PAPER
www.rsc.org/dalton
| Dalton Transactions
Synthesis and photophysical properties of multinuclear zinc-salophen
complexes: enhancement of fluorescence by fluorene termini†
Kai-Lun Kuo, Chiung-Cheng Huang and Ying-Chih Lin*
Received 29th January 2008, Accepted 6th May 2008
First published as an Advance Article on the web 18th June 2008
DOI: 10.1039/b801602j
Incorporation of fluorene groups into the salicylidene moiety significantly enhances the luminescence of a number of multinuclear alkynylated Zn(II)-salophen complexes. Preparation of these complexes was achieved by a synthetic strategy with facile handling of the reactants, simple purification of the products, and one-pot reaction process. Two synthetic methods are used for the preparation of different types of multinuclear salophen complexes. The introduction of a bis- or a tris-salicylaldehyde as a bridging unit in the presence of various alkynyl substituted monoimines in the reaction mixture containing zinc acetate resulted in the preparation of di- and tri-nuclear Zn(II)-salophen complexes of type 1, respectively. For a different type, treatment of tetraminobenzene with various
arylethynyl-substituted salicylaldehyde afforded dinuclear Zn(II) alkynylated salophen complexes of type 2 with a different structure. The photophysical behaviors of these multinuclear metal salophen complexes were investigated. Particularly, the dinuclear complex 9b of type 1 having ethynylfluorene groups in salophen moieties and dialkoxyl groups in the bridging moiety exhibits higher quantum efficiency than that of other complexes in this report. In addition, the bis-Zn(II) alkynylated salophen complex 11e bearing nitrogen donor groups displays more red-shifted pattern than those with other functional substituents both in absorption and emission spectra.
Introduction
Schiff-base complexes have received much attention recently, mainly because of their wide applications in the fields of synthesis and catalysis. Considerable research efforts are devoted to the synthesis of new complexes with transition1 and main group2
metal ions, to further develop their applications.3–7Cooperative
reactivity between multiple metal centers is commonly postulated for enzymatic systems8and has evolved into an intriguing design
principle for synthetic catalysts9including Schiff-base complexes.
Recently, Jacobsen and co-workers undertook the design of catalysts that enforce this cooperative mechanism through the construction of covalently linked bimetallic salen complexes,10and
application of such systems as highly enantioselective and efficient asymmetric ring opening catalysts has been achieved. Kinetic and enantioselectivity data provide evidence for the mechanism involving cooperative, intramolecular bimetallic catalysis. Metal salophen complexes have also been applied in the synthesis of macromolecules. The alkynylated salophen moiety was utilized to establish shape-persistent conjugated macrocycles with tunable pore diameters in the nanometer regime.11These macrocycles can
bind multiple metals, forming soluble, luminescent complexes. Moreover, Hupp and co-workers have developed a series of
Department of Chemistry, National Taiwan University, Taipei, Taiwan. E-mail: [email protected]; Fax: +(886)-2-23636359
† Electronic supplementary information (ESI) available: Fig. S1–S7:1H
NMR spectra of complexes 7b, 8b, 9a, 9b, 11a, 11d and 15a. Table S1: Comparison of the photophysical properties with energy gap for complexes
9–13 by MO calculation. Table S2: Molecular orbital calculations of
dinuclear Zn(II)-salophen complexes. See DOI: 10.1039/b801602j
bifunctional salophen type ligands to serve as building blocks for cyclic supramolecular structures.12 Previously, we reported
the synthesis of alkynylated metal salophen complexes and investigated their photophysical properties.13 Introduction of a
pyridyl group as a bridge as well as incorporation of ethynyl tethers and electron-donating groups into the salicylidene moiety of these complexes generally enhances the quantum yield of photoluminescence. These structural modifications cause a larger p delocalization over the salicylidene and the aromatic bridging rings than that in regular Zn(salophen) complexes. The rigidity of the structure and the dipole moment of the complex may thus increase. In the preparation of Mg(II)-salophen complexes we unexpectedly obtained organic monoimines13b,14 which are
considered as good precursors for the synthesis of unsymmetrical or polynuclear Schiff base complexes. Fluorenes are widely used as electron acceptors in charge transfer complexes15 and
electron transport materials.16The substituted poly(2,7-fuorene)
derivatives are particularly desirable as active constituents of organic light-emitting diodes (OLEDs) due to their thermal and chemical stability and their high emission quantum yield.17 The
fluorene structural moiety also provides a planar biphenyl unit within the molecular backbone. In this paper, we introduce fluorene group into a monoimine moiety to incorporate with various bridging units for the synthesis of two types of dinuclear Zn(II) alkynylated salophen complexes. Furthermore, we also use tris-salicylaldehydes to prepare trinuclear complexes. Upon inclusion of different substituted-arylethynyl groups including 2-ethynylfluorene derivatives and various bis-salicylaldehydes as bridge units of salophen moieties, the synthesis of these complexes and their photophysical properties are investigated.
Results and discussion
Synthesis of dinuclear Zn(II)-salophen complexes
Three bis(salicylaldehyde) compounds 1, 2 and 3 (Chart 1) were synthesized by Pd(II)/Cu(I) catalyzed Sonogashira cross-coupling reaction18 of 5-ethynylsalicylaldehyde with
1,4-bis-(dodecyloxy-2,5-diiodo)benzene, 5-bromosalicylaldehyde and 2,5-diiodothiophene, respectively.19 Additionally, a
bis-salicyl-aldehyde 4 (Chart 1) was prepared by the reaction of trioxane and salicylaldehyde in glacial acetic acid and sulfuric acid following the literature report.20These compounds were readily purified by silica
gel packed column chromatography and were isolated in about 30–80% yield. In the1H NMR spectra of these salicylaldehydes, resonances of the hydroxyl proton are observed at around dH11.1 and resonances near dH9.9 are assigned to the aldehyde group. In the13C NMR spectra, resonance of two carbonyl groups exhibits only one peak at dC 192.7 and similar results are observed for the alkynyl carbons indicating a symmetrical structure. Other spectroscopic data are also consistent with the proposed structure.
Chart 1 Structures of precusors 1–5 and 14.
A zinc-mediated, one-pot procedure provides access to a number of multinuclear Zn(II)-salophen complexes with func-tional groups displaying various steric and electronic effects. The procedure used for our preparation is slightly different from the methods reported in the literature.13b We first prepared the
organic monoimine 6a by the reaction of 5-substituted ethynyl-salicyaldehyde 5a with excess 2,3-diaminopyridine in a THF– MeOH solution. Likewise monoimine compounds 6b and 6c were also successfully prepared in high yield. To prepare complex 7a, Zn(OAc)2 was directly treated with the bis-salicylaldehyde 1 in a THF–MeOH solution at 60 ◦C. Subsequently the resulting reaction mixture was added slowly to a solution of 6a in THF, the mixture was stirred at 60◦C to give the orange product 7a which was collected by filtration in 70% yield (Scheme 1).
For 7a, two resonances of the two imine CH=N groups at dH 9.45, 9.13 in the1H NMR spectrum and at d
C164.7 and 164.4 in the13C NMR spectrum, are consistent with D
2hsymmetry for the bis-Zn(II)-salophen complex. In addition, three ethynyl carbons are observed at dC95.2, 88.7 and 81.3 in the13C NMR spectrum. The MALDI-TOF mass spectrum shows the parent ion at m/z 996.38, supporting the structure of complex 7a.
In an effort to investigate the influence of the bridge in between two salicylaldehyde groups and the substituents in ethynyl-salicylaldehyde, we used different bridging bis(salicylaldehyde)
Scheme 1 Synthesis of dinuclear Zn(II)-salophen complexes of type I.
compounds 2, 3 and 4 to react with monoimine 6a having the ethynylthiophene group to prepare three dinuclear Zn(II)-salophen complexes 8a, 9a and 10a, respectively (Chart 2). Similarly three other dinuclear salophen complexes 7b, 8b and 9b with an ethynylfluorene group were also prepared from the reactions of 6b with bis(salicylaldehyde) 1, 2 and 3, respectively. In general, these complexes were obtained in 45–70% yields. Two1H NMR absorptions of non-equivalent imine CH=N protons are observed for these complexes.
Chart 2 Structures of dinuclear Zn(II)-salophen complexes of type 1.
The one-pot reaction strategy for the preparation of mononu-clear Zn(II)-salophen complexes is used for the synthesis of dinu-clear Zn(II)-salophen complexes in which two salophen moieties are linked by a phenyl ring (see Scheme 2). The procedure involves the treatment of zinc acetate with 5-ethynylsalicylaldehyde in a mixed MeOH–THF solvent for 30 min at 60◦C. After subsequent addition of 1,2,4,5-tetraaminobenzene tetrahydrochloride, the resulting solution was further stirred for 2 days to yield a red powder identified as complex 11d in 70% isolated yield (Scheme 2).
Scheme 2 Synthesis of dinuclear Zn(II)-salophen complexes of type 2. In the1H NMR spectrum of complex 11d, a resonance of the imine groups is observed at dH9.15 and a resonance of the bridging phenyl protons adjacent to two electron-withdrawing imine groups is at dH8.30. Additionally, the singlet peak at dH3.91 is assigned to the acetylenic protons. In the13C NMR spectrum, resonances of two ethynyl carbons are found in the range of dC 74–94. Other spectroscopic data are consistent with the proposed structure of 11d. Following the same procedure, a number of bimetallic complexes 11a, 11b and 11e bearing alkynylated substituents as well as alkyne-free analogues 12 and 13 were also prepared (Chart 3).
Chart 3 Structures of complexes 12 and 13.
Reek and co-workers have reported that dinuclear Zn(II )-salophen complexes containing alkyne-free moieties can be ob-tained by a stepwise approach.14In their preparation, free salophen
ligands were first prepared by condensation of amines and aldehydes in alcohol solvent. Subsequently, metal acetate was introduced into the isolated salophen compound to afford the
bis-Zn(II)-salophen complex. Their attempts to use a one-pot approach yielded only mixtures of unidentified species. In our system, we can successfully utilize our one-pot methodology to afford dinuclear Zn(II)-salophen complexes 11, 12 and 13 with no side products such as oligomers or polymers. Therefore, our simple one-pot preparation procedure provides an alternative for the synthesis of dinuclear Zn(II)-salophen complexes.
Synthesis of trinuclear Zn(II)-salophen complexes
Under the same reaction condition as that utilized for the synthesis of dinuclear complexes, two trinuclear alkynylated salophen complexes 15a and 15b were also synthesized using suitable ratios of reactants. The precursor trisalicylaldehyde 14 is a rigid compound in which the conformational freedom is limited to the rotation about the single bonds between the ethynyl and the salicylaldehyde groups. Compound 14 was obtained from the reaction of 1,3,5-triethynylbenzene and 5-bromosalicylaldehyde using Sonogashira coupling reaction. Compound 14 dissolved in a mixture of pyridine and THF was then treated with Zn(OAc)2 and 6a in pyridine and MeOH to give the trinuclear complex 15a. By using the same procedure, complex 15b was also prepared in 50% yield (Scheme 3).
Scheme 3 Synthesis of trinuclear Zn(II)-salophen complexes.
The1H NMR spectrum of 15a shows two signals at d 9.45 and 9.13 attributed to two nonequivalent imine protons. The13C NMR spectrum reveals the two imine carbon resonances at d 163.8 and
d 162.8, and four ethynyl carbons are also observed at d 93.7,
91.8, 85.5, 79.9. No signals for aldehyde and amino protons are detected, eliminating the possibility for the formation of mono-or di-nuclear Zn(II)-salophen complexes. However, we were not able to obtain mass spectrum of these trinuclear Zn(II)-salophen complexes.
Previously, the analogous ethynyltrisalicylaldehyde, 1,3,5-tris-[(5-tert-butyl-3-formyl-4-hydroxyphenyl)ethynyl]benzene21 was
applied in the synthesis of rigid chiral Mn(II)-salophen polymers. These polymers catalyzed the asymmetric epoxidation of alkenes to give the corresponding epoxides up to 67% ee. Additionally, these complexes were reusable several times without reduced activity or selectivity.
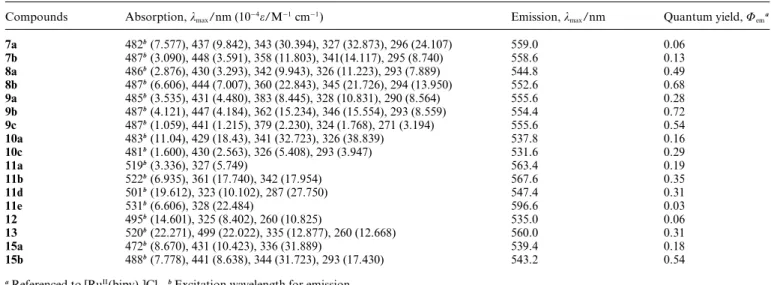
Table 1 Photophysical properties of dinuclear and trinuclear Zn(II)-salophen complexes
Compounds Absorption, kmax/nm (10−4e/M−1cm−1) Emission, kmax/nm Quantum yield, Uema
7a 482b(7.577), 437 (9.842), 343 (30.394), 327 (32.873), 296 (24.107) 559.0 0.06 7b 487b(3.090), 448 (3.591), 358 (11.803), 341(14.117), 295 (8.740) 558.6 0.13 8a 486b(2.876), 430 (3.293), 342 (9.943), 326 (11.223), 293 (7.889) 544.8 0.49 8b 487b(6.606), 444 (7.007), 360 (22.843), 345 (21.726), 294 (13.950) 552.6 0.68 9a 485b(3.535), 431 (4.480), 383 (8.445), 328 (10.831), 290 (8.564) 555.6 0.28 9b 487b(4.121), 447 (4.184), 362 (15.234), 346 (15.554), 293 (8.559) 554.4 0.72 9c 487b(1.059), 441 (1.215), 379 (2.230), 324 (1.768), 271 (3.194) 555.6 0.54 10a 483b(11.04), 429 (18.43), 341 (32.723), 326 (38.839) 537.8 0.16 10c 481b(1.600), 430 (2.563), 326 (5.408), 293 (3.947) 531.6 0.29 11a 519b(3.336), 327 (5.749) 563.4 0.19 11b 522b(6.935), 361 (17.740), 342 (17.954) 567.6 0.35 11d 501b(19.612), 323 (10.102), 287 (27.750) 547.4 0.31 11e 531b(6.606), 328 (22.484) 596.6 0.03 12 495b(14.601), 325 (8.402), 260 (10.825) 535.0 0.06 13 520b(22.271), 499 (22.022), 335 (12.877), 260 (12.668) 560.0 0.31 15a 472b(8.670), 431 (10.423), 336 (31.889) 539.4 0.18 15b 488b(7.778), 441 (8.638), 344 (31.723), 293 (17.430) 543.2 0.54
aReferenced to [RuII(bipy)
3]Cl2.bExcitation wavelength for emission.
Photophysical properties of multinuclear complexes
Relevant photophysical properties of multinuclear Zn(II)-salophen complexes studied in this work are listed in Table 1. Absorption and emission spectra of dinuclear Zn(II)-salophen complexes 7– 10 are shown in Fig. 1 and Fig. 2. Two distinct absorption bands in the range of 250–490 nm for the complexes are listed in Table 1. The high energy absorption band is assigned to the ligand-centered (LC) p–p* transition of the p-substituted phenyl ring and salicylaldimine ligand.22The low energy absorption band is
assigned to the intramolecular charge transfer (ICT) band possibly relating to the phenoxide lone pair (l) and the p* orbital of imine,
i.e. [l(phenoxide)→p*(imine))].22b
Fig. 1 Absorption spectra of the type 1 dinuclear Zn(II)-salophen complexes 7a–10a and trinuclear 15a and 15b in THF.
In general, the absorption and emission peaks of multinuclear Zn(II)-salophen complexes are more red-shifted than those of previously reported mononuclear-Zn(II) alkynylated salophen complexes. In the dinuclear Zn(II)-salophen complex of type 1, the variation of substituent group in the ethynyl side chains seems not to greatly influence the absorption and the emission
Fig. 2 Emission spectra of the type 1 dinuclear Zn(II)-salophen complexes
7a–10a and trinuclear 15a and 15b in THF.
patterns. For example, complexes 9a–9c show almost the same peaks at 487 nm in the absorption spectra and the same emission peaks at around 555 nm. The quantum yield of 0.72 for 9b with alkylfluorene and bis-alkoxylbenzene moieties is higher than those of the other complexes. However, complexes 7a and 7b, each with only one ethynyl linkage, show poorer quantum efficiency than other dinuclear Zn(II)-salophen complexes with two alkynyl linkages. For 10a and 10c in which two salophen groups are bridged by a methylene group, even though the absorption patterns are similar to those of other dinuclear complexes of type 1, the emission spectra are slightly blue-shifted 20 nm relative to those of other complexes. When the side chains are fixed with thiophene groups, the absorption spectra of complexes 7a, 8a, 9a and 10a show similar character in the low energy band with the absorption band located at around 485 nm. Especially, the absorption peaks of 11a in which two salophen moieties are linked by a phenyl ring display ca. 25 nm red-shift relative to those of type 1. Particularly, the two salophen moieties bridged with a methylene group in complex 10 lower p delocalization between two salophen moieties
relative to that of the other di-Zn(II)-salophen complexes 7, 8, 9 and 11 described in this paper. This suggests that the CH2 bridge between two Zn(II)-salophen moieties might lower the rigidity of the structure and thus decrease the dipole moment of the complex. Additionally, from theoretical calculation (see ESI†), the HOMO and LUMO frontier orbitals of complexes 7–9 generally show large electron density around the bridge moiety and salophen moiety, respectively. However, the HOMO frontier orbitals of complexes 10a and 10c exhibit major electron density in the region of ethynyl side chains and phenoxide moieties even though the LUMO frontier orbitals of them are similar to those of complexes 7–9. Moreover, the HOMO and LUMO energy gap in complexes 10a and 10c are smaller in comparison with those of complexes 7–9. Accordingly, the emission peak of complex 10a at 537 nm is slightly blue-shifted relative to those of other complexes. Attractively, complexes 7b, 8b and 9b bearing fluorene groups all exhibit relatively higher quantum yield. This means that the incorporation of ethynylfluorene chromophores into the salophen moiety slightly enhances the quantum efficiency of di-Zn(II)-salophen complexes. Similarly, this phenomenon could also be observed both in dinuclear Zn(II) complexes of type 2 and our trinuclear-Zn(II) complex systems.
Absorption and emission spectra for dinuclear Zn(II)-salophen complexes of type 2 are shown in Fig. 3 and 4, respectively. As to the ethynyl-free complexes 12 and 13, the quantum yield of 13 is higher than that of 12. The electron-donating tert-butyl groups in 13 enhance the quantum efficiency and bathochromic shift on both absorption and emission peaks relative to that of 12. The electron-donating tert-butyl group could increase the electron density in the excited states and enhance the probability of fluorescence relaxation. Additionally, four tert-butyl groups in complex 13 provide good shielding of the Zn(II)-salophen complex from solvent interactions thus leading to nonradiative deactivation. The steric bulk of the butyl group might also block the intermolecular stacking22 and decrease the self-quenching
effect thus enhancing the quantum efficiency. Moreover, the absorption peaks for the ethynyl-substituted complexes 11a, 11b, 11d and 11e are red-shifted, contrasting with those for the two ethynyl-free analogues 12 and 13. Correspondingly, this trend
Fig. 3 Absorption spectra of the type 2 dinuclear Zn(II)-salophen complexes 11–13 in THF.
Fig. 4 Emission spectra of the type 2 dinuclear Zn(II)-salophen complexes
11–13 in THF.
could also be observed by the comparison of the HOMO and LUMO energy gap for complexes 11–13 in the MO calculation (see ESI†). Concerning ethynyl-substituted complexes of type 2, both absorption and emission spectra of complexes 11a, 11b and 11e with arylethynyl-substituted groups are more blue-shifted than those of complex 11d with alkynyl groups. These complexes with a larger p delocalization over the salicylidene and aromatic rings possibly result in the enhancement of the structural rigidity. The aforementioned effects from the substituents are similar to those described in our previous reports.13 The absorption and
emission bands for dinuclear Zn(II)-salophen complexes of type 2 are observed to red shift in comparison with that in mononuclear Zn(II)-salophen complexes. Particularly, the presence of nitrogen donor groups in complex 11e results in larger red shift, ca. 35 nm in absorption spectrum and ca. 60 nm in emission spectrum relative to substituent free complex 12.
Concerning the lifetime of the excitation state, complexes 9b and 11b, of which the quantum yields are higher than other complexes in this report, were further examined to gain insights into the relaxation dynamics in solution. For electrooptical applications of Zn(II)-salophen complexes, the observed lifetimes of 9b and 11b were measured to be 2.16 and 1.43 ns, respectively, in THF.
Conclusion
In summary, we utilize various monoimines and bis- or tris-salicylaldehyde or tetraminobenzene to synthesize a series of di-and trinuclear-Zn(II)-salophen complexes. The methodology used for the synthesis of these complexes is simple and efficient. With regard to the photophysical properties, introduction of ethynylflu-orene groups in salophen moieties and dialkoxyl groups in the bridging moiety significantly enhances the quantum efficiency of these complexes. In both absorption and emission spectra, din-uclear Zn(II)-salophen complexes of type 2 i.e. complexes 11–13, exhibit slightly red-shifted bands in comparison with those of type 1 i.e. complexes 7–10. Additionally, both absorption and emission peaks of the bis-Zn(II) alkynylated salophen complex 11e having nitrogen donor groups displays a more red-shifted pattern than 11a, 11b and 11d with other functional substituents. Furthermore, multinuclear Zn(II)-salophen complexes show slightly red-shifted
bands from those of the corresponding mononuclear Zn(II )-salophen complexes in both absorption and emission spectra.
Experimental
General proceduresAll manipulations were performed under nitrogen using vacuum-line, dry-box, and standard Schlenk techniques. Et3N was dried from KOH, MeOH from Mg turnings and THF from Na/ketyl. All other solvents and reagents were of reagent grade and were used without further purification. NMR spectra were recorded on Bruker AC-300, AVANCE-400 and DMX-500SB FT-NMR spec-trometers at room temperature (unless states otherwise) and were reported in units of with residual protons in the solvent as standard (CDCl3, d 7.24; d6-methyl sulfoxide, d 2.49; d8-THF, d 3.58, 1.73).
EI and FAB mass spectra were recorded on a VG70-250S mass spectrometer and JEOL SX-102A spectrometers, respectively. MALDI-TOF mass spectra were obtained in a dithranol matrix and collected by Voyager DE-PRO (Applied Biosystem, Houston, USA) equipped with a nitrogen laser (337 nm), and operated in the delayed extraction reflector mode. Absorption spectra were obtained using a HP8453, Hewlett–Packard UV-visible spectrophotometer. Emission spectra were taken using a Hitachi F-4500 luminescence spectrometer. Luminescence quantum yield (Uem) were calculated relative to [RuII(bipy)3]Cl2in air-equilibrated aqueous solution (Uem= 0.028).23Elemental analyses were carried on a Perkin-Elmer 2400 CHN elemental analyzer. Preparation of monoimines 6a and 6c have been reported by us.13b
1,3,5-Triethynylbenzene24and compound 421were prepared by methods
reported in the literatures. Quantum mechanical calculations were performed with the Spartan 04, version 1.0.8, software package. All HOMO and LUMO orbitals energy calculations were carried out by using the AM1 semi-empirical model. Lifetime studies were performed using an Edinburgh FL 900 photon-counting system with a hydrogen-filled/or a nitrogen lamp as the excitation source. Data were analyzed using the nonlinear least squares procedure in combination with an iterative convolution method. The emission decays were analyzed by the sum of exponential functions, which allows partial removal of the instrument time broadening and consequently renders a temporal resolution of∼200 ps.
Synthesis
Compound 1. Under a nitrogen atmosphere, 5-bromosalicylal-dehyde (412 mg, 2.05 mmol), 5-ethynylsalicylal5-bromosalicylal-dehyde (100 mg, 0.68 mmol), PPh3(4 mg, 0.015 mmol) and Pd(PPh3)2Cl2(11 mg, 0.016 mmol) were dissolved in 10 mL of THF. Then 5 mL of Et3N was added, turning the solution from yellow to orange. After stirring for 20 min, CuI (3 mg, 0.016 mmol) was added, and the solution turned to dark brown. After heating at reflux at 70◦C for 48 h, the solution was cooled to room temperature. The yellow precipitate thus formed was isolated by filtration and washed with cold dichloromethane. Yield: 54 mg (30% yield based on 5-ethynylsalicylaldehyde). Spectroscopic data of 1:1H NMR (CDCl3) d 11.10 (s, 2H, OH), 9.88 (s, 2H, CHO), 7.73 (s, 2H, ArH), 7.64 (d, 2H, ArH,3J H–H = 8.4 Hz), 6.98 (d, 2H, ArH, 3J H–H= 8.4 Hz).13C NMR (CDCl3): d 192.7 (CHO), 162.6, 150.3, 136.2, 134.5, 118.9, 115.2 (Ph), 88.6 (C≡C). MS (FAB): m/z 267.2
(M++ 1). Anal. Calc. for C
16H10O4: C, 72.18; H, 3.79. Found: C, 72.26; H, 3.83%.
Compound 2. To a solution of 2,5-diiodothiophene (460 mg, 1.37 mml) in THF–Et3N (20 mL/10 mL) under nitrogen was added a mixture of 5-ethynylsalicylaldehyde (500 mg, 3.42 mmol), