E L S E V I E R Journal of Chromatography A, 771 (1997) 267-274
JOURNAL OF
CHROMATOGRAPHY A
Determination of cinnamaldehyde, cinnamic acid, paeoniflorin,
glycyrrhizin and [6]-gingerol in the traditional Chinese medicinal
preparation Kuei-chih-tang by cyclodextrin-modified micellar
electrokinetic chromatography
H s i - Y a H u a n g , K u a n g - L u n g K u o , Y o u - Z u n g H s i e h * Department of Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan, R.O.C.
Received 10 December 1996; revised 5 February 1997; accepted 7 February 1997
Abstract
This study presents a cyclodextrin-modified micellar electrokinetic chromatography (CD-MEKC) technique with a multi-wavelength detection method to determine specific components in Kuei-chih-tang. Kuei-chih-tang, a Chinese medicinal preparation, is composed of five crude herbs, i.e., Cinnarnomi ramulus, Paeoniae radix, Glycyrrhizae radix, Zingiberis rhizoma and Zizyphi fructus. The effects of SDS and ~-cyclodextrin on the analytes' migration behavior were examined. The selected components were successfully separated within 10 min using a pH 10.0 borax-NaOH buffer containing 20 mM SDS and 2 mM -y-cyclodextrin. The correlation coefficients of the linear calibration graphs for the analytes exceeded 0.993. Moreover, the optimized CD-MEKC method was employed to analyze four different Kuei-chih- tang samples. ©1997 Elsevier Science B.V.
Keywords: Kuei-chih-tang; Pharmaceutical analysis; Cinnamaldehyde; Cinnamic acid; Paeoniflorin; Glycyrrhizin; Gingerol
I. Introduction
Traditional Chinese medicine is extensively used in Asian countries. Preparing traditional Chinese medicine is time-consuming, thereby accounting for increasing use of extracted powder from crude Chinese medicinal preparations in recent years. Selecting specific components in a Chinese medicine as markers for analysis is a highly promising tech- nique for improving the quality o f Chinese medicine.
*Corresponding author.
However, experimentally studying the active ingredi- ents of concentrated preparations requires establish- ing an appropriate analytical method. Currently, investigating specific components in complicated traditional Chinese medicinal preparations is limited. The present study thus provides a promising method to explore the investigation.
Kuei-chih-tang (cinnamon combination) is a tradi- tional Chinese medicinal preparation composed of five crude herbs, i.e., Cinnamomi ramulus, Paeoniae radix, Glycyrrhizae radix, Zingiberis rhizoma and Zizyphi fructus [1]. A m o n g these herbs, Cinnamomi ramulus (Chinese name is Kuei-chih) is the major constituent in Kuei-chih-tang. The medicine is fre- 0021-9673/97/$17.00 Copyright © 1997 Elsevier Science B.V. All rights reserved
268 H.-Y. Huang et al. / J. Chromatogr. A 771 (1997) 2 6 7 - 2 7 4
quently used to treat diseases such as the common cold, abdominal pains owing to chills, neuralgia, neurasthenia and rheumatic pain. This preparation, the basis for several other preparations, is very important in traditional Chinese medicine. Therefore, its analysis is essential to further understand Kuei- chih-tang and related medicines as well.
Cinnamaldehyde / cinnamic acid, paeoniflorin, glycyrrhizin and [6]-gingerol are specific compo-
nents of Cinnamomi ramulus, Paeoniae radix,
Glycyrrhizae radix and Zingiberis rhizoma, respec- tively [2]. Oleanolic acid, a common component of sapogenins, was selected herein as a test compound for Zizyphi fructus because other specific compounds could not be obtained. Those six compounds were chosen as analytes in this study.
Thin-layer chromatography (TLC) is the conven- tional method of analyzing traditional Chinese medicinal preparations. However, this method can not be applied to simultaneously analyze several components in a single crude herb or in a medicinal preparation composed of several crude materials. Generally, high-performance liquid chromatography (HPLC) has been employed to analyze crude herbs or several marker components in a Chinese medici- nal preparation [3-6]. Nevertheless, more than 30 min are generally required for the HPLC method to analyze a Chinese medicinal preparation [5,6].
