國 立 交 通 大 學
生物科技研究所
博士論文
溶藻弧菌胺醯組胺酸雙胜肽酶之
晶體結構與突變分析
Crystal Structure and Mutational Analysis of
Aminoacylhistidine Dipeptidase (PepD) from
Vibrio alginolyticus
研究生:張晉源
Student: Chin-Yuan Chang
指導教授:吳東昆
博士
Advisor: Prof. Tung-Kung Wu Ph.D
溶藻弧菌胺醯組胺酸雙胜肽酶之
晶體結構與突變分析
Crystal Structure and Mutational Analysis of
Aminoacylhistidine Dipeptidase (PepD) from
Vibrio alginolyticus
研究生:張晉源
Student: Chin-Yuan Chang
指導教授:吳東昆 博士
Advisor: Prof. Tung-Kung Wu Ph.D
國 立 交 通 大 學
生物科技研究所
博士論文
A Dissertation
Submitted to Department of Biological Science and Technology College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Doctor of Philosophy in Biological Science and Technology
Hsinchu, Taiwan, Republic of China
July, 2011
i
摘要
胺醯組氨酸雙胜肽酶(PepD,EC 3.4.13.3)為胜肽酶家族M20中的一員,具有寬 廣受質專一性,包括可水解肌雙胜(carnosine)及相關之加長肌雙胜(homocarnosine) 以及一些三胜肽。基本上PepD可催化水解Xaa-His雙胜肽而釋放出N端的胺基酸。 在此論文中,主要研究溶藻弧菌PepD的蛋白質結構、生化特性和金屬催化機轉。 利用蛋白質結晶學解出PepD的晶體結構,結果顯示PepD為同源單體所組成的一個 二聚體,每個單體包含一個“蓋子區域”和一個雙鋅離子依存的“催化區域”。不同於 其他M20家族的二聚體,PepD二聚體結構展現一個獨特的十字構形是經由蓋子區 域接觸面間的交互作用力所形成。突變分析確定幾個重要的殘基對於蛋白質結 構、受質的辨認、與酵素的活性扮演著關鍵的角色。另一方面,銅離子取代PepD 活性中心的雙鋅離子,結果產生兒茶酚氧化活性。我們發現”雙銅−PepD”能氧化末 端帶有極性的兒茶酚衍生物,而無法氧化兒茶酚或是帶有非極性支鏈的3,5−叔丁基 鄰苯二酚(DTC)。蛋白質−配體入塢結果顯示,此類帶有極性末端的兒茶酚衍生物 可與”雙銅−PepD”結合。綜合言之,此研究對胺醯組氨酸雙胜肽酶的酵素結構-功 能-反應機制之關係提供更進一步的了解,同時加速開發抗體−酵素導向之前導藥物 治療(ADEPT)。此外,我們證實了金屬取代是造成PepD水解酵素功能酵和氧化活 性之間轉換的關鍵,因此對於酵素趨異演化開啟了一個嶄新的方向。ii
Abstract
Aminoacylhistidine dipeptidase (PepD, EC 3.4.13.3), a member of peptidase M20 family, exhibits a broad substrate specificity for unusual dipeptides carnosine (β-Ala-L-His) and homocarnosine (γ-aminobutyl-His) and a few tripeptides. Basically, PepD catalyzes the cleavage and release of N-terminal amino acid from Xaa-His dipeptide molecules. In this thesis, the PepD from Vibrio alginolyticus was physically and chemically characterized for protein 3D structure, biochemical property, and metal-catalyzed mechanism. The 3D structure was solved by X-ray crystallography, showing that PepD is a homodimer. Each monomeric subunit of the homodimer is composed of a lid domain and a catalytic domain, in which two zinc ions dwell in the active site. In distinction to other M20 family enzymes, the PepD’s dimeric structure exhibits a unique criss-cross configuration that is likely formed through interface interaction of respective lid domains. By performing mutational analysis, crucial residues were identified for maintaining PepD architecture, substrate recognition and enzymatic activity. In addition, changing the active site zinc ions with copper ions, converts PepD to catechol oxidase. The CuCu-PepD is able to oxidize catechol derivatives with a polar tail, but not catechol or 3,5-di-tert-butylcatechol (DTC) that carries non-polar side chains. This result agrees with protein-ligand docking analysis. Collectively, this study advances our overall understanding for aminoacylhistidine dipeptidase in the structure-activity relationships and facilitates future development of antibody-directed enzyme prodrug therapy (ADEPT). Most importantly, the identification of PepD functionality conversion from peptidase to oxidase has paved a new avenue for divergent enzyme evolution.
iii
誌謝
(Acknowledgement)
過去七年的研究生活,過的很充實,也覺得自己獲益良多。 而論文能順利完成,最要感謝的是我的指導教授吳東昆博士,讓一個從對生 物完全不懂的小毛頭,到現在對”酵素“這一塊一定的了解。不只是指導我實驗方 向,也教導我人生應有的態度積極!還有對研究的熱情與執著,不怕失敗的精神 更是我仿效的榜樣。我相信在未來,秉著這樣的態度將受用無窮。在此獻上最深 的敬意與謝意。同步輻射陳俊榮老師讓我有機會進入蛋白質結構的領域一探究 竟,也讓 PepD 原形畢露,也謝謝您不斷給予學生支持與討論。口試委員李耀坤 老師、李宗璘老師以及楊裕雄老師陪我度過修業期間的幾個重要考試,謝謝您們 不吝提供寶貴意見以及對於疏漏處的指正,使得論文更臻完備。也非常感謝師母 賴美伶小姐在生活上的關心與照顧,還有許多系務上的幫忙。 做研究總是問題就在下一秒、挫折就在每一天,幸運的是總能化險為夷,一 步一步的朝著目標邁進。很感謝陪著我度過這段歲月的每一個人,給我打氣,給 我支持,給我繼續向前的動力。 謝謝媛婷讓我這幾年成長很多,不厭其煩的跟我討論實驗,雖然每每都會意 見不合而吵架,但還是很謝謝妳,希望未來的日子可以繼續跟妳一起成長。程翔 學長教我正確的實驗邏輯與技巧,很感謝你從我碩一進實驗室到現在一直以來的 支持與幫忙。感謝裕國學長一路以來的支持與鼓勵,教我許多實驗,做抗體,質 譜分析等等,也帶我岀去玩,很懷念住在一起像家人般的那段日子。晉豪是我的 好室友與好學長,很謝謝你在儀器與生活上的幫忙。謝謝我的戰友文鴻,總是關 心照顧我生活的總總,也很謝謝你在細胞培養技術上的指導與幫忙。小紅妳讓我 在苦悶的研究中找到生活的樂趣。依鵑學姊平常的支持與打氣,還有討論實驗, 每次跟你聊天都有很多收穫。PepD 第一棒大鳥,也是我的好室友好戰友,謝謝你 奠定很好的實驗基礎,讓我後面做出一些很不錯的成果。還有 PepD 第二棒怡親 學妹,謝謝妳在純化養晶上的幫忙。也謝謝良偉學弟在 PepD 上的貢獻。我唯一 的徒弟奕汝,很開心可以教到一個那麼聰明可愛的”學弟“,很感謝你在我很忙的 時候一直Cover 我的實驗。文祥學弟,很開心能交到你這個好朋友,那段瘋狂的 日子,我現在還記憶猶新。謝謝宏城學長平日的關心與照顧,小八學姊在我碩一 懞懂無知的時候教導我實驗。還有跟我同屆的開心果宏明和令宗,謝謝你們在我 低潮的時候陪我談心。而分屬其它計劃的Allen、Mili、欣芳、世潁、怡臻、欣怡、 孟兒、欣樺、婉婷以及富生,謝謝你們平日的幫忙與豐富我的生活。 中研院的弘寬學長,謝謝你那麼溺愛我這位學弟,這七年來不斷的支持鼓勵,iv 在實驗上也一直幫忙,有求必應,教我蟲蟲細胞表現,還有一些data 的分析,真 的很謝謝你,還有大年初二就叫我回去作實驗,這種積極的態度,小弟我謹記在 心。同步輻射的殷程學長,謝謝你教導我從養晶到解結構的過程,每當我問一堆 很沒Sense 的問題,你都不厭其煩的跟我解惑。還有我的超級戰友加好兄弟庭蔚, 很高興在同步還可以交到你這位好朋友,總是一直在鼓勵我,直到現在拿到學位, 希望以後能繼續一起奮戰! 另外還要感謝我的好朋友們秦兄彼此的互相打氣,小胖、老徐、毒蛇常常給 予我加油、打氣,每次跟你們見完面,都會讓我更有勇氣面對下一個挑戰。 最後,特將本論文獻給我最親愛的家人在天堂的阿公,我想跟您說,我拿到 博士學位了!阿媽、爸爸、媽媽一路以來的栽培,讓我沒有後顧之憂,專心攻讀 博士學位,從小頑皮到大讓你們頭痛的我,希望以後的我可以讓您們驕傲。哥哥 和大嫂品容,謝謝你們縱容我的任性,分擔我的不愉快,還時常請我飲茶。妹妹 則是家裡的一塊寶,謝謝你讓家裡增添許多色彩。這段日子很辛苦,但我過的很 開心。感謝所有關心我,陪伴著我的人。謹記在心。 老吳說:『積極的面對每一件事』,願以此期勉自己的未來,能夠發揮所學, 對社會進一份心力。加油! 晉源 謹誌於 交通大學生物科技研究所 中華民國一OO 年八月八日
v
Table of Contents
Chapter 1. Introduction...1
1.1 Binuclear metallohydrolases...1
1.2 Metallopeptidases ...2
1.3 Aminoacylhistidine dipeptidases in the peptidase M20 family ...4
1.4 Crystal structures of M20 family peptidase ...6
1.4-1 Carboxypeptidase G2 (CPG2) from Psedomonas sp. strain RS-16...7
1.4-2 Peptidase V (PepV) from Lactobacillus delbrueckii...9
1.5 Application of M20 family peptidases in Antibody-Directed Enzyme Prodrug Therapy (ADEPT)...12
1.6 Catechol oxidase / tyrosinase...16
1.7 Crystal structure of the catechol oxidase from sweet potato ...17
1.8 Catechol oxidase activity of di-Cu2+-substituted aminopeptidase from Streptomyces griseus (SgAP) ...21
1.9 Thesis purpose ...22
Chapter 2. Materials and Methods...24
2.1 Materials ...24
2.1-1 Bacterial strains, molecular cloning/expression vectors, and resins ...24
2.1-2 Enzyme, chemicals, equipments, and reagents...24
2.2 Experimental methods ...27
vi
2.2-2 Construction of the truncated V. alginolyticus pepD catalytic domain gene
...28
2.2-3 Expression of the V. alginolyticus pepD gene, mutant pepD gene and truncated pepD gene in E. coli...29
2.2-4 Protein purification of wild-type PepD, mutant PepD and PepDCAT...29
2.2-5 Protein concentration determination...30
2.2-6 SDS-PAGE and Native-PAGE analysis...30
2.2-7 Enzymatic activity assay of PepD ...31
2.2-8 Enzyme kinetics...32
2.2-9 Circular dichroism (CD) spectroscopy ...33
2.2-10 Analytical sedimentation velocity ultracentrifugation...34
2.2-11 Crystallization and data collection of PepD crystals ...34
2.2-12 Structure determination and refinement ...35
2.2-13 Substitution of zinc ions by copper ions to form CuCu-PepD ...36
2.2-14 Oxidative activity assay of CuCu-PepD...36
