Control over Surface DNA Density on Gold Nanoparticles Allows
Selective and Sensitive Detection of Mercury(II)
Chi-Wei Liu, Chih-Ching Huang, and Huan-Tsung Chang*
Department of Chemistry, National Taiwan UniVersity, 1, Section 4, RooseVelt Road, Taipei, Taiwan ReceiVed February 24, 2008. ReVised Manuscript ReceiVed May 5, 2008
We have developed a new highly selective and sensitive technique for the detection of Hg2+using DNA-functionalized gold nanoparticles (Au NPs) and OliGreen. This system is the first that allows the detection of Hg2+based on the release of DNA molecules, induced by conformational changes on Au NP surfaces, and its sensitivity is highly dependent upon surface DNA density. When Hg2+ions interact with the thymidine units of the DNA molecules bound to the Au NPs through Au-S bonds, the conformations of these DNA derivatives change from linear to hairpin structures, causing the release of some of the DNA molecules from the surface of the Au NPs into the bulk solution to react with OliGreen. The fluorescence of OliGreen-DNA complexes increased with increasing concentration of Hg2+, and Hg2+could be detected at concentrations as low as 25 nM. A linear correlation existed between the fluorescence intensity and the concentration of Hg2+over the range 0.05-2.5µM (R2) 0.98). This simple and cost-effective probe was applied to determine the spiked Hg2+in the pond samples; the recoveries (96-102%) suggested low matrix interference and high sensitivity.
Introduction
The monitoring of mercury in aquatic ecosystems is an important issue because Hg2+contamination has severe effects on human health and the environment.1 Inductively coupled plasma mass spectrometry (ICP-MS) is a powerful technique for the determination of Hg2+; it is however expensive and not suitable for on-site analysis.2Therefore, the past few years have witnessed the development of several optical techniques for the detection of Hg2+.3–5Chemical sensors using environmentally sensitive organic molecules,3thin films of gold,3fbiocomposites,4dand polymeric materials5fhave been developed for the detection of
Hg2+. Many of these systems however displayed limitations in practical use, such as poor aqueous solubility, cross-sensitivities toward other metal ions, matrix interferences, and poor sensitivity. Thiol-functionalized gold nanoparticles (Au NPs) have become interesting nanomaterials for sensing metal ions, mainly because their surface plasmon resonance (SPR) absorbances are extremely sensitive to the nature, size, and shape of the Au NPs, their interparticle distances, and the nature of their surrounding media.6 However, their selectivity toward Hg2+over other metal ions is not excellent. In order to improve the selectivity for the detection of Hg2+, thiol-functionalized Au NPs have been used in the presence of 1.0 mM 2,6-pyridinedicarboxylic acid (PDCA), taking advantage of Hg2+-induced color changes.4aRhodamine B bound Au NPs and fluorescent Au NPs have been utilized for the selective detection of Hg2+in the presence of PDCA through energy and electron transfer mechanisms.4b,f,7Hg2+-induced aggregation of DNA-functionalized Au NPs through thymidine-Hg2+ -thymidine coordination has also been realized.5e
In this study, we present a highly selective and sensitive technique for the detection of Hg2+using DNA-functionalized Au NPs and OliGreen (a dye that binds specifically to DNA).8 The sensing mechanism of this new probe is based on the release of DNA molecules from the Au NP surface to the bulk solution (as a result of thymidine-Hg2+-thymidine coordination) and their subsequent specific interactions with OliGreen (Scheme 1). When Hg2+ions interact with the thymidine units of the DNA molecules bound to the Au NPs through Au-S bonds, the conformations of these DNA derivatives change from linear to hairpin structures,5 causing the release of some of the DNA * To whom correspondence should be addressed. Telephone and Fax:
011-886-2-33661171. E-mail: [email protected].
(1) (a) Campbell, L. M.; Dixon, D. G.; Hecky, R. E. J. Toxicol. EnViron. Health, Part B 2003, 6, 325–356. (b) Wang, Q.; Kim, D.; Dionysiou, D. D.; Sorial, G. A.; Timberlake, D. EnViron. Pollut. 2004, 131, 323–336.