Micellar electrokinetic chromatography (MEKC) is a high-performance capillary electrophoresis (CE) technique [7-11]. Cyclodextrin-modified MEKC (CD-MEKC) further enhances the separation ef- ficiency by adding various cyclodextrins in MEKC buffer. The analytical technique presented herein possesses similar advantages of CE having a high separation efficiency, short analysis time and excep- tional resolution. Consequently, MEKC has been employed to analyze Chinese crude herbs and Chi- nese medicinal preparations [12-14].
In the present study, CD-MEKC was developed to simultaneously determine selected components of the five herbs constituted Kuei-chih-tang. The effects of various separation and buffer conditions on the analytes' migration behavior were also examined. The different extraction methods and the variation of specific component concentrations in four different Kuei-chih-tang samples were discussed.
2. Experimental
2.1. Apparatus
A Beckman P/ACE 5500 capillary electrophoresis system (Fullerton, CA, USA) was used. A diode- array detector, which can scan wavelengths ranging from 190 nm to 600 nm, was connected to the system. The detection wavelength could also be programmed to change during the separation process. The P/ACE instrument was controlled using a per- sonal computer with System Gold software (Beck- man Instruments, San Ramon, CA, USA). CE was performed using a 47 cm (40 cm to detector)×50 ~xm I.D. fused-silica capillary tube (Polymicro Tech- nologies, Phoenix, AZ, USA). The capillary column was assembled in the cartridge format. The tempera- ture of the capillary during electrophoresis was maintained at 25°C. The applied voltage of the electrophoresis separation was set at 20 kV. Samples were pressure injected at 0.034 bar. Data analysis was performed on System Gold software.
2.2. Chemicals
Sodium dodecyl sulfate (SDS), borax, e~-cyclo- dextrin and [3-cyclodextrin were all purchased from Sigma (St. Louis, MO, USA). Cinnamic acid, glycyrrhizin and ~/-cyclodextrin were purchased from Nacalai Tesque (Kyoto, Japan). Cinnamaldehyde was purchased from both Nacalai Tesque and Fluka (Buchs, Switzerland). [6]-Gingerol and paeoniflorin were obtained from Yoneyama (Osaka, Japan). Oleanolic acid was bought from Extrasynthese (Genay, France). Methanol was bought from Merck (Darmstadt, Germany). Four samples of concentrated Kuei-chih-tang manufactured by different Chinese pharmaceutical companies were purchased from local drug stores in Taiwan. All other chemicals were analytical grade and were purchased from Merck. Water was purified by a Milli-Q water system (Millipore, Bedford, MA, USA) and filtered through a 0.22 I~m filter.
2.3. Procedure
H.-Y. Huang et al. / J. Chromatogr. A 771 (1997) 2 6 7 - 2 7 4 269
were prepared in methanol. Sample solutions with various concentrations were prepared by dissolving the stock solution to a methanol-water solution (7:3, v/v). Electrophoresis buffers ( b o r a x - N a O H buffer) were prepared by mixing 0.1 M borax and 0.1 M sodium hydroxide in deionized water. Each of the commercial, concentrated Kuei-chih-tang samples were accurately weighted at 2.0 g. The concentrated samples were extracted by 20 ml methanol-water solution (7:3, v/v) or pure methanol for 15 rain in an ultrasonic bath. The extracted solution was then filtered through a filter paper. The extraction and filtration procedures were repeated three times. A total of 60 ml extracted solution was concentrated to a final volume 4.0 ml and was ready for analysis. The extraction recovery study followed the same procedure described above.
3. Results and discussion
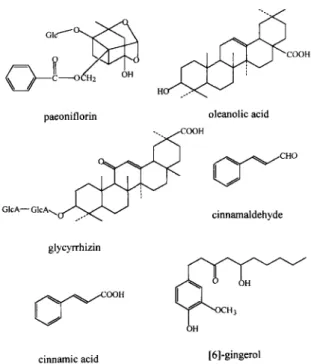
In the present study, six specific compounds were selected as the target analytes, i.e., cinnamic acid, cinnamaldehyde, [6]-gingerol, glycyrrhizin, oleanolic acid and paeoniflorin. Fig. 1 depicts their structures. Those structures markedly vary and their maximum absorbance wavelengths differ in the UV-Vis region. The maximum absorbance wavelengths of paeonifl- orin, oleanolic acid and [6]-gingerol were at 200 nm. Glycyrrhizin and cinnamaldehyde had maximum absorbances at 260 nm and 295 nm, respectively. Cinnamic acid had two similar intensity absorbances around 210 nm and 270 nm. Because of the differ- ences in absorbance wavelengths, the use of a diode- array detector for multiwavelength detection is necessary.