2.2-15 Kinetic analysis of CuCu-PepD for oxidative activity ...37
2.2-16 Protein-ligand docking of CuCu-PepD for catechol derivatives ...38
Chapter 3. Results and Discussion ...39
3.1 Expression, purification, and crystallization of V. alginolyticus PepD, and X-ray data collection of the PepD crystals...39
3.2The crystal structure of V. alginolyticus PepD...41
3.2-1 Overall structure ...41
vii
3.2-3 The lid domain...48
3.2-4 Structure comparison of V. alginolyticus PepD and related di-zinc-dependent M20/M28 family enzymes...50
3.3Mutagenesis study and enzyme kinetics of V. alginolyticus PepD...55
3.3-1 Mutational analysis on metal-binding and catalytic residues of V. alginolyticus PepD ...55
3.3-2 Mutational analysis on probable substrate C-terminal binding residues within the lid domain of V. alginolyticus PepD ...59
3.3-3 Mutational analysis on dimeric interface of V. alginolyticus PepD...61
3.4Substrate specificity alteration of the truncated PepD catalytic domain...63
3.5Proposed catalytic mechanism ...64
3.6Catechol derivatives oxidative activity of copper-substituted PepD (CuCu-PepD) ...66
3.7Protein-ligand docking between CuCu-PepD catecholamine hormones (L-dopa, dopamine, epinephrine, and norepinephrine)...68
Chapter 4. Conclusions and Future Perspectives ...70
References...74
Appendix...85
Appendix 1 Primers used in this thesis...85
viii
Appendix 3 Structure alignment of PepV and PepD with catalytic domain and lid
domain...87
Appendix 4 Analytical ultracentrifugation of PepD protein...88
Appendix 5 Cis peptide bond of PepD, PepV and CPG2...89
Appendix 6 Overall structure of (A) PepD and (B) 2QYV ...90
Appendix 7 Sequence alignment of PepD and 2QYV...91
Appendix 8 Substrate specificity of PepDCAT for various Xaa-His, His-Xaa dipeptides and two histidine-containing tripeptides...92
Appendix 9 ...94
Appendix 9.1 Expression and purifications of humans carnosinase 1 (hCN1) by baculovirus expression system...94
Appendix 9.2 Enzymatic activity assay of human CN1 ...97
Table of Figures
Figure 1.1. Binuclear metallohydrolases catalyze hydrolysis of peptide or phosphate ester bonds ...2Figure 1.2. A generalized mechanistic scheme for metallopeptidases...3
Figure 1.3. Aminoacylhistidine dipeptidase catalyzes hydrolysis of a dipeptide L-carnosine (β-Ala-L-His) into two amino acids ...5
Figure 1.4. Ribbon diagrams of the CPG2 dimers ...8
Figure 1.5. Stereo view of the metal ions binding site of CPG2...9
Figure 1.6. The crystal structure of L. delbrueckii PepV. Ribbon diagram (A) of the enzyme and local view (B) of the zinc ion binding cavity ...11
ix
Figure 1.7. Schematic of the active site of PepV...12
Figure 1.8. Schematic of Antibody-Directed Enzyme Prodrug Therapy (ADEPT) ....14
Figure 1.9. Benzoic acid mustard prodrug (4-[bis-(2-chloroethyl)-amino] benzoyl-L-glutamic acid) is cleaved by carboxypeptidase G2...15
Figure 1.10. The tyrosine metabolism pathway...17
Figure 1.11. Crystal structure of catechol oxidase from sweet potato...18
Figure 1.12. Proposed reaction pathway of catechol oxidase...20
Figure 2.1. Formation of a Schiff base by L-histidine and o-phthalaldehyde ...32
Figure 2.2. Enzyme function of catechol oxidase...36
Figure 3.1. Crystallization of V. alginolyticus PepD by the hanging-drop method ...40
Figure 3.2. Overall structure of V. alginolyticus PepD. ...42
Figure 3.3. Determination of the PepD zinc ions...43
Figure 3.4. Topological diagrams of (A) V. alginolyticus PepD and (B) the lid domain of L. delbrueckii PepV ...45
Figure 3.5. Comparisons of catalytic domain from PepD, PepV and CPG2...46
Figure 3.6. Comparison of the active sites in V. alginolyticus PepD and structural homologs...48
Figure 3.7. Comparison of the lid domain structures of PepD, PepV, and CPG2...49
Figure 3.8. PepD dimeric interface ...50
Figure 3.9. Open and closed conformations of PepD and PepV ...54
Figure 3.10. CD spectra of V. alginolyticus PepD wild-type and various mutants...56
Figure 3.11. Molecular masses determination of PepDWT and PepDS374A/S385A mutant ...62
Figure 3.12 Proposed reaction mechanism of V. alginolyticus PepD ...65
x
Figure 3.14 Structures of catechol and its derivatives ...67 Figure 3.15 Protein-ligand docking of CuCu-PepD with (A) dopamine, (B) L-dopa, (C)
epinephrine, and (D) norepinephrine ...69
Table of Tables
Table 2.1. The fluent gradient program...33 Table 3.1. Enzyme kinetics of V. alginolyticus PepD wild-type and mutant proteins .58 Table 3.2. Enzymatic studies of V. alginolyticus PepD wild-type and mutant proteins
...58 Table 3.3 Substrate specificity of PepDWT and PepDCAT for 10 Xaa-His dipeptides, 2
His-Xaa dipeptides, and 2 His-containing tripeptides ...64 Table 3.4nComparison of enzyme kinetics of PepD, CuCu-PepD, and CuCu-SgAP for
different substrates ...68
List of Abbreviations
Ala Alanine Arg Arginine Asn Asparagine Asp Aspartic acid bp Base Pair
BSA Bovine serum albumin
CD Circular dichroism Cys Cysteine
ddH2O Double Distilled Water
DMF Dimethylformamide
xi EDTA Ethylenediamine-tetraacetic acid Gln Glutamine Glu Glutamic acid Gly Glycine H, His Histidine
HPLC High Performance Liquid Chromatography Ile Isolucine IPTG Isopropyl-1-thio-β-D-galactopyranoside kb(s) kilobase(s) Lys Lysine LA Lanosterol LB Luria-Bertani Leu Leucine Met Methionine OPA o-phthaldialdehyde
PCR Polymerase Chain Reaction PEG Polyethylene glycol Phe Phenylalanine Pro Proline RT Room temperature Ser Serine T, Trp Tryptophan TEMED N,N,N’,N’-tetramethylethylenediamine Thr Threonine TLC Thin Layer Chromatography Tris base Tris(hydroxymethyl)aminomethane Tyr Tyrosine U, Ura Uracil Val Valnine WT Wild-type X-gal 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside
xii
List of Genes or proteins
Acy1 Acylase 1
ApAP Aeromonas proteolytica aminopeptidase
AS -alanine synthase
CAT Catalytic domain CN1 Carnosinase 1 CN2 Carnosinase 2 CPG2 Carboxypeptidase G2
CuCu-PepD Copper-substituted Peptidase D
CuCu-SgAP Copper-substituted Streptomyces griseus aminopeptidase
LID Lid domain
1
CHAPTER 1
Introduction
1.1 Binuclear Metallohydrolases
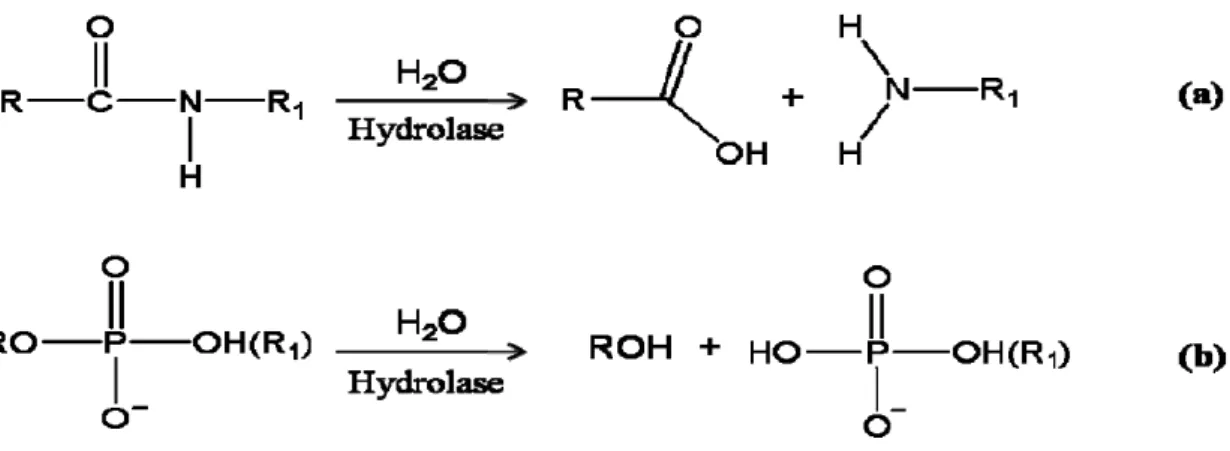
