀器使用中心。2004。核磁共振儀 Bruker Avance 400。台南: 國 立 成 功 大 學 貴 重 儀 器 使 用 中 心 。 網 址 : http://www.ncku.edu.tw/%7Efacility/facility/index.html。上網日期:2004-04-24。Chang K. L B.,G. Tsai., J. Lee., and W. R. Fu. 1997. Heterogeneous N-deacetylation of chitin in alkaline solution. Carbohydrate Research 303:327-332.
Chen, S. et al. 2004. Growth status monitoring and quality evaluation for bio-production and products using spectral sensing techniques. In “Proceedings of the Second International Symposium on Machinery and Mechatronics for Agriculture and Bio-systems Engineering”, Keynote Speech, KN-9 ~ 23. Kobe, Japan: Kobe University.
Chu K. H. 2002.Removal of copper from aqueous solution by chitosan in prawn shell: adsorption equilibrium and kinetics. Journal of Hazardous Materials 90( 1): 77-95 Duarte, M. L., M. C. Ferreira., M. R. Marvão, and J. Rocha. 2001. Determination of
the degree of acetylation of chitin materials 13C CP/MAS NMR spectroscopy. International Journal of Biological Macromolecules 28:359-363
FOSS NIRSystems. 1999. WinISI User’s Manual. Silver Spring, MD, USA:FOSS NIRSystems.
Harish, P. K. V., F. S. Kittur., and R. N. Tharanathan. 2002. Solid state structure of chitosan prepared under different N-deacetylating conditions. Carbohydrate Polymers 50:27-33
Heux, L., J Brugnerotto., J. Desbrières, M. –F Versali,. and M Rinaudo. 2000. Solid state NMR for determination of degree of acetylation of chitin and chitosan.
Biomacromolecules 1:746-751.
Martinou, A., D. Kafetzopoulos., and V. Bouriotis. 1995. Chitin deacetylation by enzymatic means: monitoring of deacetylation process. Carbohydrate Research 273:235-242.
Marvão, M. R., M. C. Ferreira., M. L. Duarte., and R. A. Pires. 1998. FR-IR spectroscopy as a tool to determine the degree of acetylation of â-chitosan from loligo pen. Advance in Chitin Science 3:123-128.
Methacanon, P., M. Prasitslp, T. Pothsree., and J. Pattaraarchachai., 2003. Heterogeneous N-deacetylation of squid chitin in alkaline solution. Carbohydrate Polymers 52:119-123.
Nil. 1996. Market of chitin and chitosan. Biol. Industry 13(3):52-61.
Nunthanid, J. S. P., K. Yamamoto, G. E. Peck. 2001. Physical properties and molecular behavior of chitosan film. Drug development and industrial pharmacy 27(2): 143-57.
Ottøy, M. H., K. M. Vårum., and O. Smidsrød. 1996. Compositional heterogeneity of heterogeneously deacetylated chitosans. Carbohydrate Polymers 29:17-24
Ravindra, R., K. R Krovvidi,. and A. A Khan,. 1998. Solubility parameter of chitin and chitosan. Carbohydrate Polymers 36:121-127.
Raymond, L., F. G. Morin., and R. H. Marchessault. 1993. Degree of deacetylation of chitosan using conductometric titration and solid-state NMR. Carbohydrate Research 246:331-336.
Shigemasa, Y., H. Matsuura, H. Sashiwa, and H. Saimoto. 1996. Evaluation od different absorbance ratios from infrared spectroscopy for analyzing the degree of deacetylation in chitin. International Journal of Biological Macromolecules 18:237-242.
Tan, S. C., E. Khor, T. K. Tan., and S. M. Wong. 1998. The degree of deacetylation of chitosan : advocating the first derivative UV-spectrophotometry method of determination. Talanta 45:713-719.
Tomihata, K., and Y. Ikada. 1997. In vitro and in vivo degradation of films of chitin and its deacetylated derivatives. Biomaterials 18:567-575.
Vårum, K. J., M. W. Anthonsen,, H. Grasdalen, and O. Smidsrød, 1991. Determination of the degree of N-acetylation and the distribution of N-acetylated chitins (chitosans) by high-field n.m.r. spectroscopy. Carbohydrate Research 211:17-23.
Ylitalo, R., S. Lehtinen., E. Wuolijoki., P. Ylitalo., and T. Lehtimaki, 2002. Cholesterol-lowering properties and safety of chitosan. Arzneimittel- Forschung 52(1):1-7.
計 畫 成 果 自 評
本研究成果內容與原提計畫相符,達成預期目標:(1)建立以近紅外光技 術檢測幾丁質與幾丁聚醣之定量分析模式; (2)以近紅外光分析模式檢測並分 析幾丁類物質之去乙醯率。 本研究所建立的幾丁類物質去乙醯率之近紅外線檢測模式,可以有效地應 用於幾丁類物質乙醯率之即時線上檢測,並藉以改善幾丁類物質製程之品管監 控。研究成果適合於學術期刊發表,目前正在改寫投稿中,其中部份研究成果已 於 2004 年 9 月在“ International Symposium on Machinery and Mechatronics for Agriculture and Bio-systems Engineering (ISMAB 2004) ”(日本神戶)及 2004 年 9 月在「2004 生機與農機論文發表會」(宜蘭)發表:Chen, S. et al. 2004. Growth status monitoring and quality evaluation for bio-production and products using spectral sensing techniques. In “Proceedings of the Second International Symposium on Machinery and Mechatronics for Agriculture and Bio-systems Engineering”, Keynote Speech, KN-9 ~ 23. Kobe, Japan: Kobe University.
蔡志成、陳世銘、楊宜璋、黃怡靜。2004。以近紅外光檢測幾丁類物質去乙醯率 之研究。2004 生機與農機論文發表會論文摘要集。123-124。台北:台灣生物機 電學會。