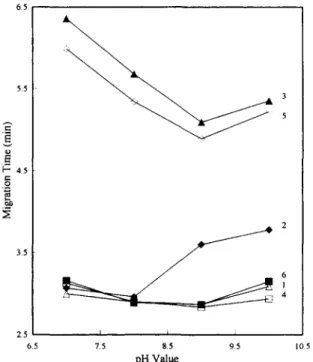
Several buffer systems, such as phosphate buffer (pH 7.0 or pH 8.0) and borate buffer (pH 9.0 or pH 10.0), were preliminarily tested for separating the analytes. Fig. 2 illustrates the influence of pH values on the separation of selected compounds. Cinnamic acid and glycyrrhizin could be resolved from pH 7.0 to pH 10.0. The migration time of oleanolic acid dramatically changed when the pH value exceeded 8.0 because more oleanolic acid molecules were dissociated. Thus, a good separation efficiency and resolution for oleanolic acid could be achieved in a
paeoniflorin
~
COOH
GIcA GlcA-~o~-,~I
H
~
COOH
oleanolic acid,../CHO
cinnamaldehyde glycyrrhizin~
OOH
cinnamic acid [6]-gingerol
Fig. 1. Molecular structures of the six analytes.
pH 9.0 or pH 10.0 buffer. However, cinnamal- dehyde, [6]-gingerol and paeoniflorin had similar velocities as the electroosmotic flow velocities at each of the given pH values. Therefore, the three analytes could not be satisfactorily resolved in those pH values. Since the best separation efficiencies and resolutions for the analytes were achieved at pH 10.0, modifiers were added to the pH 10.0 borate buffer for subsequent analysis.
3.1. Effects of SDS on the separation
Various concentrations of SDS, ranging from 10 mM to 60 mM, were added to the pH 10.0 borate buffer. The migration behavior of cinnamaldehyde, [6]-gingerol and oleanolic acid were significantly altered when the running buffer contained SDS. Specifically, their migration times increased with increasing SDS concentration. Experimental results thus indicated that those three compounds strongly interacted with SDS micelles. Fig. 3 depicts two electropherograms obtained from two different buffer solutions. One of the running buffers contained 20 mM SDS (Fig. 3b) and the other did not contain SDS
270 H.-Y. Huang et al. / J. Chromatogr. A 771 (1997) 267-274 65 5.5 ['~ 4 5 ~ s 2 3.5 6 1 4 2.5 I I i 6.5 7,5 8.5 9,5 105 pH Value
Fig. 2. Effects of buffer pH values on the analytes' migration times, l=Paeoniflorin; 2=oleanolic acid; 3=glycyrrhizin; 4= cinnamaldehyde; 5=cinnamic acid; 6=[61-gingerol. Conditions: capillary, 47 cm (40 cm to detector)×50 ~m I.D.; applied voltage, 20 kV; detection wavelength, 200 nm; column temperature, 25°C.
(Fig. 3a). As those results indicate, adding SDS micelles significantly decreased the migration veloci- ties of cinnamaldehyde, [6]-gingerol and oleanolic acid. Such a decrease is due to the compounds' hydrophobic properties. In contrast with the strong influence of SDS micelles on the migration behaviors of those three compounds, the migration velocities of paeoniflorin, glycyrrhizin and cinnamic acid altered only slightly in the SDS buffer. This phenomenon can be accounted for by the molecular structure of the compound. More specifically, the latter three compounds are too polar to interact with SDS.