Nearly all metabolic and signaling pathways involve at least one event of hydrolytic cleavage of peptides or phosphate ester bonds. Although both types of bonds are considered as thermodynamically sensitive to hydrolysis, significant kinetic barriers exist that help keep these reactions tightly regulated under normal conditions1. For example, several hydrolases contain a co-catalytic metallo-active site that mediates reactions by lowering to the transition energy state. The binuclear metallohydrolases catalyze a diverse set of reactions including degradation of DNA, RNA, phospholipid, phosphoester, and polypeptides (Fig. 1.1). These enzymes are involved in a wide array of physiologic processes, such as hormone level regulation, tissue repair, protein maturation and degradation, and cell-cycle control1-4; likewise, perturbing the activities of binuclear metallohydrolases has been implicated in the early onset and progression of disease, such as carcinogenesis. Binuclear metallohydrolases have also been shown to mediate degradation of agricultural neurotoxins, urea, β-lactam-containing antibiotics, and several phosphorus materials used in chemical weaponry4. The majority of binuclear metallohydrolases are Zn2+-dependent proteins, but not exclusively, as the enzymes containing other divalent metal ions such as Mn2+, Co2+, Ni2+, and Cu2+ also exist.
2
Figure 1.1. Binuclear metallohydrolases catalyze hydrolysis of peptide or phosphate
ester bonds. Molecular illustrations for (a) peptidases that catalyze the hydrolysis of
peptide bonds in polypeptides, and (b) phosphoesterases and nucleases that catalyze the hydrolysis of phosphoester bonds in phosphorylated amino acids and saccharides, nucleotides, DNA, and RNA.
1.2 Metallopeptidases
Peptidase that requires a metal ion as a cofactor for catalytic activity was designated as metallopeptidase. The classification of metallopeptidases is based on the given functional groups within their active sites. The MEROPS database (http://merops.sanger.ac.uk) grouped the entire set of metallopeptidases into 15 evolutionarily-related clans (based on commonalities among the structural fold) and more than 50 families5, 6. Depending on the number of metal ions required for catalysis, metallopeptidases can be divided into two broad types: in many metallopeptidases: one which requires only one metal ion (in the major metallopeptidase) and the other one that requires two metal ions in “co-catalysis” (a special sub-set in the family). All known co-catalytic metallopeptidase functions as exopeptidase, including aminopeptidase, carboxypeptidase, dipeptidase, and tripeptidase. In contract, metallopeptidase that contains only one catalytic metal ion can act as either exopeptidases or endopeptidases,
3
in spite of existing some exceptions exist.
In metallopeptidase, the metal ions that coordinates with amino acid residues often serves as Lewis acid. These residues in coordination are usually charged residue, such as His, Glu, Asp, or Lys. In addition, at least one other residue is essential for catalysis, which has been theorized to act as a nucleophile that activates the metal ions and binds substrates. Results from crystallographic studies of solved metallopeptidases suggested that about one-half belongs to three clans, MA, MB and MX/MBA, and that each of these family members contains the HEXXH pentapeptide for chelating zinc ions5, 7.
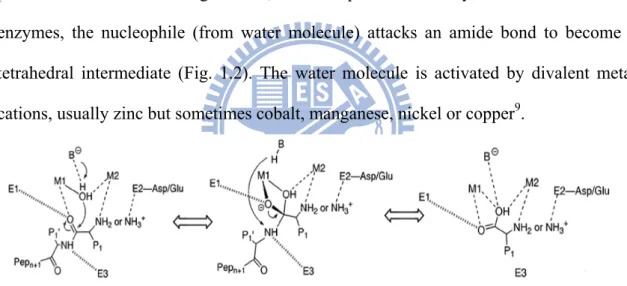
Metallopeptidases play fundamental roles in certain biochemical events, such as protein maturation and degradation, tissue repair and cell-cycle control8. In these enzymes, the nucleophile (from water molecule) attacks an amide bond to become a tetrahedral intermediate (Fig. 1.2). The water molecule is activated by divalent metal cations, usually zinc but sometimes cobalt, manganese, nickel or copper9.
Figure 1.2. A generalized mechanistic scheme for metallopeptidases. First, the
substrate binds to the metallopeptidase active site, with the carbonyl group of the scissile peptide bond interacting with M1 and a conserved enzyme residue, E1. The N-terminus either interacts with M2 or with one or more acidic enzyme residues in the vicinity (indicated in the middle reaction by E2). Additional enzymatic histidine or backbone carbonyl group interactions, at E3, facilitate substrate binding in the correct register. The scissile peptide bond is then attacked by a solvent molecule that has been activated by its interaction with the metal ions, and by an enzyme residue that functions as a general
4
base, B−. Whether or not the subsequent tetrahedral intermediate is stabilized by interactions with both metal ions and E2 side chains depends upon the particular enzyme system. Breakdown of the intermediate is most likely promoted by the addition of a proton to the amine group that has departed from the former general base, B-H, as first suggested in studies of thermolysin9.
1.3 Aminoacylhistidine dipeptidases in the peptidase M20 family
The 15 clans of metallopeptidases in the MEROPS database
(http://merops.sanger.ac.uk)10 are: MA, MC, MD, ME, MF, MG, MH, MU, MK, MM,
MN, MO, MP, MQ, and M-. The clan MH contains a variety of co-catalytic zinc-dependent peptidases that bind two atoms of zinc per monomer. The zinc atoms are held by five amino acid ligands, and binding can be inhibited by the presence of a general metal chelator, such as ethylenediamine-tetraacetic acid (EDTA) or orthophenanthroline (1,10-phenanthroline)6. The peptidase clan MH is further divided into four families: M18, M20, M28, and M42. The M20 peptidases, in particular, were characterized by their property of binding a water molecule along with by two zinc ions which are held by five residues at the active site, in the order of His/Asp, Asp, Glu, Glu/Asp, and His. Additional Asp and Glu residues adjacent to metal-binding residues are also are thought to be important for catalysis5. The Asp residue between two catalytic residues binds both metal ions. Several kinds of M20 family enzymes exhibit a binuclear-binding domain, including the dipeptidases (PepD11, PepT12, PepV13), aminopeptidases (Streptomyces griseus, SgAP)14, carnosinases (CN1, CN2)15, carboxypeptidases (CPG2)16, β-alanine synthases (βAS)17, desuccinylases (DapE)18 and
deacetylases (ArgE)19. Variations exist in the individual sub-families that are revealed upon amino acid sequence alignment. Peptidase family M20 has been further divided
5
into four sub-families, based upon the distinct active site residues: M20A, M20B, M20C, and M20D.
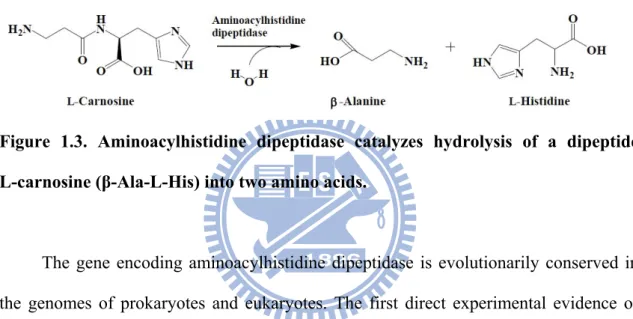
Aminoacylhistidine dipeptidases (EC 3.4.13.3, published under the names Xaa-His dipeptidase, X-His dipeptidase, carnosinase, and PepD) belong to the M20C sub-family. These zinc-containing metallopeptidases catalyze the cleavage and release of an N-terminal amino acid, usually a neutral or hydrophobic residue, from Xaa-His dipeptides or polypeptides (Fig. 1.3)5.
Figure 1.3. Aminoacylhistidine dipeptidase catalyzes hydrolysis of a dipeptide
L-carnosine (β-Ala-L-His) into two amino acids.
The gene encoding aminoacylhistidine dipeptidase is evolutionarily conserved in the genomes of prokaryotes and eukaryotes. The first direct experimental evidence of aminoacylhistidine dipeptidase activity was obtained in 1974 when the carnosinase from the bacteria Pseudomonas aeruginosa was demonstrated to hydrolyze the dipeptide L-carnosine20. Over the next few years, more of these enzymes were discovered in several other bacterial species or higher order organisms; however, only the PepD from
Escherichia coli has been fully characterized using genetic and biochemical
approaches21.
The E. coli pepD gene encodes a 52 kDa protein and is active as a homodimer, having a molecular mass of 100 kDa22. The pure enzyme exhibits a pH and temperature optimum of pH 9.0 and 37°C, respectively. E. coli PepD appears to function principally as an aminoacylhistidine dipeptidase with broad substrate specificity, which is activated
6
by Co2+ and Zn2+ and deactivated by metal chelators21. The previous report described how the expression of pepD negatively affects biofilm formation, a necessary process for infection to be established in a host system; in this case, a fish system was used to determine the molecular mechanisms by which bacterial infection may be significantly impacting fish mortality, and consequently marine economy23. The specific role of PepD in infection makes it a promising therapeutic target to control bacterial pathogenic infectious disease.
In more general terms, however, dipeptidases are involved in the final breakdown of protein degradation fragments produced by the targeted actions of other peptidases, or of dipeptides being processed for subsequent utilization of their amino acid components. Indeed, studies involving PepD-deficient mutants of E. coli24 and Salmonella typhimurium25, have indicated that PepD can hydrolyze dipeptides needed as an amino acid source. However, the full biological impact of PepD remains unclear.
1.4 Crystal structures of M20 family peptidases
To date, several of the M20 family enzymes have been crystallized and reported in the literature. These enzymes have exhibited an overall two-domain organization, including a di-zinc binding catalytic domain and a structurally similar but smaller domain. Nearly all M20 enzymes exist as homodimers. The Peptidase V (PepV) from
Lactobacillus delbrueckii, however, represents a notable exception as it appears to exist
7
1.4-1 Carboxypeptidase G2 (CPG2) from Pseudomonas sp. strain RS-16
Enzymes of the carboxypeptidase G class catalyze the C-terminal glutamate moiety hydrolyzation from folic acid and its analogues such as methotrexate27. In 1997, Brick and colleagues reported the first crystal structure of an M20 metallopeptidase28, 29. To obtain the crystal structure of carboxypeptidase G2 (CPG2) from Pseudomonas sp.
strain RS-16 at 2.5 Å resolution, the group used multiple isomorphous replacement (MIR) methods. CPG2 was determined to be a typical homodimer of the peptidase M20
family, in which each subunit is a component of a larger catalytic domain containing two zinc ions in the active site. The smaller domain forms the dimer interface by means of hydrophobic interactions between helices, as well as through hydrogen bonding between two β strands from each subunit (Fig. 1.4).
The catalytic domain of CPG2 comprises residues 23-213 and 326-415, which
contain the N- and C-terminal of the polypeptide chain, respectively. The dimerization domain of CPG2 consists of a single, 110 residues insertion in the catalytic domain
sequence, comprising residues 214-325. In the metal ions binding site within the catalytic domain of CPG2, two zinc ions (Zinc 1 and Zinc 2) are present, which are 3.3 Å
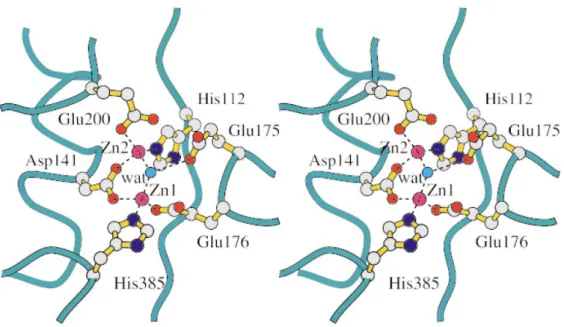
away from each other. Zinc 1 is bound by Asp141, Glu176 and His385, while Zinc 2 is bound by the other carboxylate oxygen of Asp141, Glu200 and His112. Furthermore, a water molecule is bound by Glu175 underneath and in the middle of the two zinc ions; this molecule is activated by the zinc ions in order to facilitate the nucleophilic attack on the substrate, which in turn forms the tetrahedral intermediate (Fig. 1.5).
8
Figure 1.4. Ribbon diagrams of the CPG2 dimers. Views from different orientations
are presented in (a) and (b). The two subunits are coloured in blue and yellow, respectively. The magenta spheres represent bound zinc ions. In (b), the continuous β sheet is visible across the two subunits forming the dimer interface29.
9
Figure 1.5. Stereo view of the metal ions binding site in CPG2. Ball-and-stick
representation of the two zinc ions and their binding residues are shown. Atoms are in standard colours. The bridging active-site water molecule (wat) is indicated as a light
blue sphere. The Glu175 residue is presumed to promote the attack of the water molecule on the substrate29.
1.4-2 Peptidase V (PepV) from Lactobacillus delbrueckii
The L. delbruekii pepV gene is 1413 nucleotides in length and consists of 470 amino acids that encode a protein with predicted molecular mass of 52 kDa. PepV functions as an aminoacylhistidine dipeptidase or carnosinase, cleaving Xaa-His dipeptide to generate a source of histidine. It has been characterized as a relatively non-specific dipeptidase, with demonstrated ability to cleave a variety of dipeptides. The broad dipeptide species is able to be hydrolyzed, especially those with an unusual β-alanyl residue in the N-terminus; in addition, the N-terminal amino acid has been shown to be targeted for removal from a few distinct tripeptides30.
10
(ArgE, EC 3.5.1.16) and succinyldiaminopimelate desuccinylase (DapE, EC 3.5.1.18). Most recently, it has been described as a member of the aminoacylase-1 family31, which can hydrolyze amide bonds in a zinc-dependent manner. Interestingly, when the dipeptidase pepV gene was deleted from Lactobacilli a significant decrease in growth rates was observed, but the final cell density remained unaffected.
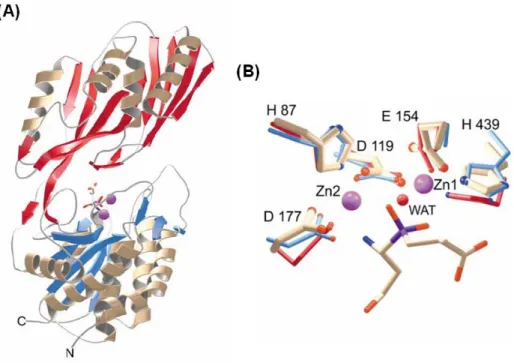
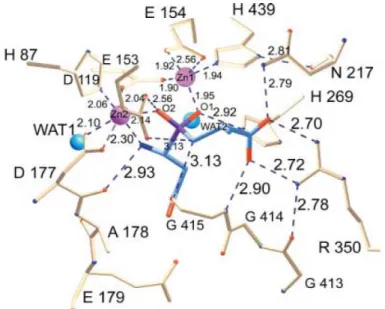
PepV from L. delbruekii was the first discovered crystallized dinuclear dipeptidase with carnosine-hydrolyzing enzymatic activity in the M20 family26. To date, PepV remains the only monomeric among all of the M20 family aminopeptidases. Based on its crystal structure, PepV was recognized as a metallopeptidase haboring two zinc ions in a single monomer. Therefore, it was theorized and demonstrated that PepV could be inhibited by metal chelation agents, such as 1,10-phenanthroline or EDTA. The 3D structure of PepV protein reveals two distinct domains, designated as the catalytic domain and the lid domain (Fig. 1.6A). The catalytic domain encompasses residues from Met1 to Gly185 and from Ser388 to Glu468, whereas the lid domain comprises the residues from Glu186 to Gly387. In the catalytic domain, five residues – His87, Asp119, Glu154, Asp177 and His439 – are critically responsible for chelating the two zinc ions (Fig. 1.6B). The mechanisms of substrate binding and hydrolysis are mediated by Glu153, Asn217, His269, Arg350 and Glu415 (Fig. 1.7).
11
Figure 1.6. The crystal structure of L. delbrueckii PepV. Ribbon diagram (A) of the
enzyme and local view (B) of the zinc ion binding cavity. In Fig. B, the inhibitor of
PepV (beige) is superimposed upon the zinc binding residues of Aeromonas proteolytica
ApAP (blue) and CPG2 (red). Residues are numbered according to PepV sequence. The
catalytic water molecule of CPG2 is depicted in red (WAT)26.