(2) Wang, M; Feng, W; Shi, J; Zhang, F; Wang, B; Zhu, M; Li, B; Zhao, Y; Chai, Z Talanta 2007, 71, 2034–2039.
(3) (a) Nolan, E. M.; Lippard, S. J. J. Am. Chem. Soc. 2003, 125, 14270– 14271. (b) Yang, Y.-K.; Yook, K.-J.; Tae, J. J. Am. Chem. Soc. 2005, 127, 16760– 16761. (c) Yoon, S.; Albers, A. E.; Wong, A. P.; Chang, C. J. J. Am. Chem. Soc.
2005, 127, 16030–16031. (d) Moon, S.-Y.; Youn, N. J.; Park, S. M.; Chang, S.-K.
J. Org. Chem. 2005, 70, 2394–2397. (e) Nolan, E. M.; Racine, M. E.; Lippard, S. J. Inorg. Chem. 2006, 45, 2742–2749. (f) Morris, T.; Szulczewski, G. Langmuir
2002, 18, 5823–5829. (g) Hennrich, G.; Sonnenschein, H.; Resch-Genger, U.
J. Am. Chem. Soc. 1999, 121, 5073–5074. (h) Prodi, L.; Bargossi, C.; Montalti, M.; Zaccheroni, N.; Su, N.; Bradshaw, J. S.; Izatt, R. M.; Savage, P. B. J. Am. Chem. Soc. 2000, 122, 6769–6770. (i) Rurack, K.; Kollmannsberger, M.; Resch-Genger, U.; Daub, J. J. Am. Chem. Soc. 2000, 122, 968–969. (j) Caballero, A.; Martinez, R.; Lloveras, V.; Ratera, I.; Vidal-Gancedo, J.; Wurst, K.; Tarraga, A.; Molina, P.; Veciana, J. J. Am. Chem. Soc. 2005, 127, 15666–15667. (k) Coskun, A.; Akkaya, E. U. J. Am. Chem. Soc. 2006, 128, 14474–14475. (l) Zhao, Y.; Zhong, Z. J. Am. Chem. Soc. 2006, 128, 9988–9989.
(4) (a) Huang, C.-C.; Chang, H.-T. Chem. Commun. 2007, 12, 1215–1217. (b) Huang, C.-C.; Chang, H.-T. Anal. Chem. 2006, 78, 8332–8338. (c) Thomas, J. M.; Ting, R.; Perrin, D. M. Org. Biomol. Chem. 2004, 2, 307–312. (d) Ono, A.; Togashi, H. Angew. Chem., Int. Ed. 2004, 43, 4300–4302. (e) Kim, I.-B.; Bunz, U. H. F. J. Am. Chem. Soc. 2006, 128, 2818–2819. (f) Huang, C.-C.; Yang, Z.; Lee, K.-H.; Chang, H.-T. Angew. Chem., Int. Ed. 2007, 46, 6824–6828.
(5) (a) Katz, S. J. Am. Chem. Soc. 1952, 74, 2238–2245. (b) Yamane, T.; Davidson, N. J. Am. Chem. Soc. 1961, 83, 2599–2607. (c) Miyake, Y.; Togashi, H.; Tashiro, M.; Yamaguchi, H.; Oda, S.; Kudo, M.; Tanaka, Y.; Kondo, Y.; Sawa, R.; Fujimoto, T.; Machinami, T.; Ono, A. J. Am. Chem. Soc. 2006, 128, 2172–2173. (d) Tanaka, Y.; Oda, S.; Yamaguchi, H.; Kondo, Y.; Kojima, C.; Ono, A. J. Am. Chem. Soc. 2007, 129, 244–245. (e) Lee, J.-S.; Han, M. S.; Mirkin, C. A. Angew. Chem., Int. Ed. 2007, 46, 4093–4096. (f) Tang, Y.; He, F.; Yu, M.; Feng, F.; An, L.; Sun, H.; Wang, S.; Li, Y.; Zhu, D. Macromol. Rapid Commun.
2006, 27, 389–392.
(6) (a) Moores, A.; Goettmann, F. New J. Chem. 2006, 30, 1121–1132. (b) Kim, Y.; Johnson, R. C.; Hupp, J. T. Nano Lett. 2001, 1, 165–167.