As Fig. 3a reveals, the cinnamaldehyde signal was extremely weak due to the simultaneous emergence of the electroosmotic flow. According to experimen- tal results, cinnamaldehyde had only one peak at various pH values (ranging from pH 7.0 to pH 10.0) of the running buffer without SDS. However, as Fig. 3b shows, the cinnamaldehyde peak split into two unresolved peaks as the buffer contained 20 mM SDS. Interestingly, the unresolved two peaks had
< (a) 4 (b) O.Ol U i 2 5 5 I I I I 4 6 8 10 12 Time (rain)
Fig. 3. Electropherograms of the analytes in (a) pH 10.0 borax- NaOH buffer and (b) pH 10.0 borax-NaOH buffer containing 20 mM SDS. Other conditions as in Fig. 2.
identical UV-Vis spectra. Also, cinnamaldehyde obtained from two different manufacturers had identical results. Since both manufacturers claimed that the cinnamaldehyde's purity exceeded 98%, the split peaks are not likely caused by the chemicals' impurity. In contrast, the split peaks are more likely due to the fact that cinnamaldehyde can interact either with SDS micelles or SDS monomers, thereby yielding different migration velocities. Notably the splitting condition decreased with an increasing SDS concentration. The unresolved two peaks became one peak again after addition of more than 50 mM SDS to the running buffer.
Fig. 3b indicated that all six analytes were resolved within 11 min. However, as shown in this figure, the peaks of glycyrrhizin, cinnamic acid and cinnamaldehyde were close. To easily identify the three compounds in actual samples, the separation buffer must be further modified to enhance the resolution of the analytes. Experimental results re- vealed that the separation time became longer with an increasing SDS concentration, but not all of the separation resolutions increased with an increasing SDS concentration. Therefore, we selected a buffer
H.-Y. Huang et al. I J. Chromatogr. A 771 (1997) 267-274 271
containing 20 mM SDS to enhance the resolution and minimize the separation time.
3.2. Effects of cyclodextrins on the separation
The migration times of glycyrrhizin were shor- tened as 13-cyclodextrin or ~/-cyclodextrin was added to a 20 mM SDS buffer. Whereas the migration times of glycyrrhizin exhibited no significant changes when the buffer contained ct-cyclodextrin. In addi- tion, the migration times of oleanolic acid only significantly altered in a ~/-cyclodextrin modified buffer. From those results, we can infer that those two analytes can form inclusion complexes with ~/-cyclodextrin. The fact that size-matching between cyclodextrin and analytes plays a prominent role in forming inclusion complexes suggests that only ~t- cyclodextrin is sufficiently large for glycyrrhizin and oleanolic acid. Therefore, "y-cyclodextrin was added into the borate buffer containing 20 mM SDS to enhance the resolution.
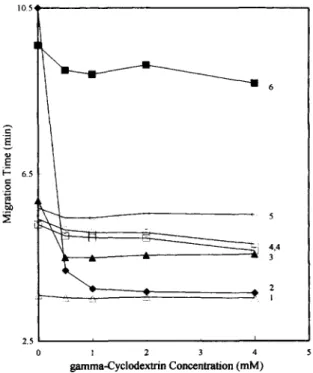
For the six analytes' migration behavior, Fig. 4 shows the effects of adding various concentrations of ~/-cyclodextrin to a 20 mM SDS buffer. According to this figure, the migration times of oleanolic acid and glycyrrhizin markedly changed. More specifically, oleanolic acids migration time shortened from 10.5 min to 3.7 min, even when only 0.5 mM ~/-cyclo- dextrin was added. This enormous decrease in migra- tion time indicated that oleanolic acid had the highest degree of inclusion complexation with ~/-cyclodex- trin among the analytes. Fig. 3b reveals that oleanolic acid strongly interacted with SDS micelles. However, that interaction became weak with the presence of ~/-cyclodextrin in the buffer. In the case of the other four analytes, paeoniflorin, cinnamic acid, cinnamaldehyde and [6]-gingerol, adding ~/- cyclodextrin in the buffer only slightly affected them.