The two zinc ions, as described by Jozic et al., are presumed to play two different roles in the hydrolysis of substrates, that which stabilizes the substrate-enzyme tetrahedral intermediate and that which activates the catalytic water molecule (Fig. 1.7). Zinc 1, which is associated with the imidazole group of His439 and the carboxylate oxygen of Glu154 and Asp119, appears to primarily facilitate binding with His269 via an “oxyanion binding hole”. Such binding results in polarization of the scissile carbonyl group and consequently promotes nucleophilic attack by the catalytic water molecule. Zinc 2 is coordinated by His87, the carboxylate oxygen of Asp177, and the bridging Asp119. This ions appears to primarily function in the activation of the catalytic water molecule,
12
and acts as the strong Lewis acid promoting binding and hydrolysis.
Figure 1.7. Schematic of the active site of PepV. The Asp-Ala phosphinate inhibitor
mimics the dipeptide substrate, as shown in blue. The bridging catalytic water attacks the carbonyl carbon of the scissile peptide bond, forming the sp3-orbital substrate-enzyme tetrahedral intermediate26.
1.5 Application of M20 family peptidases in Antibody-Directed Enzyme Prodrug
Therapy (ADEPT)
M20 family enzymes hold great potential for biotechnological applications and therapeutic significance13, 26, 30-34 based upon their crucial roles in a wide variety of the most basic physiologic processes. For example, Lactobacillus sp. PepV13 and S.
typhimurium peptidase T (PepT)32 function in amino acid utilization, whereas E. coli allantoate amidohydrolase and yeast Saccharomyces kluyveri β-alanine synthase (βAS)33 are involved in the nucleotide catabolic pathways. In addition, other species-specific dipeptidases represent promising molecular tools to manipulate a desired physiologic state; these include the E. coli K12 PepD21, human brain-specific carnosinase (CN1) and
13
nonspecific carnosinase (CN2)15, and mouse CN235, that can regulate unusual dipeptides, such as L-carnosine (β-Ala-His) and L-homocarnosine (γ-amino-butyl-His), as well as a few other distinct tripeptides. Epidemiologic studies investigating the underlying molecular mechanisms of signal transduction-related diseases by evaluating sibships have indicated that deficiencies in serum carnosinase are significantly associated with tremor, myoclonic seizures, hypotonia, and profound psychomotor retardation36-40. The gastric ulcer- and cancer- causing Helicobacter pylori bacteria expresses a succinyldiaminopimelate desuccinylase that may be an effective target of antimicrobial agents41 to clear infection. Likewise, Pseudomonas sp. strain RS-16 carboxypeptidase G2 (CPG2) has been proposed for use in antibody-directed enzyme prodrug therapy for
the development of a rescue agent in cases of methotrexate overdoses16, 29, 42-44.
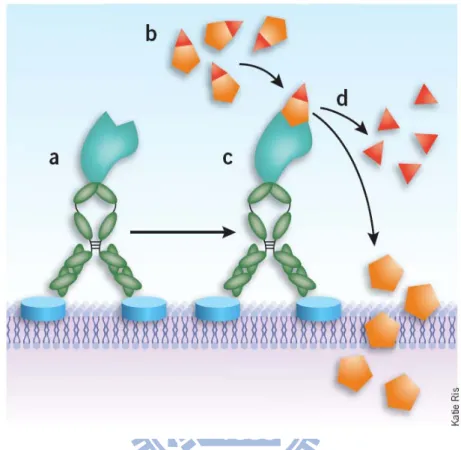
The idea of using enzymes as amplifiers to convert relatively nontoxic prodrugs into active cytotoxic agents was first explored in the 1960s. This strategy, is known as ADEPT45, 46, has been successfully applied to elimination of cancer cells47, 48. In principle (Fig. 1.8), an enzyme-monoclonal antibody (or fragment) conjugate, known as an antibody-enzyme conjugate (AEC), is administered systemically or locally at the tumor site. The molecule-specific antibody acts as a guidance system to target a specific antigen present the surface of the tumor cell (membrane) or in the extracellular fluid surrounding the tumors. Thus, the AEC is trapped in the region of the tumor, and in theory will not detrimentally affect normal tissues. The unbound AEC is expected, and has been demonstrated, to be cleared from the body through the standard urinary route.
Following administration of the AEC, the prodrug, an exogenous substrate for a particular endogenous enzyme, is administered systemically. In this manner, the prodrugs are only converted to active cytotoxic drugs when they reach the targeted tumor sites and diffuse through the tumor mass to enter cells. This approach is
14
advantageous in that it does not require a specific antigenic marker to be present on the tumor cells. Such site-selective prodrug activation has been demonstrated to result in reduced side effects to remote non-cancerous tissues.
Figure 1.8. Schematic of Antibody-Directed Enzyme Prodrug Therapy (ADEPT)42.
(a) First, the mAb-enzyme conjugate binds to its target tumor cell-surface antigen. (b-d) After unbound mAb-enzyme is actively or passively cleared from circulation, the cytotoxic agent is administered in an inactive (prodrug) form (b), which is selectively bound by the mAb-enzyme on the tumor cell surface (c), causing cleavage of the inactivating sequence from the prodrug and releasing multiple copies of active drug into the tumor microenvironment48.
ADEPT has been used in animal tumor models of human choriocarcinoma and colonic and breast carcinoma49-51. Recently, Francis et al. reported the results of a phase I ADEPT trial, in which a murine F(ab’)2 anti-carcinoembryonic (anti-CEA) antigen
15
fragment linked to the bacterial enzyme carboxypeptidase G2 (CPG2) was used in
conjunction with the bis-iodo phenol mustard prodrug in patients with advanced colorectal carcinoma or other CEA-expression tumors52. Although this trial did not result in tumor regressions, it did demonstrate that a potent prodrug could be administered with acceptable toxicities and that were primarily limited to myelosuppression.
In 1985, the Charing Cross group developed monoclonal antibodies (W14 and SB10) to human β chorionic gonadotrophin and anti-CEA antibody (A5B7)45. They designed their studies and reagents based on the fact that target antigens should be expressed on the membrane of tumor cells or secreted into the extracellular space of tumors. Oncogen products or overexpressed gene products that have an external domain on the cell membrane, such as some growth factor receptors, should also be considered as useful targets. The synthetic benzoic acid mustard prodrug (4-[bis-(2-chloroethyl)-amino] benzoyl-L-glutamic acid), the investigators chose to use is known to be targeted by CPG2 for cleavage of its terminal glutamic acid residue to
generate the active alkylating agent 4-[(2chloroethyl)[2(mesylozy)ethyl]-amino] benzoic acid (Fig. 1.9), and subsequently be actived in cancer cells.
Figure 1.9. Benzoic acid mustard prodrug (4-[bis-(2-chloroethyl)-amino]
16
1.6 Catechol oxidase / tyrosinase
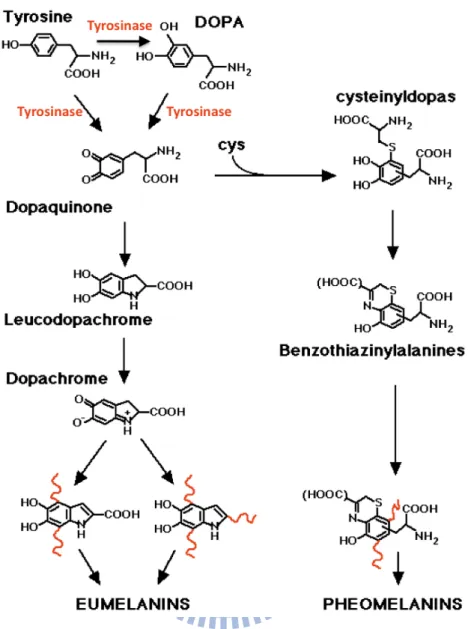
Catechol oxidases (EC 1.10.3.1) and tyrosinase (EC 1.14.18.1) are structurally similar enzymes and both belong to the type-3 copper proteins53. These enzymes are widely distributed among plant, animal and fungal species, in which they facilitate production of melanin derivatives (Fig. 1.10)54. Two-electron transfer reactions are carried out during the oxidation step involving a broad range of o-diphenols and the corresponding o-quinones55, 56. Once activated, the quinones are auto-polymerized and are able to form the brown polyphenolic catechol melanins, which act to protect animal skin from sun damage and plants from penetration by pathogens or insects57. Furthermore, tyrosine is able to be oxidized by tyrosinase to produce L-3,4-dihydroxyphenylalanine (L-Dopa) in animals, and L-dopa is the upstream substance of several neurotransmitters, such as dopamine and epinephrine. Thus, catechol oxidase/tyrosinase is not only the key enzyme in synthesis of melanin derivatives, but also regulates the biosynthesis and metabolism of neurotransmitters58, 59.
17
Figure 1.10. The tyrosine metabolism pathway. Tyrosinase catalyzes the
o-hydroxylation of phenols, and further oxidation of the resultant catechol leads to
processing of o-quinone.