(7) (a) Kamat, P. V.; Barazzouk, S.; Hotchandani, S. Angew. Chem., Int. Ed.
2002, 41, 2764–2767. (b) Dulkeith, E.; Morteani, A. C.; Niedereichholz, T.; Klar,
T. A.; Feldmann, J.; Levi, S. A.; van Veggel, F. C. J. M.; Reinhoudt, D. N.; Mo¨ller, M.; Gittins, D. I. Phys. ReV. Lett. 2002, 89, 203002. (c) Huang, T.; Murray, R. W. Langmuir 2002, 18, 7077–7081. (d) Fan, C.; Wang, S.; Hong, J. W.; Bazan, G. C.; Plaxco, K. W.; Heeger, A. J. Proc. Natl. Acad. Sci. U.S.A.
2003, 100, 6297–6301. (e) Chen, S.-J.; Chang, H.-T. Anal. Chem. 2004, 76,
3727–3734. (f) Jennings, T. L.; Singh, M. P.; Strouse, G. F. J. Am. Chem. Soc.
2006, 128, 5462–5467. (g) Ipe, B. I.; Thomas, K. G.; Barazzouk, S.; Hotchandani,
S.; Kamat, P. V. J. Phys. Chem. B 2002, 106, 18–21.
(8) The Handbook, http://www.probes.com/handbook/sections/0803.html. 10.1021/la800589m CCC: $40.75 2008 American Chemical Society
Published on Web 06/26/2008
Downloaded by NATIONAL TAIWAN UNIV on July 30, 2009
molecules from the surface of the Au NPs, primarily as a result of steric effects, and into the bulk solution. OliGreen molecules then interact with these free DNA species, resulting in an increase in the fluorescence at 525 nm upon excitation at 480 nm. The DNA-OliGreen complexes fluoresce∼1000-fold more intensely than does the free OliGreen, which is only weakly fluorescent. Although the DNA molecules on the surfaces of the Au NPs also interact with OliGreen, a negligible degree of fluorescence occurs because of the Au NPs, which are efficient fluorescence quenchers.
Experimental Section
Chemicals. Trisodium citrate, tris(hydroxymethyl)aminomethane (Tris), and all of the metal salts used in this study were purchased from Aldrich (Milwaukee, WI). Hydrogen tetrachloroaurate(III) trihydrate was obtained from Acros (Geel, Belgium). The OliGreen ssDNA quantitation reagent and kit was obtained from Molecular Probes (Portland, OR). Because the concentration of OliGreen is unknown, for simplicity, its concentration was represented as 100× in this study. The 5′-thiol-modified probe (5′-T16 AGCCATC-TCTTCTCCGAGCCGGTCGAAATAGTGAGT-3′) and control (5′
-C15TAGC
CATCTCTTCTCCGAGCCGGTCGAAATAGTGAGT-3′) DNA samples were purchased from Integrated DNA Technology, Inc. (Coralville, IA).