The migration sequence of the analytes remained the same while adding 0.5 mM to 4 mM ~/-cyclo- dextrin to the buffer. The migration times of glycyrrhizin and oleanolic acid were only slightly influenced when ~/-cyclodextrin concentration ex- ceeded 2 mM. In addition, the resolution between cinnamaldehyde and glycyrrhizin decreased as the ~/-cyclodextrin concentration exceeded 2 mM. Con- sequently, the optimum condition for separating the
105 6 6: I 5 4,4 3 2 2.5 I ~ I 0 1 2 3 4 5
gamma-Cyclodcxtrin Concentration (raM)
Fig. 4. Effects o f 3,-cyclodextrin concentrations on the analytes' migration times. Conditions: separation solution, ~/-cyclodextrin in 38 m M b o r a x - N a O H buffer containing 20 m M SDS, pH 10.0. Other conditions as in Fig. 2.
six analytes could be achieved with the pH 10.0 borate buffer containing 20 mM SDS and 2 mM ~/-cyclodextrin. Fig. 5 depicts the separation of the target analytes in optimum conditions. Resolutions among glycyrrhizin, cinnamaldehyde and cinnamic acid were more enhanced under this condition than with no ~/-cyclodextrin in the buffer. Also, the entire separation was completed within 10 min.
Table 1 lists the average migration times, re- producibilities, correlation coefficients of linear cali- bration graphs and limits of detection for the six analytes in optimum conditions. The relative stan- dard deviations (R.S.D.s) of the migration times were less than 0.42%. The concentration ranges for cali- bration graphs were 105-420 Ixg/ml for paeoni- florin, 150-600 l~g/ml for oleanolic acid, 200-800 ixg/ml for glycyrrhizin, 40-160 i~g/ml for cin- namaldehyde, 8-32 I~g/ml for cinnamic acid and 10-40 ixg/ml for [6]-gingerol. The peak area of the electropherogram was employed for quantitation of the analytes. The correlation coefficients of the
272 H.-Y. Huang et al. / J. Chromatogr. A 771 (1997) 267-274
g
ca g < R 0 0 0 5 A U 1 4 n i I I 0 2 4 6 8 10 Time (min)Fig. 5. Separation of specific analytes in the optimum condition. Conditions: separation solution, 38 mM borax-NaOH buffer containing 20 mM SDS and 2 mM ~/-cyclodextrin, pH 10.0; pressure injection, 3 s. Other conditions as in Fig. 2.
g
<I
0 0 2 A U 1_J
4 5 M - , - - - . - I I I 2 4 6 8 10 12 Time (min)Fig. 6. Separation of Kuei-chih-tang by CD-MEKC. Conditions: detection wavelength: 200 nm (before 4.0 min), 270 nm (after 4.0 min), 295 nm (after 4.8 min), 270 nm (after 5.3 min) and 200 nm (after 8.0 min). Conditions as in Fig. 5.
c a l i b r a t i o n g r a p h s w e r e g r e a t e r than 0.993. In addi- tion, the limits o f d e t e c t i o n for t h o s e a n a l y t e s r a n g e d f r o m 0.77 to 28.43 }xg/ml. O u r e x p e r i m e n t a l results thus c o n f i r m that the C D - M E K C m e t h o d definitely p o s s e s s e s the a d v a n t a g e s o f h i g h precision, h i g h r e s o l u t i o n and short analysis t i m e f o r a n a l y z i n g those analytes.
3.3. E x t r a c t i o n a n d d e t e r m i n a t i o n o f specific c o m p o n e n t s in K u e i - c h i h - t a n g
Fig. 6 s h o w s the e l e c t r o p h e r o g r a m o f K u e i - c h i h - tang. B e c a u s e those analytes had m a x i m u m absor- b a n c e s at d i f f e r e n t w a v e l e n g t h s , the d e t e c t i o n w a v e - l e n g t h was p r o g r a m m e d to c h a n g e during the sepa-
Table 1
Average migration times, relative standard deviations, correlation coefficients of calibration graphs and limits of detection of six analytes
Analyte Migration time (min) ~ R.S.D. (%)4 Correlation coefficient Limit of detection
of calibration graph (r) ( Ixg / ml )
Paeoniflorin 3.49 0.31 0.999 6.97 Oleanolic acid 3.65 0.36 0.993 28.43 Glycyrrhizin 4.56 0.42 0.999 15.01 Cinnamaldehyde 4.86 0.32 0.997 2.83 Cinnamic acid 5.43 0.27 0.999 1.46 [6]-Gingerol 8.82 0.25 0.995 0.77 n = 2 0 .