1.7 Crystal structure of the catechol oxidase from sweet potato
In 1998, the first crystal structure of catechol oxidase from sweet potato was determined by Klabunde and colleagues.60 The structure revealed a monomer of ~39,000
Mr that formed an ellipsoid shape with dimensions of 55 × 45 × 45 Å (Fig. 1.11A). In
18
coordinated by three histidine residues. Specifically, CuA is chelated by His88, His109 and His118, while CuB is chelated by His240, His244 and His274 (Fig. 1.11B). Notably, there is a covalent cycteine-histidine bond that exists between the Cε atom of His109, one of the copper ions, and the sulfur atom of Cys92 in metal ions center. The covalent cysteine-histidine bond has been suggested to maintain an enatic state that stabilizes the electronic structure of the metal to facilitate oxidation of the o-diphenol substrate and the following rapid electron transfer of the redox processes.
Figure 1.11. Crystal structure of catechol oxidase from sweet potato60. (A) Ribbon
diagrams of overall structure. (B) Diagram of the electron density in the oxidized catalytic dinuclear copper site. The sulfur atom of Cys92 presented here does not ligate to the copper center, but represents that covalently bound to the Cε atom of His109.
Based on the crystal structure of catechol oxidase in the resting dicupric Cu(II)-Cu(II) state, it appears that oxidation of the reduced dicuprous Cu(II)-Cu(II) form, as well as that in complex with the inhibitor – phenylthiourea (PTU), is carried out in the dicopper center by means of a four-electron reduction of the molecular oxygen to water (Fig. 1.12a). In the reduced enzyme state, the dioxygen molecule binds to the dicuprous metal center, replacing the water molecule bonded to CuA (Fig. 1.12b, and top of Fig.
19
1.12a). Next, the dicopper center opens via the rotation of the side chain of Phe261 to allow the catechol substrate (CAT) access to the active site. The crystal structure of the catechol oxidase-PTU complex (Fig. 1.12e, and bottom of Fig. 1.12a) suggested that simultaneous binding of CAT and dioxygen is feasible. Furthermore, it appears that the complex favors a mono-dentate binding model (Fig. 1.12c, and second from the top in Fig. 1.12a) in which the ortho-hydroxyl groups from CAT bind to CuB after deprotonation. The finding of the Glu236 hydrogen bound to a solvent molecule proximal to the dicopper center suggested a potential role for this residue in substrate deprotonation. In the proposed ternary catechol oxidase-O22-CAT complex, protonation
of the peroxide group and cleavage of O-O bond are carried out by two-electrons transfer from the substrate to the peroxide. Glu236 along with the second non-coordinating hydroxyl group of the substrate might donate a proton and promote the loss of water and the eventual production of o-quinone. Finally, protonation of the bridging group by solvent leads to the active site entering into a resting hydroxide-bridged dicupric state (Fig. 1.12d, and second from the bottom in Fig. 1.12a).
20
Figure 1.12. Proposed reaction pathway of catechol oxidase. (a) Schematic diagram
of the reaction catalyzed by catechol oxidase. (b-d) The 3D structure is presented for each of the reaction steps, as derived from crystallographic analysis. Note that the ternary catechol oxidase-O22-CAT complex shown in (c) is a model structure, guided by
21
1.8 Catechol oxidase activity of di-Cu2+-substituted aminopeptidase from
Streptomyces griseus (SgAP)
It is generally believed that enzyme catalysis has evolved in order to carry out specific chemical changes in stable transition states of a given substrate that is required for normal physiologic processes. One such example is that of the peptide-bond hydrolysis mediated by metallopeptidases that occurs during the tetrahedral transition state61. However, enzymatic promiscuity is a common occurrence in nature, that being when more than one type of reaction is catalyzed by a single enzyme, and is believed to have resulted from distinct routes of evolution among members of a superfamily62-65. In recent years, the novel approaches to explore and discover the new metalloprotein systems were carried out to regulate their activities with the active-site metals that using apo metalloprotein molecules as natural ligands and observing the effects of binding. To date, however, only a few papers have reported results from altering catalytic specificity by simple metal substitution of metalloenzymes66.
Recently, the dinuclear aminopeptidase from Streptomyces griseus, SgAP, was found to exhibit a high efficiency of catalytic promiscuity toward phosphoester hydrolysis under different physiological conditions67-69. Moreover, the peptide hydrolysis activity of SgAP70, 71 was able to be converted to catechol oxidative activity by performing a di-Cu2+ substitution of the di-Zn2+ in the metal binding center72, 73. The resultant CuCu-SgAP could effectively catalyze oxidation of catechol and catechol derivatives, such as 3,5-di-tert-butylcatechol (DTC), dopamine, and 4,5-dichlorocatechol (DCC). In fact, the oxidative efficiency of CuCu-SgAP for DTC (kcat/Km = 3295 M-1s-1) was much better than that of a number of artificial synthetic
metal complexes and was only ~10 times smaller than the natural catechol oxidase from
22
catechol oxidase were structurally compared, it was determined that they both harbor a “metlike” di-metal ions site with a bridging OH or H2O between the two metal ions
centers that likely plays a critical role in catalysis75. Although the active sites in these two enzymes are quite different (one being a mixed His/carboxylate environment (Fig. 1.5 and Fig. 1.6B), the other an all-His environment (Fig. 1.11B)), the catalytic promiscuity of SgAP reflects that the active site does not need to be restricted to one structural pattern. This finding revealed a new direction of oxidation catalysis for rational design for ligands and proteins. To date, SgAP and its metal-substituted derivatives remain the only protein system with obvious multiple and diverse catalytic activities, those being phosphodiesterase and catechol oxidase. Therefore, this enzyme and its metal-substituted derivatives are considered amenable to preparations as unique dinuclear systems that may provide further detailed understanding of the correlation between structure and mechanism of metal-centered hydrolytic and oxidation/oxygenation chemistry. In addition, this study may also serve as a “living fossil” system to untangle the mysteries of divergent enzyme evolution.
1.9 Thesis purpose
Aminoacylhistidine dipeptidase is widely distributed among prokaryotes and eukaryotes, and its biological significance and function have been evolutionarily conserved as well. PepD is well known to hydrolyze physiologically-important dipeptides into two amino acids, such as carnosine and homocarnosine, but the structure-reaction relationship of PepD has yet to be studied in detail. V. alginolyticus PepD shares only 20% amino acid sequence identity with the several other M20 family enzymes whose crystal structures have been reported76. In our previous studies, we had cloned and expressed the V. alginolyticus pepD gene and were able to carry out detailed
23
biochemical characterization, including defining the enzyme kinetics of the produced recombinant protein76. The subsequent studies that comprised this thesis aimed at investigating the structure-function relationship by using protein X-ray crystallography coupled with site-directed mutagenesis77, 78. Enzyme kinetic studies were also carried out to determine the reaction rate and underlying catalytic mechanism. Ultimately, our characterization of the active-site architecture of PepD enzyme will aid in future studies to identify residues that may be modified to yield alternative substrate recognition properties and improve the potential therapeutic value of this protein and its closely related family members.
On the other hand, previous studies have reported that substituting zinc ions for copper ions in the active site of aminopeptidases allowed for the enzyme function to be effectively converted from peptide hydrolase to catechol oxidase72. Therefore, the copper-substituted PepD (CuCu-PepD) was prepared to investigate whether conversion of enzyme function could be attained. Moreover, the robustness of the new activity was evaluated, as well as the new substrate selectivity, by using several catechol derivatives. The findings from this study are expected to provide further insights into the general enzyme activities of peptide hydrolysis and catechol oxidation, and advance our understanding of the correlation between specific metal ions and enzyme function. Finally, this study might also reveal a new direction for divergent enzyme evolution.
24
CHAPTER 2
Materials and Methods
2.1 Materials
2.1-1 Bacterial strains, molecular cloning/expression vectors, and resin
In this study, bacterial strains Escherichia coli XL1-Blue, BL21(DE3) and BL21(DE3) pLysS were purchased from Novagen. Plasmid vector pET-28a(+) and pCR®2.1-TOPO were purchased from Invitrogen and Novagen, respectively. Bacterial growth condition, agar plate preparation, recombinant DNA purification, DNA sequence determination, agarose gel electrophoresis and protein concentration determination were performed according to standard procedures or the commercial kit. Ni-NTA His-Band® Resin was purchased from Amersham Pharmacia Biotech.
2.1-2 Enzymes, chemicals, equipments, and reagents
(1) Enzymes: All restriction enzymes were purchased from New England BioLabs lnc. The pfu DNA polymerase was purchased from Stratagene. The T4 DNA ligase was purchased from Promega. All enzymes were used according to the recommended protocols.
(2) Chemicals: The following section lists the chemicals utilized in this study. The manufacturers were included in the square bracket. Acetic acid [Merck], Acrylamide [GE Healthcare], Agarose [USB], APS [GE Healthcare], α-Ala-L-His [Sigma],
25
β-Asp-L-His [Sigma], Bestatin [MP Biomedicals], BactoTM Agar [DIFCO], L-carnosine [ICN Biomedicals, Inc.], Citric acid [Sigma], Coomassie® Brilliant blue R250 [Merck], Catechol [Alfa Aeser], Deoxyribonucleotide triphosphate (dNTP) 100mM Solutions [GE Healthcare], DNA 10kb Ladder [BioBasic Inc.], L-Dopa [Sigma], Dopamine [Sigma], Ethanol (95% and 99%) [Merck], GABA-His [Sigma], Glycine [Merck], Gly-Gly-His [Sigma], Gly-His-Gly [Sigma], Gly-His [Sigma], Glycerol [Merck], L-Histidine [Sigma], His-His [Bachem], His-Val [Bachem], His-Ile [Bachem], L-homocarnosine [Sigma], Hydrogen chloride [Merck], Ile-His [Bachem], Imidazole [USB], IPTG [GeneMark, Taiwan], Kanamycin sulfate [USB], LB Broth (Miller) [DIFCO], Leu-His [Bachem], Methanol [Merck], Oligonucletide Primers [BioBasic Inc.], Potassium chloride [Merck], Potassium diphosphate [Merck], Ser-His [Bachem], Sodium chloride [AMRESCO], Sodium hydroxide [Merck], SYBR®
Green I [Roche], TEMED [GE Healthcare], Tris base [Merck], Tyr-His [Bachem], Val-His [Bachem], X-gal [GeneMerck, Taiwan], Yeast Extra [DIFCO].