Synthesis of Au NPs. Au NPs were prepared through citrate-mediated reduction of HAuCl4.9aAqueous 1 mM HAuCl4(250 mL) was brought to a vigorous boil with stirring in a round-bottom flask fitted with a reflux condenser, and 38.8 mM trisodium citrate (25 mL) was then added rapidly to the solution. The mixture was heated under reflux for another 15 min, during which time its color changed from pale yellow to deep red. The solution was cooled to room temperature while stirring continuously. The sizes of the nanoparticles were verified through transmission electron microscopy (TEM) analysis (H7100, Hitachi High-Technologies Corporation, Tokyo, Japan); the Au NPs appeared to be nearly monodisperse, with an average size of 13.3 ( 0.6 nm. The particle concentration of the Au NPs (ca. 15 nM) was determined according to Beer’s law using an extinction coefficient of ∼108M-1cm-1at 520 nm9b(double-beam UV-vis spectrophotometer,
Cintra 10e, GBC, Victoria, Australia) for Au NPs of 13.3 nm diameter. Preparation of DNA-Au NPs. The thiol-modified DNA oligo-nucleotides were attached to the Au NPs according to modified literature procedures.9cThe 5′-thiol-modified oligonucleotides were received in
the disulfide form HOCH3(CH2)5S-S-5′-oligo. These oligonucleotides were reacted directly with the Au NPs through attachment of both the
Scheme 1.Schematic Representation of Hg2+Nanosensors at Various DNA-to-Au NP Molar Ratios: <30 (A), 30-50 (B), and g 60 (C)
SensitiVe and SelectiVe Detection of Hg Ions Langmuir, Vol. 24, No. 15, 2008 8347
Downloaded by NATIONAL TAIWAN UNIV on July 30, 2009
HO(CH2)6S- and oligo-S- units onto the Au NP surfaces. Aliquots of aqueous Au NP solutions (990µL) in 1.5 mL tubes were mixed with the thiooligonucleotides (7.5-200 µM, 10 µL) to obtain a final concentration of 15 nM Au NPs and 0.075-2.0µM oligonucleotides. After reaction for 16 h at room temperature, 100 mM NaCl (salt aging) was added to the solution and incubated for 48 h to form DNA-Au NPs. The mixtures were centrifuged for 25 min at 16 000 rpm to remove the excess thiol-DNA. Following removal of the supernatants, the oily precipitates were washed with 4 mM trisodium citrate. After three centrifuge/wash cycles, the colloids were resuspended separately in 4 mM trisodium citrate and stored in a refrigerator (4°C). To determine the number of DNA molecules on each Au NP, the amount of DNA in the supernatant after centrifugation was measured using OliGreen. Analysis of Samples. Aliquots (40µL) of 25 mM Tris-HCl (pH 8.2) solutions containing Hg2+(0-15µM) and DNA-Au NPs (0.75 nM) were maintained at room temperature for 1 h. OliGreen (with a final concentration of 0.025×) was added to each solution and incubated for 10 min prior to measuring the fluorescence. A water sample from a pond on the NTU campus was filtered through a 0.2 µm membrane. Aliquots of the pond water (18 µL) were spiked with standard solutions (2µL) of Hg2+at concentrations over the range 0.05-15µM. The mixtures were diluted to 40 µL using 25 mM Tris-HCl (pH 8.2) buffer (20µL) and then analyzed using the present approach by employing the 0.75 nM DNA-Au NP probe solution. Lifetime studies were performed by using an Edinburgh FL 900 photon-counting system with a hydrogen-filled lamp as the excitation source. The emission decays were analyzed by the sum of exponential functions, which allows partial removal of the instrument time broadening and consequently renders a temporal resolution of ∼200 ps.
Results and Discussion
Preparation of DNA-Au NPs. To prepare DNA-Au NP complexes through Au-S bonding, we mixed 15 nM Au NP (size: 13.3 ( 0.6 nm) solutions individually with 75, 150, 1000, and 2000 nM 5′-thiol-modified oligonucleotides (5′ - T16AGCCATCTCTTCTCCGAGCCGGTCGAAATAGTGAGT-3′). We prepared the first two of these solutions (i.e., the 75 and 150 nM solutions) without salt aging; we prepared two solutions containing the 1000 nM 5′-thiol-modified oligonucleotides (one with and one without salt aging); and we prepared the last one (2000 nM) with salt aging. The Au NP and DNA-Au NP solutions both had rose-red hues, with surface plasmon resonance (SPR) absorption bands at∼520 nm. TEM images of both of these samples
(Figure S1, Supporting Information) displayed well-dispersed NPs. We used OliGreen to detect the presence of unbound DNA molecules in the supernatant after centrifugation (16 000 rpm, 25 min); we estimated that there were 5, 10, 30, 60, and 90 DNA
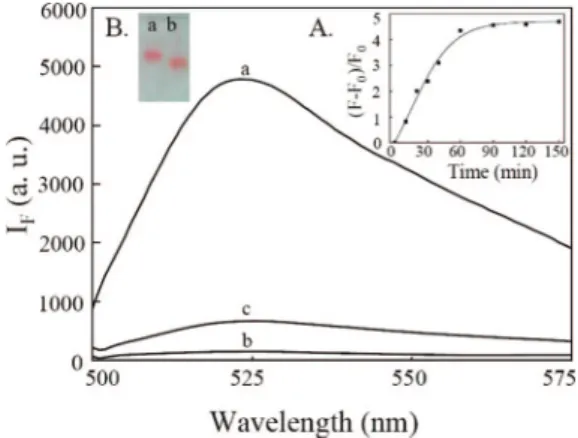
Figure 1.Fluorescence spectra of solutions of (a) OliGreen-DNA (45 nM) and (b,c) OliGreen-DNA-Au NPs in the (b) absence and (c) presence of Hg2+(5.0µM). Inset A: Time course measurement of the fluorescence intensity (525 nm) of DNA-Au NPs (0.75 nM) upon addition of Hg2+(5.0µM). Inset B: Agarose gel electrophoresis images of (a,b) 60DNA-Au NPs (7.5 nM) in the (a) absence and (b) presence of Hg2+(50.0µM). Slab gel separation was performed in 1.0% agarose under a constant electric field (-8.3 V/cm) for 20 min. Buffer, 25 mM Tris-HCl, pH 8.2; excitation wavelength, 480 nm. The fluorescence intensities (IF) are plotted in arbitrary units (a.u.).