H,-Y. Huang et al. / J. Chromatogr. A 771 (1997) 2 6 7 - 2 7 4 273
Table 2
The extraction recoveries of the analytes spiked in Kuei-chih-tang
Analyte Recovery (%)a R.S,D. (%)
Paeoniflorin 99.7 2.79 Oleanolic acid 99.2 1.21 Glycyrrhizin 85.3 3.33 Cinnamaldehyde 90,1 3.88 Cinnamic acid 93.6 3.49 [6]-Gingerol 94.4 4.66
Values are means of triplicate determinations.
ration of Kuei-chih-tang. The detection wavelength was initially set at 200 nm, then was changed to 270 nm at 4.0 min, to 295 nm at 4.8 min, to 270 nm at 5.3 min and to 200 nm at 8.0 min. As shown in Fig. 6, more than ten peaks appeared in the elec- tropherogram. However, paeoniflorin, glycyrrhizin, cinnamaldehyde, cinnamic acid and [6]-gingerol were adequately resolved from other unknown com- pounds and could be clearly identified.
The specific components in Kuei-chih-tang sam- ples were identified by comparing both the migration times and the UV spectra of standards with those in actual samples. The analytes were further confirmed by spiking standards in actual samples. The specific components can be adequately identified through those processes.
Next, the influences of extraction solutions on the extraction of Kuei-chih-tang were investigated be- cause the six analytes had different solubilities in the extraction solutions. The methanol-water solution
and the methanol solution were employed as ex- traction solutions in this study. The methanol-water solution was an appropriate extraction solution for all the analytes except cinnamaldehyde. The cinnamal- dehyde extracted by methanol exhibited a signifi- cantly larger signal than when extracted by the methanol-water solution. This difference implies that the former has a better solubility and extraction power than the latter for cinnamaldehyde. Therefore, methanol was used as the extraction solvent for quantitation of cinnamaldehyde. The six analytes were successfully extracted by methanol-water or methanol solution with ultrasonic bath. Table 2 presents the recoveries of standards spiked in Kuei- chih-tang. The recoveries of the analytes ranged from 85.3% to 99.7%. The R.S.D.s of those re- coveries were smaller than 5.0%. The results demon- strated that the extraction method was adequate for the analysis.
The comparisons of six analytes in four Kuei-chih° tang samples manufactured by different companies are shown in Table 3. Notably, oleanolic acid could not be found in Kuei-chih-tang. As shown in Table 3, the amounts of each analyte in these four different samples were quite different. The large variation in the specific component concentrations is probably due to either different manufacturing processes or different sources of crude herbs. The relatively large R.S.D.s of actual samples compared with standards is probably due to the complexity of the Chinese medicinal preparations and the heterogeneity of the concentrated powder. However, the R.S.D. values
Table 3
Contents of specific components in four different Kuei-chih-tang samples
Analyte Sample I a Sample 2" Sample 3 a Sample 4 a
Mean R.S.D. Mean R.S.D. Mean R.S.D. Mean R.S.D.
(mg/g) (%) (mg/g) (%) (mg/g) (%) (mg/g) (%) Paeoniflorin 18.09 0,88 19.23 1.93 10.14 0,23 3.99 2.62 Oleanolic acid _b _~ _h _b Glycyrrhizin 13.87 1,47 13.43 3.82 8.09 3.09 6.56 1.94 Cinnamaldehyde 3.04 5.33 0.83 3.88 0.39 5,38 0.56 4.36 Cinnamic acid 0.27 3.71 0.19 3.01 0.32 6.17 0.22 4.82 [6]-Gingerol 0,036 1.89 0.108 1.43 0.008 6.35 0.026 5.35 n = 3 . Not detected.
274 H.-Y. Huang et al. / J. Chromatogr. A 771 (1997) 267-274 were in the same range as those obtained from HPLC
or MEKC analysis of the Chinese medicinal prepara- tions [5,6,12].
Our results demonstrated that the amount of [6]- gingerol in Kuei-chih-tang was quite low. Since [6]-gingerol is a specific component in
Zingiberis
rhizoma,
the small quantity of [6]-gingerol is proba- bly due to the fact that the concentration of [6]- gingerol is markedly lower than other analytes. As mentioned earlier, oleanolic acid could not be found in Kuei-chih-tang. Thus, oleanolic acid was tested for a in a preparation containing onlyZizyphi
fructus.
However, no oleanolic acid peak appeared. Its absence suggests that oleanolic acid is not a specific component of extractedZizyphi fructus.
Therefore, searching for other specific components of
Zizyphi fructus
is necessary for future analysis.Simultaneously determining specific components of crude herbs constituting Chinese medicine is difficult because of the complexity of the matrix and the concentration differences among various specific components. Compared with the conventional TLC method, the five specific components of Kuei-chih- tang could be simultaneously determined by the CD-MEKC method. Therefore, the analysis method introduced herein is quite appropriate for analyzing Kuei-chih-tang. This investigation not only provides quantitative information of Kuei-chih-tang, but ana- lytical information as well to improve its preparation process. Furthermore, the MEKC technique can be employed to analyze other traditional Chinese medicinal preparations, shedding further light into the extremely complex preparation of Chinese medi- cine.
4. Conclusion
The present study successfully develops a CD- MEKC method to analyze specific components in Kuei-chih-tang. Those analytes can be completely separated with the borate buffer containing 20 mM SDS and 2 mM ~/-cyclodextrin. Specific components in Kuei-chih-tang can be determined by the CD- MEKC method coupled with a relatively simple and
efficient extraction method. Results of this inves- tigation provide valuable information regarding actu- al components in concentrated Chinese medicine. The method serves as a highly promising alternative for quantitatively analyzing concentrated Chinese medicinal preparations as well. Further study in this area is underway.
Acknowledgments
This research was supported by Grand NSC 86- 2113-M-009-016 from the National Science Council of Taiwan, R.O.C.
References
[1] H.-Y. Hsu and C.-S. Hsu, Commonly Used Chinese Herb Formulas with Illustrations, Oriental Healing Arts Institute, Long Beach, 1990.
[2] The Analysis Methods for Chinese Medicine, Vols. 7 and 8, National Laboratories of Foods and Drugs, Department of Health, Taipei, 1995.
[3] K. Sagara, T. Oshima, T. Yoshida, Y.-Y. Tong, G. Zhang and Y.-H. Chen, J. Chromatogr., 409 (1987) 365.
[4] K. Yoneda, E. Yamagata and M. Tsujimura, Shoyakugaku Zasshi, 45 (1991) 220.
[5l Y.-C. Lee, C.-Y. Huang, K.-C. Wen and T.-T. Suen, J. Chromatogr. A, 660 (1994) 299.
[6] Y.-C. Lee, C.-Y. Huang, K.-C. Wen and T.-T. Suen, J. Chromatogr. A, 692 (1995) 137.
[7] S. Terabe, K. Otsuka and T. Ando, Anal. Chem., 57 (1985) 834.
[8] M.J. Sepaniak, A.C. Powell, D.F. Swaile and R.O. Cole, in P.D. Grossman and J.C. Colburn (Editors), Capillary Electro- phoresis--Theory and Practice, Academic Press, San Diego, CA, 1992, Ch. 6.
[9] F. Foret, L. Kriwinkov~i and P. Bocek, Capillary Zone Electrophoresis, VCH, Weinheim, 1993.
ll0l S.F.Y. Li, Capillary Electrophoresis, Elsevier, Amsterdam, 1993.
[11] N.A. Guzman (Editor), Capillary Electropboresis Technolo- gy, Marcel Dekker, New York, 1993.
[12] Z. Iwagami, Y. Sawabe and I. Nakagawa, Shoyakugaku Zasshi, 46 (1992) 49.
[13] Y.-Z. Hsieh and H.-Y. Huang, J. Chromatogr. A, 759 (1997) 193.
[14] S.-J. Sheu and C.-F. Lu, J. High Resolut. Chromatogr., 18 (1995) 269.