(3) Kits: The following experimental kits were used in this study. BCA Protein Assay Reagent and Albumin Standard [PIERCE], GFXTM PCR DNA and Gel Band purification Kit [GE Healthcare], Plasmid Miniprep Purification Kit [GeneMark], QIAamp DNA Mini Kit [Qiagen], TOPO TA Cloning® Kit [Invitrogen], Plasmid Miniprep Purification Kit [GeneMark].
(4) Equipments: The following instruments were used in this thesis: ABI PRISM® 3100 Genetic Analyzer [Applied Biosystems], Amicon® Ultra [Millipore], Centrifuges 5415R [eppendorf], Electrophoresis Power Supply EPS 301 [GE Healthcare], Fluoroskan Ascent FL Microplate Reader [Thermo], GeneAmp® PCR System 9700 Thermal Cycler [Applied Biosystems], High-Performance Liquid Chromatography (HPLC) [Agilent], Kodak Electrophoresis Documentation an Analysis System 120 [Kodak], Mighty Small
26
II for 8×7 cm gels electrophoresis instruments [GE Heathcare], Millex®-GS 0.22 μm Filter Unit [Millipore], Millex®-GS 0.45 μm Filter Unit [Millipore], Multiskan Ascent Microplate Reader [Thermo], SteritopTM 0.22μm Filter Unit [Millipore], Ultrasonic Processor VCX 500/750 [Sonics], UV-visible spectrometer [Amersham Biosciences]. (5) Reagents
Ampicillin stock solution (100mg/mL):
Dissolve 1 g Ampicillin sulfate in 10 mL double distilled water (ddH2O). Filter it
through a 0.22 μm pore size filter and stock it at -20°C.
Destain buffer I:
Mix 400 mL methanol, 100 mL acetic acid and distilled water (dH2O) to 1 L. Store at
room temperature.
Destain buffer II:
Mix 50 mL methanol, 120 mL acetic acid and dH2O to 1 L. Store at room temperature.
6 X DNA loading dye:
0.25% Bromophenol blue and 30% glycerol in ddH2O. Store at -20 °C.
IPTG stock solution:
Dissolve 4.086 g IPTG in 10 mL ddH2O. Filter through 0.22 μm pore size filter and
store at -20 °C.
Kanamycin stock solution:
Dissolve 250 mg kanamycin sulfate in 10 mL ddH2O. Filter through 0.22 μm pore size
filter and store at -20 °C.
LB medium:
27
LB plate:
25 g LB Broth and 20 g BactoTM Agar was dissolved in 1 L ddH2O and sterilized. The
sterile solution was poured and dispersed in Petri dishes before coagulation.
OPA reagent (for enzymatic activity assay):
Dissolve 50 mg OPA in 5 mL methanol first and then mix with 20 mL borate buffer. The borate buffer was mixed by 0.2 M boric acid (dissolved in 0.2 M potassium chloride solution) and 0.2 M sodium hydroxide solution (50 : 50, v/v).
10 X SYBR Green solution:
10,000 X SYBR®
Green I was diluted to 10 X with DMSO. Store it under darkness.
50 X TAE buffer:
Add 242 g Tris-base, 57.1 mL Acetic acid, and 0.5 M EDTA into 800 mL ddH2O. Adjust
the total volume into 1 L and pH value into pH 8.5. Store it at room temperature. Dilute the concentration to 1 X with ddH2O. Adjust the pH value to 7.5~7.8 before utlization.
X-gal stock solution:
Dissolve 400 mg X-gal in 10 mL dimethylformamide (DMF) and store in the darkness at -20 °C
2.2 Experimental methods
2.2-1 Site-directed mutagenesis of V. alginolyticus pepD
Site-directed mutagenesis of the V. alginolyticus pepD gene was carried out using the Stratagene QuikChange site-directed mutagenesis kit (Agilent Technologies, Inc., Santa Clara, CA). Mutagenesis primers were designed for use with the pET-28a(+)-pepD plasmid (wild-type) template (Appendix 1). The PCR reaction was carried out via the nonstrand-displacing action of pfuTurbo DNA polymerase to extend and incorporate the
28
mutagenic primers, resulting in the nicked circular strands. The PCR products with wild-type and mutant plasmids were incubated with Dpn I for 4 hrs at 37 °C to selectively digest the methylated non-mutated parental wild-type plasmids. After Dpn I digestion, the mutant plasmids were transformed into E. coli XL-1 Blue competent cells, with selection for kanamycin resistance. Mutations were confirmed by restriction enzymes and DNA sequencing. The recombinant mutant plasmids were transformed into
E. coli BL21(DE3) competent cells for expression of the mutated PepD proteins.
2.2-2 Construction of the truncated V. alginolyticus pepD catalytic domain gene
The truncated V. alginolyticus PepD catalytic domain gene (pepDCAT) was composed of the pepD gene sequence of nucleotides 1-558 and 1203-1470. An 826-bp fragment, which included 558 bp of the 5’-end and 268 bp of the 3’-end of the pepD gene, was amplified by PCR using the following primer pairs: CYC-PepD-BamHI-1 (sense primer, 5’-CGGGATCCGTGTCTGAGTTCCATTCTG-3’) and CYC-PepDcat-4 (antisense primer, 5’-TCCAGCCTGGTCCTGCACAACCCATGTACAC-3’) and by
CYC-PepDcat-3 (sense primer, 5’-TGGGTTGTGCAGGACCAGGCTGGAAACCAGATG-3’) and CYC-PepD-XhoI-2
(antisense primer, 5’-CGCTCGAGTTACGCCTTTTCAGGAATG-3’). The PCR product was subcloned into pET-28a(+)-pepDCAT, which was then transformed into E. coli BL21(DE3) pLysS competent cells for the production of the PepD catalytic domain protein (PepDCAT).
29
2.2-3 Expression of the V. alginolyticus pepD gene, mutant pepD gene and truncated
pepD gene in E. coli
The wild-type PepD, mutant PepD and PepDCAT proteins were produced in the same manner. Colonies grown on an LB plate were inoculated into LB broth supplemented with 50 μg/mL of kanamycin and grown at 37 °C until A600 of 0.5-0.6 was
reached. At this point, protein production was induced by the addition of isopropyl thio-β-D-galactoside to a final concentration of 0.5 mM, and the culture was incubated at 37 °C for an additional 4 hrs before harvest. The cells were collected by centrifugation and then resuspended in 15 mL of 20 mM Tris-HCl (pH 7.0) buffer containing 0.5 M NaCl. The mixture was sonicated, and the cell debris was removed by centrifugation at 11,000 ×g for 30 min at 4 °C.
2.2-4 Portein purification of wild-type PepD, mutant PepD and PepDCAT
The wild-type PepD, mutant PepD and PepDCAT proteins were purified in the same protocol. The supernatant containing recombinant protein was loaded onto a Ni-SepharoseTM 6 Fast Flow column previously prepared by washing with 10 column volumes of buffer A (20 mM Tris-HCl, 0.5 M NaCl, pH 7.0) containing 20 mM imidazole. The protein-loaded column was first washed with 5 column volumes of buffer A + 20 mM imidazole, then with 5 column volumes of buffer A containing 70, 200, or 500 mM imidazole. Fractions of 1 mL each were collected, and the protein concentration in each fraction was determined using the PIERCE BCA Protein Assay Reagent with BSA as the standard. In addition, the eluted fractions were collected for SDS-PAGE analysis and enzymatic activity assay. By SDS-PAGE analysis, the high-purity eluted fractions were collected and dialyzed with 2 L of 50 mM Tris-HCl
30
(pH 7.0) buffer for 2 hrs, followed by 3 L buffer for 8 hrs. After enzymatic activity assay, the purified recombinant proteins were stored at -80 °C for up to 6 months without loss of activity.
2.2-5 Protein concentration determination
The protein concentrations of purified proteins were measured using BCA Protein Assay Reagent. To each well of the F96 MicroWellTM plate was added a 20 μL sample mixed with 200 μL BCATM Working Reagents (BCATM Reagent A : BCATM Reagent B = 50 : 1). The reactions were incubated at 37 °C for 30 min in the dark. The absorbances of samples were measured at 562 nm on a Multiskan Ascent Microplate Reader. 2 mg/mL bovine serum albumin (BSA) stock and successive dilutions (1.5, 1.0, 0.75, 0.5, 0.25, 0.125, 0.025 mg/mL) served as standards, following the same procedure described above.
