行政院國家科學委員會專題研究計畫 成果報告
負載型奈米貴金屬對含氯有機物的脫氯反應(2/2)
計畫類別: 個別型計畫 計畫編號: NSC92-2211-E-002-063- 執行期間: 92 年 08 月 01 日至 93 年 07 月 31 日 執行單位: 國立臺灣大學環境工程學研究所 計畫主持人: 駱尚廉 計畫參與人員: 林進榮,歐信宏,強國倫 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 93 年 11 月 22 日
中文摘要 零價金屬自發性釋放電子還原水中含氯有機物已引起廣泛研究與運用,過去 十年,許多反應機制與動力學的研究建立了基礎的數據與認識,反應途徑的確認 有利於預估有毒中間產物的濃度與種類分布,速率影響因子的探討改善處理系統 的設計能力。目前技術的研究重點在於有效提高脫氯速率與降低毒性副產物的累 積,雙金屬與粒徑奈米化兩者是成效最顯著的技術,但是活性快速損失的特性限 制其運用的價值,因此,高活性的獲得與再生是突破瓶頸的關鍵。 本研究導入離子交換樹脂為載體,負載奈米金屬顆粒(nano-M0 )兼具回收水 溶性金屬離子(Mn+)的功能,接續地,中高溫的氫氣轉化 Mn+為 nano-M0提供反 應活性的再生。就還原方法而言:氣態還原的成本遠低於液相還原法(NaBH4 或 N2H4為還原劑)且熱處理可提高 nano-M0在載體上的機械穩定度;就功能而言:以 離子交換樹脂為載體可侷限 Mn+成核結晶的程度,降低 nano-M0的粒徑,同時可 避免 nano-M0 在反應過程中發生團聚作用,此外,樹脂的陽離子交換功能可吸 附反應產物- Mn+,避免出流水金屬濃度高於法規標準,同時釋出強酸離子(H+) 降低反應系統 pH 值; 就實用性而言: 質量輕、可塑性高的高分子載體具有安裝 與更新簡便的特性。 本研究分為三個部分,第一部分為材料的製備與特性分析;第二部分為含氯 有機物的破壞行為;第三部分為管柱實驗。第一部分的研究內容有載體上金屬顆 粒的粒徑分佈與表面積、金屬氧化態變化以及載體對金屬離子與目標污染物的吸 脫附行為;第二部分研究內容有三氯乙烯在 nano-M0 /resin(Cu、Sn、Sn-Pd)的 降解動力學反應、含氯中間產物與最終產物的產率;第三部分研究內容有材料活 性的壽命與再生以及水質條件導致的失活反應。 目前已完成的研究進度包括 nano-Cu0 /resin 的製備、特性分析與破壞四氯化碳 的反應動力學與機制研究,其結果如以下敘述:零價銅顆粒奈米化程度對反應 活性有相當大的影響,當銅顆粒平均粒徑為 33 nm,一階的比表面積反應速率 常數(surface-area-normalized rate coefficient, kSA)為粉末狀銅元素(0.04mm)的 19
倍; 銅顆粒平均粒徑為 8 nm,kSA遽增為 125 倍。利用正戊烷萃取殘存在水樣與 載體中四氯化碳的量,分辨 nano-Cu0/resin 對四氯化碳的吸附與破壞作用,其 中破壞的反應階數為 0.91-0.94 相當接近一階反應,樹脂對於 CCl4的吸附容量 約商業銅粉的 70 倍。產物的分析發現 30-40%碳回收率的 CO2濃度,極低量的 甲烷但無含氯甲烷類,ESCA 分析反應後複合材料發現主要為 CuO 與些微 CuCO3,故奈米銅將四氯化碳完全無毒化成二氧化碳為可能的反應機制。由於 強酸型陽離子交換樹脂以 H+交換水中反應產物 Cu2+,因此反應後水溶液 pH 約 3-4、殘餘銅離子濃度低於 0.1mg/L。負載奈米金屬於離子交換樹脂將可提供安 裝與更換便捷的系統設計,兼具反應金屬離子濃度的控制與回收。
Abstract
Nanoscale zerovalent copper supported on a cation resin was successfully synthesized to enhance the removal of carbon tetrachloride (CCl4) from contaminated water. The use of the cation resin as a support prevents the reduction of surface area due to agglomeration of nanoscale zero valent copper particles. Moreover, the cation resin recycles the copper ions resulting from the reaction between CCl4 and Cu0 by simultaneous ion exchange. The decline in the amount of CCl4 in aqueous solution results from the combined effects of degradation by nanoscale zero valent copper and sorption by the cation resin; thus the amount of CCl4 both in aqueous solution and sorbed onto the resin were measured. The pseudo-first-order rate constant normalized by the surface-area and the mass concentration of nanoscale zero valent copper (kSA)
was 2.1 0.1 ± ×10−2 L h-1 m-2, approximately twenty times that of commercial powdered zero valent copper (0.04 mm). Due to the exchange between Cu2+ and the strongly acidic ions (H+ or Na+), the pH was between 3 and 4 in unbuffered solution and Cu2+ at the concentration of less than 0.1 mg L-1 was measured after the dechlorination reaction. In the above-ground application, resin as a support would facilitate the development of a process that could be designed for convenient emplacement and regeneration of porous reductive medium.
KEYWORDS: nanoscale, zerovalent metal, copper, dechlorination, carbon
tetrachloride
1. Introduction
Chlorinated aliphatic organic compounds, particularly containing one to three carbon atoms, such as carbon tetrachloride (CCl4), trichloroethylene (TCE), and tetrachloroethylene (PCE), are widely used as industrial solvents in degreasing, washing, extraction, foaming, spraying, and manufacturing (Muftikian et al., 1995; Cheng and Wu, 2000). Improper use and disposal of these solvents has resulted in wide-spread occurrence in groundwater. In recent years, treatment of aqueous solution containing chlorinated solvents has attracted much attention. Conventional physicochemical methods accompanying the use of expensive chemicals or high energies largely restrict application at the field scale. Biological processes may be effective, but conditions must be well controlled to enable microorganisms to grow. Thus, it may not always be practical. Phytoremediation, using the ability of plants to remove and degrade chlorinated solvents, may offer a cost-effective, in situ, and safe alternative to conventional physicochemical methods (McCutcheon and Schnoor, 2003).
The use of zero valent metals as reductants to degrade chlorinated organic compounds is cost-effective and effectively reduces the concentration of target contaminants (Gillham and O’Hannesin, 1994; Matheson and Tratnyek, 1994; Agrawal and Tratnyek, 1996; Orth and Gillham, 1996; Gu et al., 1999; Phillips et al., 2000). Being inexpensive and benign in environment, zero valent iron (Fe0)is the most applied reactive metal; the potential for reducing more complex anthropogenic chemicals such as pentachlorophenol (Kim and Garraway, 2000), pesticides (Sayles et al., 1997; Eykholt and Davenport, 1998) and azo dyes (Nam and Tratnyek; 2000) has been demonstrated in the past few years. Recently, however, several drawbacks associated with the reaction between Fe0 and chlorinated organics included (1) as the
reaction proceeds, the pH in the reaction zone slightly rises (Matheson and Tratnyek, 1994; Phillips et al., 2000; Doong et al., 2003), caused by the net result of a number of chemical processes including iron corrosion and subsequent precipitation of ferrous iron (Su and Puls, 2004). At elevated pH, the precipitation of iron hydroxides and iron carbonates form and decrease reactivity by reducing the exposure of the iron surface to aqueous solution (Chen et al., 2001). The media can become plugged and cemented into pipes. (2) High concentration of iron ions, coming from several oxidation reactions, in the effluent of a above-ground reactor leads to the sludge disposal and treatment at additional cost and the potential blocking of pipes of the treatment system. (3) The difficulty and frequent replacement or the chemical reconditioning of the massive iron particles in the above-ground reactor results in higher costs.
The available surface area of iron is the most important factor that governs the reduction rate. Studies have demonstrated the reductive degradation rates for several target compounds are proportional to the total surface area of iron (Matheson and Tratnyek, 1994; Agrawal and Tratnyek, 1996; Siantar et al., 1996; Huang et al., 1998; Su and Puls, 1999). A nanoparticle is conventionally defined as the diameter of a particle in the 1 to 100 nm range (Klabunde, 2001). Reducing the particle size of an equivalent mass of Fe0 into the nanoscale range sharply increases reactivity (Boronina et al., 1995; Wang and Zhang, 1997; Choe et al., 2000). Zhang et al. (1998) reported that the surface-area-normalized rate constant of the reaction between nanoscale Fe0 and TCE or CCl4 can be increased by a factor of 10 to100 compared to commercial Fe0 powder (< 10 m). The dramatically increased reactivity results from a higher ratio of surface area to volume of nanoscale Fe0, higher levels of stepped surface (stepped surface is also known as high Miller index which usually has a step structure), and higher surface energies compared to powdered Fe0 (Zhang et al., 1998). Nanoscale Fe0 particle suspensions can be applied in an above-ground treatment reactor or directly injected into contaminated aquifers. However, the critical obstacles in the application of nanoscale Fe0 to reduce chlorinated solvents are (1) the immediate oxidation in air or water due to the active surface of nanoscale Fe0, (2) immediate agglomeration into micrometer particles due to the tendency to reduce the high surface energies, and (3) difficulty in separating such small particles from the effluent of the above-ground reactor when nonmagnetic iron oxides such as goethite and hematite form.
Use of a support medium to stabilize and isolate a metal particle avoids some problems with nanoscale particles (Ziolo et al., 1992). In this study, attachment to a commercial cation resin not only prevents nanoscale zero valent metal agglomeration but also recycles metal ions (the product of the dechlorination reaction) by simultaneous ion exchange. Furthermore, attachment to a fixed medium prevents metal ions from being washed out of the treatment system. Moreover, acidic cation resins release H+ or Na+ to exchange equivalent metal ionsand cause acidic conditions which can inhibit precipitation of metal salts. The polymer matrix is also light and easily deformable. The concept for the use of nanoscale zero valent metal coated resin mesh in an above-ground reactor and the reactions are shown in Figure 1.
In this work, zero valent copper was chosen as an electron donor to degrade CCl4. The use of copper as a reductant was preferred due to (1) the reliable in situ measurement of the surface area of resin-supported copper particles by temperature-programmed reduction (TPR) has been demonstrated (Grift et al., 1991), and (2) nanoscale zero valent copper exhibits relative stability in water, avoiding undesired competitive reactions. Based on these reasons, this study compared the deholgenation kinetics of nanoscale and powdered copper particles. This study also
evaluated feasibility of using reductive nanoscal zero valent metal coated resin to degrade chlorinated organics in an above-ground reactor.
2. Experimental Section 2.1. Chemicals
All aqueous solutions were made in water purified with a Milli-Q system (18.2 MΩ/cm). High-pressure liquid chromatography grade carbon tetrachloride, chloroform, and dichloromethane were obtained from Aldrich (USA). Copper (Π) nitrate trihydrate was from Alfa (USA). Copper powder was obtained from Riedel-de Haen (approx. 0.04 mm, > 99.5%, GR grade, USA).
2.2. Preparation of Nanoscale Cu0 Coated Resin
The polymer matrix used in this study is a commercial ion-exchange resin manufactured by Dow Chemical Company (USA) and marketed under the trade name Dowex TM. The sulfonated polystyrene matrix is composed of spheres with diameters of 30 to 60 m. The resin was first loaded with the desired concentration of Cu2+ from an aqueous solution of Cu(NO3)2·3H2O, followed by thorough washing to remove excess physisorbed copper ions. After washing, the fresh samples were dried in air at 120 °C for 18 h. The dried samples were then reduced under a flow of H2 and N2 gas (20 vol. %, 100 mL min-1). During the reduction, the temperature was increased from ambient to 300 °C at a rate of 10 °C min-1 and then maintained at 300 °C for 3 h. The samples were finally cooled to room temperature in the reducing gas atmosphere.
2.3. Characterization of Nanoscale Cu0 Coated Resin
TPR was used to study the required reduction temperature with the apparatus similar to one described previously (Bond and Namijo, 1989). The fresh sample of Cu2+ on the resin was dried in air at 120 ℃ for 18 h. Then, the resin was heated in a flow of He (50 mL min-1) from room temperature to 250 ℃ at 10 ℃ min-1, then maintained at 250 ℃ for 30 min to remove the volatiles. A flow of H2 and N2 gas [20 vol. %, 100 mL min-1, Hoekloos (Netherlands)] was used as the reducing gas. The oven temperature was programmed to rise from ambient to 350 ℃ at 10 ℃ min-1 and then staying at 350 ℃ for 1 h.
The oxidation states of copper on the resin were identified by electron spectroscopy for chemical analysis (ESCA). The ESCA measurements were perform by using a Vacuum Generators ECSALAB MKΠ photoelectron spectrometer (East Grinsted, U.K.) with an ALKα1,2 (1486.6 eV) X-ray source and a hemispherical 150 mm mean radius electron analyzer with a take-off angle of 900. During the data acquisition, the pressure in the sample chamber did not exceed 6.7×10−8 Pa.
The morphology and size of the resulting nanoscale Cu0 coated resin was viewed with a transmission electron microscopy. The localized elemental information from the chose region was determined with energy disperse X-ray spectroscopy (EDX) in conjunction with electron microscopy.
2.4. Surface Area of attached Copper Particles
As described by Grift et al. (1991), the determination of the surface area of attached copper particles is based on the measurement of hydrogen consumption after surface oxidation of the copper by N2O.
2Cu + N2O Æ Cu2O + N2 (copper surface atoms only) (1) Cu2O + H2 Æ 2Cu + H2O (2) According to the surface oxidation measurements using N2O at various temperatures (Evans et al., 1983), the average site density is 1.4 ×1019 Cu-atoms m-2. Compared to the area of the thermal conductivity detector signals for 1 mL H2 (40.9 mol at 1 atm,
25 ℃) undergoing temperature programmed reduction (A1ml,H2), the H
N
2 consumption of various copper ions on the resin undergoing temperature programmed reduction was obtained. The numbers of copper atoms at the surface ( ) were calculated from the stoichiometry of Equation (2).
Cu surf , s H mL Cu surf Cu surf N A A N = × × −6× , 1 , , 40.9 10 2 (mol) (3) where is the area of the thermal conductivity detector signal for the second TPR, and N
Cu surf
A ,
s is 2 from the stoichiometry of Equation (2). Copper surface area (SCu) on
the resin is sin 19 , ) 10 4 . 1 1 ( re av Cu surf Cu W N N S × × × = (m2-Cu g-resin-1) (4) where Nav is Avogadro constant (6.023×1023mol-1), and Wresin is the mass (g) of the
resin.
2.5. Reactor System
All dynamic experiments were performed for the degradation and sorption of CCl4 by Cu0 coated resin in a closed batch system with zero headspace (initial pH = 7.1 to 7.3). In these systems, the desired amount of Cu0 coated resin was added into a 15 mL amber serum vial, sequentially filled up with Ar-purged unbuffered Milli-QTM water. A 100 µL aliquot of CCl4 (700 mg L-1) was then added under the water surface. Immediately after CCl4 addition, the vials were capped with Teflon silicone septa and aluminum seals and then mixed on a rotary shaker (50 rmp) at room temperature (24± 1 °C) in the dark.
2.6. Extraction and Analysis
Aqueous phase and total system concentration of CCl4 were determined by liquid-liquid and liquid-solid extraction using n-hexane as a solvent. The sampling method and extraction process was similar to the method reported by Burris et al. (1995). There are two stages to extract CCl4 from residual aqueous phase and sorbed phase on the resin. Firstly, 5 mL of aqueous solution was collected from the amber serum vial by a gas-tight syringe through the septa, and simultaneously another disposable needle was used to inject 13 mL of helium to replace the liquid removed. Liquid-liquid extraction of the sample was performed with 5 mL n-hexane by axial rotation on a roller drum at 10 rpm, at room temperature, and in the dark for 30 min. The successive withdrawal of 8 mL of solution using the second gas-tight syringe was discarded. Secondly, another syringe of 5 mL n-hexane was spiked to the same vial to extract CCl4 from residual aqueous solution and the resin by axial rotation on a roller drum under the conditions described above. Two separate 0.5 mL samples of the liquid-liquid and the liquid-solid extractions were measured using a HP5890 GC equipped with a DB-624 capillary column and an electron capture detector operated in the splitless mode. Temperature conditions were programmed as follows: oven temperature at 40 ℃; injection port temperature at 180 ℃; detector temperature at 300 ℃. Ultrapure nitrogen was the GC carrier gas at a flow rate of 4.16 mL min-1. Peaks were quantified by comparing retention time and peak areas with standards. The total CCl4 mass (mg vial-1) come from the sum of both the measured masses in the liquid-liquid and the liquid-solid extractions. The sorbed CCl4 concentration on resin (Cs, g-CCl4 g-resin-1) was determined by the difference of total and aqueous
s W w T s m V C C C = −( × ) (5) where CT denotes the total mass, Cw is the aqueous phase concentration, VW is the
volume of aqueous solution, and ms is the mass of the added resin. All experiments
were duplicated or triplicated.
The pH was measured with a Beckman Model 71 pH meter. The concentration of copper ions in aqueous solution was determined by an atomic absorption spectrophotometry (PerkinElmer AAnalyst 800TM). Chloride was analyzed using an ion chromatograph (Dionex DX-100 TM).
3. Results and Discussion 3.1. Reduction Temperature
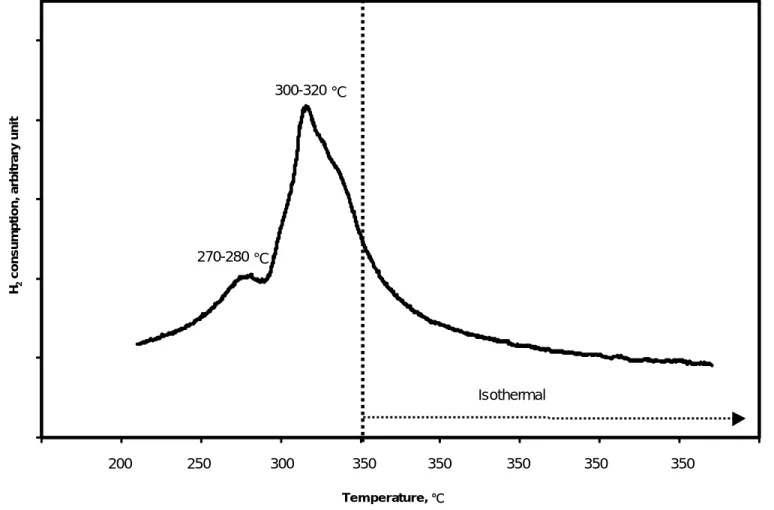
The temperature programmed reduction of Cu2+ on the resin in a flow of H2 and Ar (20 vol. %, 60 mL min-1) indicated two peaks of hydrogen consumption between 270 to 280 ℃, and between 300 to 320 ℃ (Figure 2). These peaks correspond to the reduction of Cu2+ to Cu+ and the reduction of Cu+ to zero valent copper. After a dried sample of Cu2+ on the resin was reduced in a flow of H2 and N2 gas (20 vol. %, 50 mL min-1) at 300 ℃ for 3 h, all copper on the resin was reduced to zero valence. Figure 3 shows that only zero valent copper remained. Thus, this ESCA established that the temperature was high enough to ensure complete reduction. Compared to the common aqueous-phase reduction with NaBH4 or N2H4 as reduction agents, the use of H2 and N2 gas to reduce copper ions to zero valent copper decreases the manufacturing expense because the more expensive chemicals are not required.
3.2. Characterization of Nanoscale Zero Valent Copper Coatings on Resin
Based on transmission electron microscopy, Figure 4 shows the distribution of 10 mg of zero valent copper per g of resin has a size in the range of 5 to 25 nm. The ion-exchange resin surface as analyzed by EDX was dominant by thionyl, sodium, and carbon. Thus, the resin easily held zero valent copper in nanoscale patches or particles and did not destroy the ion-exchange groups during the process.
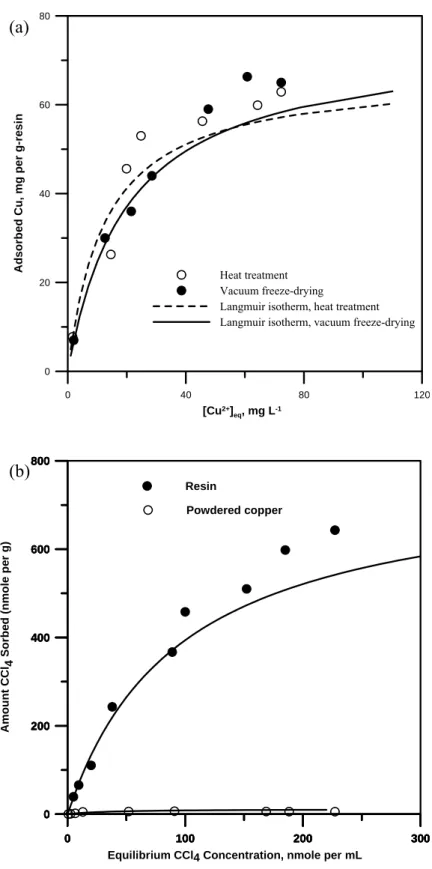
The effect of heat pretreatment (under a flow of 20 vol. % H2 and N2 gas at 300 ℃ for 3 h) on the sorption of Cu2+ onto the resin was evaluated using the Langmuir isotherm. Another sample of resin was dried by a vacuum freeze-drying technique (26.8 Pa and -55 ℃ for 24 h). As shown in Figure 5 (a) and Table 1, the estimated sorption capacities of Langmuir isotherm for both of the resin with the different pretreatments were 67.6± 10.9 nmol g-1 for heat pretreatment and 79.5 8.9 nmol g± -1 for vacuum freeze-drying. The results indicated the ion-exchange capability of the resin after heat pretreatment (a complete reduction procedure) nearly remained intact (the difference of two sorption capacities was not statistically significant with 95% confidence and 2 degrees of freedom using the independent t-test). Elemental analysis with EDX for the resin surface (magnification by 5000 times) revealed typical total copper contents of 1.5 to 2.0% (atomic percentage of total elements including carbon, oxygen, sulfur, sodium, and copper) before and after the dechlorination reaction, indicating little dislodging and few copper ions in solution. Therefore, nanoscale zero valent copper coated resin possessed high mechanical stability in water. After reaction, the copper on the resin was dominantly oxidized into CuO as identified by ESCA (Figure 3).
3.3. Degradation and Sorption of Carbon Tetrachloride Using Resin Coated with Nanoscale Zero Valent Copper
The disappearance of CCl4 from aqueous solutions may be due to degradation reactions by zero valent copper or sorption onto the resin. Sorption of CCl4 from solution onto the resin can be described by the Langmuir isotherm [Figure 5 (b)].
Table 1 provides that the observed maxima as 11.5± 6.2 and 770.1 41.5 nmol g± -1 for CCl4 on the powdered copper particles and the resin, respectively. Thus, the sorption of aqueous CCl4 onto the resin dominates soprtion onto the zero valent copper.
A conceptual model of the nanoscale zero valent copper-resin-CCl4-water system incorporating the degradation reaction by zero valent copper and sorption and desorption to the resin is similar to that proposed by Burris et al. (1998). The loss of total mass of CCl4 in a closed reactor was fitted with a first-order rate equation of the form w m s SA w a T k C k a C dt dC × × × = × = − ρ (6) where CT, Cw and Cs are the total, aqueous, and sorbed-phase CCl4 concentrations g
per vial, respectively; t is the reaction time (h). Ka is the degradation rate constant (h-1);
kSA is the surface-area-normalized rate coefficient (L h-1 m-2); aS is the specific surface
area of metal (m2 g-1); and ρm is the mass concentration of the metal (g L-1); The
disappearance rate of aqueous mass with respect to time was due sorption, desorption and degradation reaction.
s d w s w a w k C C C dt dC χ χ − + = − (7) where χsand χd are the soprtion and desorption rate constants, respectively.
Despite the sorption onto the zero valent copper surface, the CCl4 concentrations were measured as aqueous phase in solution and sorbed phase on the resin. Each vial without headspace contained 0.2 or 0.5 g resin coated with nanoscale zero valent copper (25 mg-Cu per g-resin), or 0.3 g commercial powdered copper (0.04 mm). In separate controls, total concentrations (mass per vial) were stable during the same period. The extraction recoveries for CCl4 from aqueous and sorbed phases in the blank resin vials (0.2 g-resin per vial) ranged from 91 to 98%.
Initially, the specific surface area of nanoscale zero valent copper supported on the resin and for the powdered copper were measured by hydrogen consumption during temperature programmed reduction. The values are shown in Table 2. The surface area of nanoscale zero valent copper is approximately 22 times that of powdered copper.
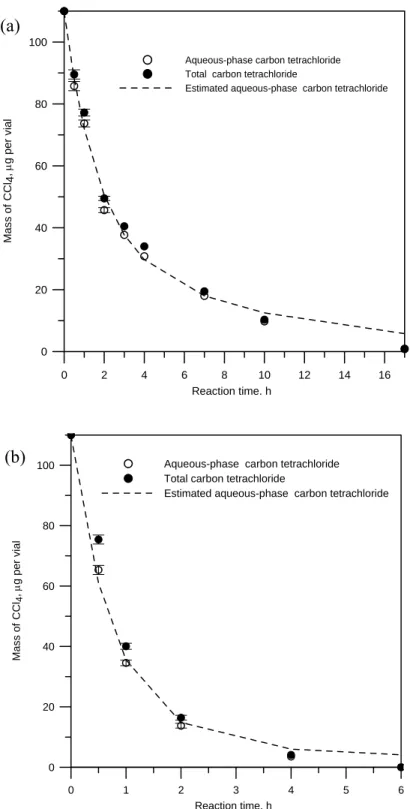
As shown in the Figure 6(a) and (b), complete degradation of CCl4 in 0.2 and 0.5 g nanoscale zero valent copper coated resin per vial took about 16 to 17 h and 5 to 6 h, respectively. The observed degradation rates (kobs) were 0.24± 0.02 h-1 for 0.2 g
nanoscale zero valent copper coated resin per vial and 0.80± 0.03 h-1 for 0.5 g nanoscale zero valent copper coated resin per vial and then normalized with the specific surface area (1.12 0.02 m±
±
2-Cu/g-resin) and the mass concentration of nanoscale zero valent copper coated resin (13.3 g L-1) to kSA, 0.016 0.002 and
0.021 0.001 L h
± ±
±
-1 m-2, respectively. The k
obs and kSA for powdered copper were
0.028 0.004 h-1 and 0.0010 0.0001 L h-1 m-2, respectively. Reducing the size of copper particles to a nanoscale would sharply increase kSA by a factor of about 24.
Other experiments (0.5 g zero valent copper coated resin per vial) revealed that the
kobs for nanoscale zero valent copper coated resin submerged continuously in
Ar-purged Milli-Q water for 7 d was 0.73± 0.004 h-1. Then, the same nanoscale zero valent copper coated resin was regenerated by H2 at 300 ℃ for 3 h, increasing the kobs
to 0.85 0.003 h± -1. Thus the reactivity of nanoscale zero valent copper coated resin was decreased in the long-term reaction, but was restored by H2 treatment (95% confidence and 4 degrees of freedom using the independent t-test).
The sorption and desorption rate constants χsand χd were estimated by fitting the observed aqueous CCl4 concentration Cw with the exact solution of Eq. 7 derived
by Burris et al. (1998). The sorption and desorption rate constants χsand χd were respectively 0.21± 0.002 and 0.28 ± 0.002 h-1 for 0.2 g nanoscale zero valent copper coated resin per vial; these were 0.41± 0.03and 0.30 ± 0.03 h-1 for 0.5 g nanoscale zero valent copper coated resin per via. Increasing nanoscale zero valent copper coated resin loading per vial increased the sorption rate constant but not the desorption rate constant. As shown in Figure 6, the initial Cw data were matched by
the estimated values of the exact solution with values of constant χsand χd(within 7 and 2 h, respectively). The apparent differences between the observed and estimated data in the final period of reaction implies the desorption rate may be not first-order (the estimated deposition rate is slower than the actual rate).
3.4. Residual Copper Ions Concentration and pH
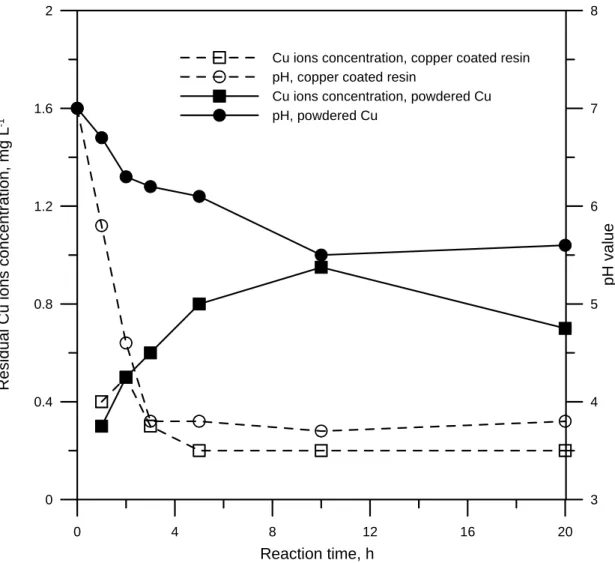
The residual copper ions in the effluent are the product of the reaction between CCl4 and Cu0. These are considered to be hazardous species and the concentrations must be reduced below those required by effluent standards. Figure 7 shows the residual copper ions concentration and the pH of both commercial powdered copper and nanoscale zero valent copper coated resin. For a powdered Cu-CCl4-H2O system, the residual copper ions exceeded 1.0 mg L-1, falling to 0.6 mg L-1 for a long period, due to the precipitation of metal carbonate or oxide (Figure 3 shows the build up of CuO). However, these concentrations are typically under 0.1 mg L-1 and the pH dropped to 3 to 4 in nanoscale zero valent copper coated resin system. The difference in copper ion concentration in aqueous solution between copper powder and nanoscale zero valent copper coated resin results from the ion exchange of the resin as a support. On the nanoscale zero valent copper coated resin, when zero valent copper is oxidized into copper ion, H+ or Na+ are released from the resin to exchange copper ion. Thus, the use of the resin as a support not only removes copper ions from the solution but also drops pH to acidic conditions.
The possible copper contamination resulting from the displacement by other multivalent cations present in groundwater limits the potential in situ application of nanoscale zero valent copper coated resin. This competition depends on the composition of groundwater is inevitable and mainly causes the decrease in copper recovery over time. Considering metal toxicity and the required reduction temperature of metal ions, alternative metals like tin may to be more applicable in situ.
4. Conclusions
Nanoscale zero valent copper coated resin, synthesized in a flow of H2 and N2 gas (20 vol %, 50 mL min-1) at 300 ℃ for 3 h, is an efficient and mechanically stable reductant for the degradation of CCl4 in water. Reducing the size of the copper particles to a nanoscale increases the reactivity by a factor of about 24. The use of the cation-resin as a support decreases the copper ions in the effluent at a concentration of less 0.1 mg/L and simultaneously maintains the solution pH at 3 to 4. Over the long term, this reductant can be restored to reactivity by H2. Thus, advantages of the application of nanoscale zero valent copper coated resin as reductive material to degrade chlorinated solvents can be attributed to (1) the increase in the rate constant due to the nano-size of metal particles; (2) the combined effect of degradation and sorption; (3) the acid pH range during the dechlorination process; (4) the recycling of metal ions in the solution. In the above-ground application, this reductant would facilitate the development of a process that could be designed for convenient
emplacement and regeneration of porous reductive medium. In situ, metal coated
resin may not be as useful until the effects of competitive ions in local ground water can be related to the release of toxic metals.
References
Agrawal, A.; Tratnyek, P. G., 1996. Reduction of nitro aromatic compounds by zero-valent iron metal. Environ. Sci. Technol. 30, 153-160.
Bond, G. C., Namijo, S. N., 1989. An improved procedure for estimating the metal surface area of supported copper catalysts. J. Catal. 118, 511-512.
Boronina, T., Klabunde, K. j., Sergeev, G. B., 1995. Destruction of organohalides in water using metal particles: carbon tetrachloride/water reaction with magnesium, tin, and zinc. Environ. Sci. Technol. 29, 1511-1517.
Burris, D. R., Campbell, T. J., Manoranjan, V. S., 1995. Sorption of trichloroethylene and tetrachloroethlyene in a batch reactive metallic iron-water system. Environ.
Sci. Technol. 29, 2850-2855.
Burris, D. R., Allen-King, R. M., Manoranjan, V. S., Campbell, T. J., Loraine, G. A., and Deng, B., 1998. Chlorinated ethene reduction by cast iron: sorption and mass transfer. J. Envr. Engr. 1012-1019.
Chen, J. L., Al-Abed, S. R., Ryan, J. A., Li, Z., 2001. Effects of pH on dechlorination of trichloroethylene by zero-valent iron. Journal of Hazardous Materials B83, 243-254.
Cheng, S. F., Wu, S. C., 2000. The enhancement methods for the degradation of TCE by zero-valent metals. Chemosphere 41, 1263-1270.
Choe, S., Chang, Y. Y., Hwang, K. Y., Khim, J., 2000. Kinetic of reductive denitrification by nanoscale zero-valent iron. Chemosphere, 41, 1307-1311.
Doong, R. A., Chen, K. T., Tsai, H. C., 2003. Reductive dechlorination of carbon tetrachloride and tetrachloroethylene by zerovalent silicon-iron reductants.
Environ. Sci. Technol. 37, 2575-2581.
Evans, J. W., Wainwright, M. S., Bridgewater, A. J., Young, D. J., 1983. On the determination of copper surface area by reaction with nitrous oxide. Appl. Catal. 7, 75-83.
Eykholt, G. R., Davenport, D. T., 1998. Dechlorination of the chloroacetanilide herbicides alachlor and metolachlor by iron metal. Environ. Sci. Technol. 32, 1482-1487.
Gillham, R. W., O’Hannesin, S. F., 1994. Enhanced degradation of halogenated aliphatics by zero-valent iron. Ground Water 32, 958-967.
Grift, C. J. G., Wielers, A. F. H., Joghi, B. P. J., Beijnum, J., Boer, M., Versluus-Helder, M., Gues, J. W. J., 1991. Effect of the reduction treatment on the structure and reactivity of silica-supported copper particles. Catal. 131, 178-189. Gu, B., Phelps, T. J., Liang, L., Dickey, M. J., Ron, Y., Kinsall, B. L., Palumbo, A. V.,
Jacobs, G. K., 1999. Biogeochemical dynamics in zerovalent iron columns: Implications for permeable reductive barriers. Environ. Sci. Technol. 33, 2170-2177.
Huang, C. P., Wang, H. W., Chiu, P. C., 1998. Nitrate reduction by metallic iron. Wat.
Res. 32, 2257-2264.
Klabunde, K. J., 2001. Nanoscale materials in chemistry. Wiley, New York, p. 11. Kim, Y. H., Carraway, E. R., 2000. Dechlorination of pentachlorophenol by zero
valent iron and modified zero valent irons. Environ. Sci. Technol. 34, 2014-2017. Matheson, L. J., Tratnydk, P. G., 1994. Reductive dehalogenation of chlorinated
methanes by iron metal. Environ. Sci. Technol. 28, 2045-2053.
McCutcheon, S. C., Schnoor, J. L., 2003. Phytoremediation: Transformation and Control of Contaminants. Wiley, New York, p. 1-10.
Muftikian, R., Fernando, Q, Korte, N., 1995. A method for rapid dechlorination of low molecular weight chlorinated hydrocarbons in water. Wat. Res. 29, 2434-2439.
Nam, S.; Tratnyek, P. G., 2000. Reduction of azo dyes with zero-valent iron. Wat. Res. 34, 1837-1845.
Orth, W. S., Gillham, R. W., 1996. Dechlorination of trichloroethene in aqueous solution using Fe0. Environ. Sci. Technol. 30, 66-71.
Phillips, D. H., Gu, B., Watson, D. B., Roh, Y., Liang, L., Lee, S. Y., 2000. Performance evaluation of a zerovalent iron reactive barrier: mineralogical characteristics. Environ. Sci. Technol., 34, 4169-4176.
Sayles, G. D., You, G., Wang, M., Kupferle, M. J., 1997. DDT, DDD, and DDE dechlorination by zero-valent iron. Environ. Sci. Technol. 31, 3448-3454.
Siantar, D. P., Schreier, C. G., Chou, C. S., Reinhard, M., 1996. Treatment of 1,2-dibromo-3-chloropropane and nitrate-contaminated water with zero-valent iron or hydrogen/palladium catalysts. Wat. Res. 30, 2315-2322.
Su, C., Puls, R. W., 1999. Kinetics of trichloroethene reduction by zerovalent iron and tin: pretreatment effect, apparent activation energy, and intermediate products.
Environ. Sci. Technol. 33, 163-168.
Su, C., Puls, R. W., 2004. Nitrate reduction by zerovalent iron: effects of formate, oxalate, citrate, chloride, sulfate, borate, and phosphate. Environ. Sci. Technol. 38, 2715-2720.
Wang, C. B., Zhang, W. X, 1997. Synthesizing nanoscale iron particles for rapid and complete dechlorination of TCE and PCBs. Environ. Sci. Technol. 31, 2154-2156. Zhang, W. X., Wang, C. B., Lien, H. L., 1998. Treatment of chlorinated organic
contaminants with nanoscale bimetallic particles. Cata. Today 40, 387-395.
Ziolo, R. F., Giannelis, E. P., Weinstein, B. A., O’Horo, M. P., Ganguly, B. N., Mehrotra, V., Russell, M. W., Huffman, D. R., 1992. Matrix-mediated synthesis of nanocrystalline γ–Fe2O3: A new optically transparent magnetic material.
Flow of contaminant Resin nanoscale M0 [Org-Cl]aq [Org-Cl]sorb sorption desorption Products ne -degradation SO3-H+ Mn+ H+
Nanoscale zero valent metal coated resin mesh (a)
(b)
Tem perature, ℃ H2
consumption, arbitrary unit
Isothermal 270-280 ℃
300-320 ℃
250 300 350
200 350 350 350 350
Figure 2. Consumption of hydrogen during temperature programmed reduction of nanoscale zero valent copper coated resin. The metal coating was 40 mg-Cu per g-resin
22500 23000 23500 24000 24500 25000 25500 26000 26500 27000 27500 925 930 935 940 945 950 955 960 Binding Energy, eV
Intensity, arbitrary unit
Before reaction After reaction Cu CuO 2P1/2 2P1/2
67 nm
Cu
0Figure 4. The TEM image of resin with a coating of 10 mg of zero valent copper per g resin. The metallic particles appear as spots with high contrast. The inset EDX graph shows the elements on the surface.
0 40 80 0 [Cu2+] eq, mg L-1 0 20 40 60 80 A d so rb ed Cu, mg p e r g-r e s in Heat treatment Vacuum freeze-drying
Langmuir isotherm, heat treatment Langmuir isotherm, vacuum freeze-drying (a) 12 0 100 200 300 0 200 400 600 0 Powdered copper 0 100 200 300 0 200 400 600 0 Resin 0 100 200 300 0 200 400 600 0 0 100 200 300
Equilibrium CCl4 Concentration, nmole per mL 0 200 400 600 0 Amount C C l 4 So rbe d (nmole pe r g) 80 80 80 80 (b)
Figure 5. Sorption isotherms (Averaged results from replicate tests were reported.): (a) Cu2+ on the resin pretreated by vacuum freeze-drying and heat pretreatment. (b) CCl4 on the resin and the powdered Cu particles. Fitted lines are based on the Langmuir equation using the parameters in Table 1.
0 2 4 6 8 10 12 14 16 Reaction time, h 0 20 40 60 80 100 Mas s of CCl 4 , µg per v ial
Aqueous-phase carbon tetrachloride Total carbon tetrachloride
Estimated aqueous-phase carbon tetrachloride
(a) 0 1 2 3 4 5 6 Reaction time, h 0 20 40 60 80 100 Ma ss o f CCl 4 , µg pe r vial
Aqueous-phase carbon tetrachloride Total carbon tetrachloride
Estimated aqueous-phase carbon tetrachloride (b)
Figure 6. Disappearance of CCl4 with 95% confidence interval in a well-mixed batch system (15 mL vial with no headspace). (a) 0.2 g nanoscale zero valent copper coated resin,χs: 0.21± 0.02 h-1,
d
χ : 0.28± 0.02 h-1. (b) 0.5 g nanoscale zero valent copper coated resin,χs: 0.41 0.03 h± -1,
d
0 4 8 12 16 20 Reaction time, h 0 0.4 0.8 1.2 1.6 2 -1 3 4 5 6 7 8 pH v a lu e
Cu ions concentration, copper coated resin pH, copper coated resin
Cu ions concentration, powdered Cu pH, powdered Cu R e sidua l Cu ions concent ration, mg L
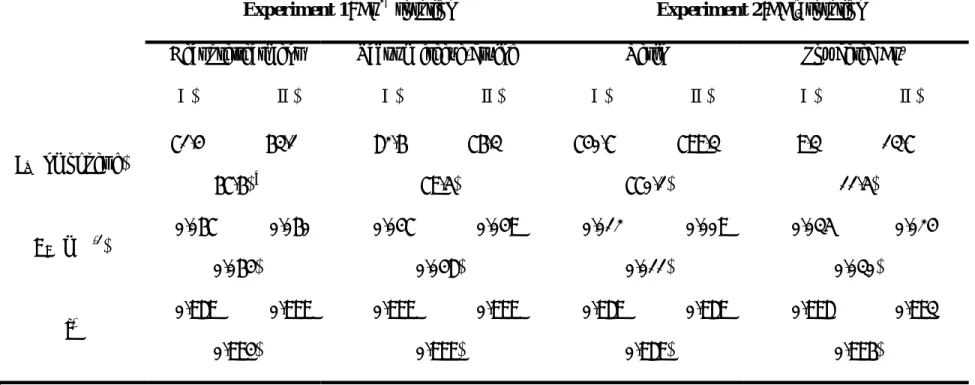
TABLE 1. Fitted Langmuir parameters for sorption of Cu2+ by the resin with different pretreatments (Experiment 1) and for sorption of CCl4 by the resin and the powdered copper particles (Experiment 2).
Experiment 1: Cu2+ sorption Experiment 2:CCl4 sorption
Heat pretreatment (Ⅰ) (Ⅱ) Vacuum freeze-drying (Ⅰ) (Ⅱ) Resin (Ⅰ) (Ⅱ) Powdered Cu0 (Ⅰ) (Ⅱ) Sm (nmol per g) 71.4 63.1 (67.6) a 82.6 76.3 (79.5) 740.7 799.3 (770.1) 9.3 13.7 (11.5) KL (mM-1) 0.067 0.060 (0.064) 0.047 0.049 (0.048) 0.012 0.009 (0.011) 0.035 0.024 (0.030) r2 0.989 0.999 (0.994) 0.999 0.999 (0.999) 0.989 0.989 (0.989) 0.998 0.993 (0.996)
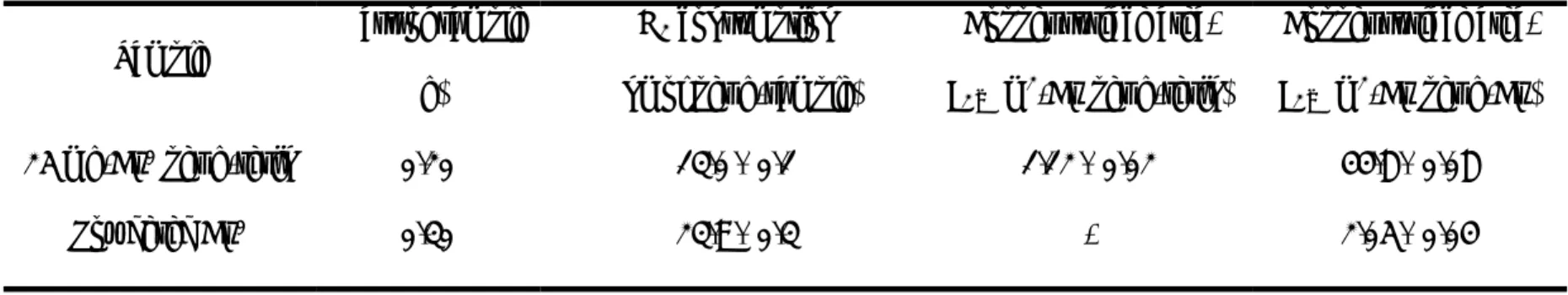
TABLE 2. Specific Surface Areas of Resin Coated with Nanoscale Zero Valent Copper and Powdered Copper.
Sample
Mass of sample (g)
H2 consumption (nmol per g-sample)
Copper surface area, SCu (m2-Cu per g-resin)
Copper surface area, SCu (m2-Cu per g-Cu) 25 mg-Cu0 per g-resin 0.20 13.0± 0.1 1.12 ± 0.02 44.8 ± 0.08
Evaluation
Three papers have been accepted by Chemosphere, Water research, and Journal of Hazardous Materials until now. The titles are:
(1) Chin Jung Lin, Shang-Lien Lo and Ya Hsuan Liou (2004) Degradation of Aqueous Carbon Tetrachloride by Nanoscale Zero Valent
Copper on a Cation Resin. Chemosphere (accepted).
(2) Chin Jung Lin, Shang-Lien Lo and Ya Hsuan Liou (2004) Dechlorination of Trichloroethylene in Aqueous Solution by Noble
Metal-Modified Iron. Journal of Hazardous Materials (accepted).
(3) Chin Jung Lin*, Shang-Lien Lo (2004) Effects of Iron Surface Pretreatment on Sorption and Reduction Kinetics of Trichloroethylene