Figure 2.(A) Quenching efficiency (1 - F0/Ff) of the fluorescence intensity (525 nm) at different DNA-to-Au NP ratios. (B) Enhanced ratios [(F - F0)/F0] of the fluorescence intensities (525 nm) of solutions at different DNA-to-Au NP ratios after addition of Hg2+(5.0µM). F
f, fluorescence intensity of free OliGreen-labeled DNA; F0, fluorescence intensity of OliGreen-labeled DNA-Au NPs; F, fluorescence intensity of OliGreen-labeled DNA-Au NPs in the presence of Hg2+(5.0µM). Concentration of Au NPs: 0.75 nM. All other conditions were the same as those described in Figure 1.
Figure 3. Metal ion-induced fluorescence changes (525 nm) of the OliGreen-DNA-Au NPs in 25 mM Tris-HCl at pH 8.2 in the (A) absence and (B) presence of 1.0 mM NaCl. Concentrations of the other metal ions: 5.0µM. All other conditions were the same as those described in Figure 1.
Downloaded by NATIONAL TAIWAN UNIV on July 30, 2009
molecules per Au NP after the 75, 150, 1000 (nonaged and salt-aged solutions), and 2000 nM DNA solutions, respectively, had been added to the 15 nM Au NP solutions. We found that the surface amounts of DNA increased upon increasing DNA concentration in the solutions and reached a plateau at 2000 nM DNA when using 15 nM Au NPs. Salt aging weakens hydrogen bonding between bases, causing the formation of greater numbers of linear DNA molecules, which bind better to the Au NPs.10 For simplicity, we denote the five DNA-Au NPs having 90, 60, 30, 10, and 5 DNA molecules per Au NP as 90DNA-Au NPs, 60DNA-Au NPs, 30DNA-Au NPs, 10DNA-Au NPs, and 5DNA-Au NPs, respectively. On the basis of our results, we suggest that the concentration of DNA and salt aging are two important factors controlling the surface density of DNA in each Au NP.
Sensing Strategy. To test our new sensing system’s ability to detect Hg2+, we conducted fluorescence measurements of a solution containing the 60DNA-Au NPs and OliGreen before and after the addition of Hg2+. Curve (a) in Figure 1 indicates that the fluorescence of the solution was negligible in the absence of Hg2+, consistent with the fact that Au NPs are efficient quenchers. After adding Hg2+, the solution fluoresced weakly at
525 nm (ca. 3.8-fold greater than that in curve (c)) upon excitation at 480 nm. The quantum yields of free OliGreen and DNA-OliGreen complexes are less than 1% and 95% (fluorescein was used as a reference), respectively. Although the fluorescence of the solutions increased upon increasing the concentration of OliGreen, we found that the DNA-Au NPs aggregated in the presence of greater than 0.025× OliGreen, which caused significant loss in the sensitivity and reproducibility of the sensor system. The optimum concentration of OliGreen was 0.025× in the study system. Inset (A) to Figure 1 suggests that the reaction reached completion within 60 min. Because we believe that the addition of Hg2+ induced immediate changes in the DNA conformations, the long equilibrium time presumably arose as a result of the slow release of DNA molecules into the bulk solution. After centrifugation, we determined the concentration of DNA molecules in the supernatant, from which we estimated that 14.6% of the DNA molecules had been displaced from the Au NPs. The fluorescence (curve (c)) is weaker than that (curve (a)) of a solution containing 45 nM DNA and OliGreen, revealing that only few DNA molecules were released to the bulk solution. Based on the fluorescence intensities in curves (a) and (b), we estimate that greater than 97% of the fluorescence of the DNA-OliGreen complexes was quenched by the Au NPs. In addition to a change in fluorescence, the SPR absorption increased (ca. 1.1-fold) and underwent a blue shift from 522 to 517 nm after the addition of 5.0 µM Hg2+ (Figure S2, Supporting Information). The changes in UV-vis absorption provide strong evidence that Hg2+ions were adsorbed onto the surface.11To further investigate the role that Hg2+played in the fluorescence and absorption changes of the 60DNA-Au NP solution, we performed gel electrophoresis using 1.0% agarose gels. The image depicted in inset B of Figure 1 indicates that DNA-Au NPs in the presence of Hg2+migrated faster toward the anode than they did in the absence of Hg2+. Because the surface negative charge density of the 60DNA-Au NPs in the absence of Hg2+ was higher than that in the presence of Hg2+, the faster electrophoretic migration of the latter indicates that the surface morphology of 60DNA-Au NPs must have changed; note that the diameter of the 60DNA-Au NPs in the presence of Hg2+is smaller than that in the absence of Hg2+.12The circular dichroism (CD) spectra (Figure S3, Supporting Information) confirm the formation of the folding structure of probe DNA mediated by Hg2+.5cWe further supported our reasoning by adding the mixtures of the probe DNA (30 nM) and Hg2+(0-10µM) to Au NP solutions (1 nM) and then measuring the unbound DNA molecules using OliGreen after centrifugation. Upon increasing the concentration of Hg2+, the percentages of DNA molecules bound to Au NPs decreased from 34.8% to 2.2% (Figure S4, Supporting Informa-tion). We also performed a control experiment utilizing Au NPs that had been treated with a random 5′-thiol-modified oligo-nucleotide lacking a polythymidine component at the 5′ end (5′ -C15TAGCCATCTCTTCTCCGAGCCGGTCGAAATAGT-GAGT-3′). We found that only 1% of these DNA molecules were released (Figure S5, Supporting Information) after adding 5.0µM Hg2+under the same conditions used to obtain the data in Figure 1.
Effects of the Surface DNA Density. To further support our hypothesis that the surface DNA density on the Au NPs played an important role in determining the sensitivity, we compared the fluorescence intensities of five different DNA-Au NP solutions (9) (a) Mucic, R. C.; Storhoff, J. J.; Mirkin, C. A.; Letsinger, R. L. J. Am.
Chem. Soc. 1998, 120, 12674–12675. (b) Link, S.; El-Sayed, M. A. J. Phys. Chem. B 1999, 103, 8410–8426. (c) Storhoff, J. J.; Elghanian, R.; Mucic, R. C.; Mirkin, C. A.; Letsinger, R. L. J. Am. Chem. Soc. 1998, 120, 1959–1964.
(10) (a) Hurst, S. J.; Lytton-Jean, A. K. R.; Mirkin, C. A. Anal. Chem. 2006, 78, 8313–8318. (b) Demers, L. M.; Mirkin, C. A.; Mucic, R. C.; Reynolds, R. A.; Letsinger, R. L.; Elghanian, R.; Viswanadham, G. Anal. Chem. 2000, 72, 5535– 5541.
(11) Yang, Z.; Lin, Y.-W.; Tseng, W.-L.; Chang, H.-T. J. Mater. Chem. 2005, 15, 2450–2454.
(12) (a) Liang, X.; Kuhn, H.; Frank-Kamenetskii, M. D. Biophys. J. 2006, 90, 2877–2889. (b) Parak, W. J.; Pellegrino, T.; Micheel, C. M.; Gerion, D.; Williams, S. C.; Alivisatos, A. P. Nano Lett. 2003, 3, 33–36.
Figure 4. (A) Fluorescence responses of OliGreen-DNA-Au NPs (0.75
nM) after the addition of Hg2+ions (0, 0.05, 0.1, 0.25, 0.5, 1.0, and 2.5
µM). (B) Plot of fluorescence intensity (525 nm) versus Hg2+ concentration. All other conditions were the same as those described in Figure 1.
SensitiVe and SelectiVe Detection of Hg Ions Langmuir, Vol. 24, No. 15, 2008 8349
Downloaded by NATIONAL TAIWAN UNIV on July 30, 2009
(0.75 nM) in the presence of 5.0µM Hg2+. Figure 2 indicates that the surface DNA density did have a strong impact on the Hg2+ -induced fluorescence changes. When there were 5 or 10 DNA molecules per Au NP, the fluorescence intensity at 525 nm was quenched completely by the Au NPs. At such low DNA surface densities, the DNA species on the surface were more likely to exist in flattened structures as a result of strong interactions with the Au surface.13Thus, the addition of Hg2+did not induce the release of great amounts of DNA molecules from these Au NP surfaces to the bulk solution (Scheme 1A). When there were 30 DNA molecules per Au NP, the fluorescence was not quenched completely by the Au NPs. We suggest that these DNA structures were not as flat as those in the low-DNA-surface-density systems, mainly because of repulsion among DNA molecules, based on the fact that energy and electron transfer efficiencies are inversely proportional to the 6 and 4 powers, respectively, of the distance between the donor and acceptor. In the presence of 5.0µM Hg2+, the fluorescence was weaker (by 10%) than that in the absence of Hg2+, suggesting that almost none of the DNA molecules had been released into the bulk solution. The changes in fluorescence suggest that the DNA structures on the Au NP surfaces were different in the presence and absence of Hg2+ (Scheme 1B). When there were 60 or 90 DNA molecules per Au NP (60- and 90DNA-Au NPs), stretched, linear DNA structures dominated on the Au NP surfaces, mainly because of steric effects and strong electrostatic repulsion among DNA molecules. As a result, the quenching efficiencies of the Au NPs with respect to the fluorescence of the DNA-OliGreen complexes were lower (higher background). In the presence of 5.0 µM Hg2+, the fluorescence increases observed for the 60- and 90DNA-Au NPs were approximately 3.8- and 3.6-fold, respectively. We suggest that Hg2+ions induced the formation of hairpin DNA structures and, thus, some of the DNA molecules were released to the bulk solution (Scheme 1C). After centrifugation, we used OliGreen to determine the concentration of DNA in the supernatants. Correspondingly, we estimated that approximately 14.6% and 21.0% of the DNA molecules had been released from the surfaces of the 60- and 90DNA-Au NPs, respectively, and into their bulk solutions. We also measured the lifetimes of the solutions to further support our reasoning. The lifetimes of the DNA-OliGreen and OliGreen-DNA-Au NPs were 3.08 and 1.10 ns, respectively. In the presence of Hg2+(10 µM), we determined the lifetime of the solution to be 2.25 ns, which again supports that OliGreen-DNA-Hg2+complexes were released from Au NP surfaces to the bulk solution. Because of their lower background signal, the 60DNA-Au NPs provided a higher sensitivity than the 90DNA-Au NPs did; therefore, we used the former sample for subsequent studies.
Specificity. To investigate the specificity of the 60DNA-Au NPs toward Hg2+, we tested their behavior toward the metal ions (each 5.0µM) Mg2+, Ca2+, Co2+, Ni2+, Cu2+, Zn2+, Cd2+, Pb2+, Fe2+, Au3+, and Fe3+. Figure 3A indicates that only the addition of Hg2+caused a fluorescence increase, revealing the exceptional specificity of this present method. The decreased fluorescence observed for the 60DNA-Au NPs in the presence of the other metals ions is most likely due to decreased electrostatic repulsion among the DNA molecules through a process of charge screening; that is, the DNA structures in the presence of the metal ions were more compact, causing the DNA-OliGreen complexes to be positioned closer to the Au NP surfaces, leading to a greater quenching effect and, thus, lower fluorescence. Figure 3B
indicates that the metal-induced changes in fluorescence in the presence of salt (1.0 mM NaCl) were smaller to those in the absence of salt. In the presence of 1.0 mM NaCl, the metal-induced changes in fluorescence are smaller, resulting in less interference. We note that the sensitivity of the present approach toward Hg2+was at least 150-fold greater than that toward any of the other tested heavy metal ions and more than 200-fold higher than that toward alkaline and alkaline earth metal ions. Figure 4A indicates that the fluorescence of the 60DNA-Au NPs increased upon increasing the Hg2+concentration. A linear correlation existed between the fluorescence intensity and the concentration of Hg2+over the range 0.05-2.5µM (R2) 0.98). The limit of detection (LOD), at a signal-to-noise ratio of 3, for Hg2+was 25 nM. Because the number of DNA molecules on Au NPs was well controlled under different conditions and the DNA-Au NPs were stable at 4°C for at least 1 month, the linear ranges were the same when using five different batches of 60DNA-Au NPs. To test the practicality of this present approach, we used it to analyze a water sample obtained from a pond on the National Taiwan University (NTU) campus. To determine the concentration of Hg2+ions, we applied a standard addition method. A linear correlation existed between the fluorescence intensity and the concentration of Hg2+ ions (over the range 0.05-1.0µM (R2) 0.97)) spiked into the pond water. We did not detect the presence of Hg2+ions in any of the pond water samples, in good agreement with ICP-MS data. The recoveries of these measurements were 96%-102% (Figure S6, Supporting Information).
Conclusion
We have devised a new assay for the sensitive and selective detection of Hg2+ions. To the best of our knowledge, this system is the first that allows the detection of Hg2+based on the release of DNA molecules, induced by conformational changes on Au NP surfaces. The sensitivity of these DNA-Au NPs toward Hg2+is highly dependent upon their surface DNA density. Our new sensing mechanism is different from that of Hg2+-induced aggregation of DNA-functionalized Au NPs from at least three aspects: (1) Hg2+-induced dissociation of DNA molecules versus Hg2+-induced aggregation of Au NPs; (2) use of DNA-Au NPs versus two-cDNA-Au NPs; and (3) fluorescence versus colo-rimetry. Although our approach provides a lower LOD (25 vs 100 nM) at room temperature (vs 50°C), use of OliGreen to label DNA is required. In future studies, we will investigate the effects that the DNA size and sequence, the Au NP size, and reaction temperature have on the sensitivity of the DNA-Au NPs toward Hg2+. On the basis of the fact that different DNA sequences have various affinities toward different metal ions, we believe that this present approach also holds great promise as a means of detecting metals other than Hg2+.
Acknowledgment. This study was supported by the National Science Council of Taiwan under Contract NSC 96-2627-M-002-013.
Supporting Information Available:TEM images of solutions containing Au NPs and DNA-Au NPs (Figure S1). UV-vis absorbance spectra of solutions containing Au NPs and DNA-Au NPs in the absence and presence of Hg2+(Figure S2). Ellipicity versus Hg2+ion concentration (Figure S3). Plot of binding percentage versus Hg2+concentration (Figure S4). Fluorescence response of control OliGreen-DNA-Au NPs (0.75 nM) upon addition of 5.0µM Hg2+ions (Figure S5). Fluorescence response of OliGreen-DNA-Au NPs upon addition of Hg2+ions to a matrix of pond water (Figure S6). This material is available free of charge via the Internet at http://pubs.acs.org.
LA800589M (13) (a) Storhoff, J. J.; Elghanian, R.; Mirkin, C. A.; Letsinger, R. L. Langmuir
2002, 18, 6666–6670. (b) Li, H.; Rothberg, L. Proc. Natl. Acad. Sci. U.S.A. 2004,
101, 14036–14039. (c) Wang, L.; Liu, X.; Hu, X.; Song, S.; Fan, C. Chem. Commun. 2006, 36, 3780–3782.
Downloaded by NATIONAL TAIWAN UNIV on July 30, 2009