2.2-6 SDS-PAGE and Native-PAGE analysis
After protein purification, gel electrophoresis was used to confirm for protein expression level, purity, and molecular weight. The samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 12.5% gels. Each 10 μL sample was mixed with 2 μL 5X SDS-PAGE sample buffer and incubated at 95ºC for 5 min to denature proteins. Electrophoresis was performed with 1X SDS-PAGE running buffer at 90 Volts for 30 min, followed by 120 Volts for 1.5 hrs. The SDS-PAGE gel was stained with a stain buffer containing Coomassie Brilliant blue R-250 for 30 min and destained with destain buffer I (methanol/acetic acid/water = 4:1:5, v/v/v) for 20 min, followed by destain buffer II (methanol/acetic acid/water = 1.2:0.05:8.75)
31
overnight.
Native-PAGE was performed to examine the native form of PepD. The purified and dialyzed protein fractions were separated by Native-PAGE on 7.5% gels. The experimental steps were similar to SDS-PAGE analysis, except that the gel contained no SDS and there was no denaturing treatment. Each 10 μL sample was mixed with 2 μL 5X Native-PAGE sample buffer, and immediately followed by an iced 1X Native-PAGE running buffer at 90 Volts for 3 hrs in a 4ºC circulating water bath. The proteins were stained and destained in the same way as for SDS-PAGE analysis.
2.2-7 Enzymatic activity assay of PepD
PepD activity assay was according to a method described by Teufel et al15. on the
basis of measurement of histidine derived with o-phthaldialdehyde (OPA) (Fig. 2.1). The reaction was initiated by addition of substrate and stopped by adding trichloroacetic acid (TCA) after 15 min incubation at 37 C. Histidine was produced from the substrate hydrolyzing by the enzyme. Then, fluorescence of OPA-derived L-histidine was measured using Fluoroskan Ascent FL (Exc: 355 nm and Em: 460 nm). The reaction
with L-histidine and L-carnosine only solution were treated in the same way to serve as the positive and negative control, respectively. All reactions were carried out in triplicate.
32 L-Carnosine N NH O OH H N H2N O OH H PepD HO NH2 O -Alanine L-Histidine N HN NH2 OH O N HN N N H N
OPA (Schiff base)
NH2 OH O L-Histidine H O H O OH O
Figure 2.1. Formation of a Schiff base by L-histidine and o-phthalaldehyde.
2.2-8 Enzyme kinetics
For determination of Vmax, Km, and kcat of V. alginolyticus PepD wild-type and mutant
proteins, the method described by Csámpai et al. was slightly modified for use with High Performance Liquid Chromatography (HPLC) and fluorescence detection. A system consisting of an Agilent 1100 Series Quaternary pump, Autosampler, Fluorescence Detector and Inertsil ODS-3 (7 μm, 7.6 mm×250 mm) column was used. The eluent system consisted of two components: eluent A was 0.05 M sodium acetate at pH 7.2, while eluent B was prepared from 0.1 M sodium acetate–acetonitrile–methanol (46:44:10, v/v/v) (titrated with glacial acetic acid or 1 M sodium hydroxide to pH 7.2). The gradient program was as described in Table 2.1. The fluent flow-rate was 0.8 mL/min at 30 ºC.
33
Table 2.1: The fluent gradient program
Step Time (min) A (%) B (%) 1 0 100 0 2 5 50 50 3 15 25 75 4 20 0 100
Different concentrations of L-carnosine (0.25, 0.5, 1, 1.5, 2, 5, and 10 mM) were added as to a nanomolar concentration of enzyme solution in 200 μL at pH 7.0 for 20 min at 37 °C. The liberated histidine was derivatized with 100 μL OPA reagent for 5 min at 37 °C, and the fluorescence was detected as described previously. Fluorescence of the histidine with derivatived OPA was measured by FLD (λExc: 355 nm and λEm: 460 nm).
Various concentrations of L-histidine solution derivatived with OPA reagent were detected, using the method described above, to serve as standards. The data collected were applied to the Lineweaver-Burk equation. The kcat/Km values reflect values
assuming 100% activity of the enzyme preparation.
2.2-9 Circular dichroism (CD) spectroscopy
The secondary structure of the wild-type and the mutant PepD proteins were confirmed by monitoring CD spectra. The protein sample concentration was 0.2 mg/mL in 50 mM Tris-HCl, pH 7.0 buffer. The CD spectra were recorded every 1 nm between 200 to 300 nm wavelength used a quartz cuvette of 1 mm path-length in a Jasco J-715 spectropolarimeter, Only 50 mM Tris-HCl, pH 7.0 buffer was as the control. The results were scanned 4 times and averaged. Converted the data into mean residue ellipticity (MRE) by using the equation : [θ]MRE = (MRW × θobs/c × d). θobs is the observed
34
the enzyme (MRW = M/n, M = 53548.8 g/mole, n = 490 amino acid residues), d is the cuvette path-length in cm, and c is the protein concentration in mg/mL.
2.2-10 Analytical sedimentation velocity ultracentrifugation
Sedimentation velocity is an analytical ultracentrifugation (AUC) method that measures the molecular moved rate for providing both the molecular mass and the shape of molecules. This technique can distinguish the native state of the protein in either a monomer, dimmer, or even tetramer form. The data were evaluated according to the g*(s) method developed by Walter Stafford. Since the g*(s) analysis yields both the sedimentation coefficient s from the peak of the curve, the apparent molecular weight can also be determined. Depending on the application and optical system, the protein concentration ranging 0.1 mg/mL to 0.5 mg/mL was used and the sample volume was about 500 μL. Sample was equilibrated with 20 mM Tris-HCl pH 7.0 buffer and this equilibrated buffer was used as another reference control into the reference sector. The sedimentation velocity analysis was performed at National Tsing Hua University.
2.2-11 Crystallization and data collection of PepD crystals
Crystallization of PepD was performed at 291 K by the hanging-drop vapor-diffusion method against a reservoir solution containing PEG 400 (28%, v/v), 0.2 M CaCl2 and 0.1M Na-HEPES buffer (pH 7.5), Crystals of a diamond shape appeared
within six months and grew to maximum dimensions of 0.3 × 0.2 × 0.1 mm3. The protein crystals were transferred to the cryo protectant solution containing glycerol (15%, v/v) prior to the X-ray diffraction experiment. Diffraction data were collected to 3.0 Å resolution on SPXF beamline BL13B1 at the National Synchrotron Radiation Research
35
Center (NSRRC) in Taiwan and beamline BL12B2 at SPring-8 in Japan. The data were processed using the HKL2000 suite79. The redundancy independent merging R factor (Rr.i.m.) and the precision indicating merging R factor (Rp.i.m.) were calculated using the
program RMERGE80, 81. The crystals belong to space group P65 with unit cell parameters
a = 80.42 Å and c = 303.11 Å. The asymmetric unit contained two protein molecules,
corresponding to a solvent content of 53.4%.
2.2-12 Structure determination and refinement
The structure was solved by molecular replacement with MOLREP (CCP4) using the structure of Xaa-His dipeptidase from Haemophilus somnus 129PT (PDB code 2QYV) as the search model. The orientation of the lid domain was first located and fixed, subsequently leading to the determination of the relative position of the single catalytic domain. For structural refinement, the model was built using WinCOOT and refined using REFMAC5 (CCP4) to give the final Rwork = 0.231 and Rfree = 0.27482,
respectively. The Ramachandran results were determined using MOLPROBITY, and the percentage of residues in favored, allowed, and disallowed were 94.5, 98.6, and 1.4%, respectively83. The structure found to have good stereochemistry was fully defined from Glu3 to Glu488, with all main chain angles in the most favorable or generally allowed regions84. All figures were produced using PyMOL. The atomic coordinates and structure factor data have been deposited in the Protein Data Bank (www.rcsb.org): PDB ID codes 3MRU.
36
2.2-13 Substitution of zinc ions by copper ions to form CuCu-PepD
PepD protein first was dialyzed overnight with 50 mM Tris-HCl buffer at pH 7.5 containing 200 mM NaCl and 10 mM EDTA to remove divalent zin ions (apo-PepD). The apo-PepD was dialyzed twice with the same buffer but without EDTA and exchanged with 50 mM Tris-HCl buffer at pH7.5 before adding 2mM CuCl2. After
dialyzing overnight, the pooled CuCu-PepD protein then were dialyzed with a 50 mM Tris buffer (pH7.0) and stored at -20 °C.
2.2-14 Oxidative activity assay of CuCu-PepD
Oxidation of various catechol derivatives by CuCu-PepD were determined via the measurement of the products of o-quinone moiety (Fig. 2.2A). The oxidative products of dopamine, L-dopa, epinephrine and norepinephrine, which would be tautomerized to from the aminechrome derivertives (Fig. 2.2B) were detected at λmax: 480 nm, 475 nm,
475 nm and 490 nm, respectively.
Figure 2.2. Enzyme function of catechol oxidase. (A) Oxidation of catechol
derivatives by catechol oxidase to form o-quinone moieties. (B) Overall reaction catalyzed by catechol oxidase with the substrate L-dopa. L-dopa undergoes oxidation to dopaquinone. Tautomerization of the dopaquinone ring by intramolecular nuclerophilic attck results in the formation of dopachrome.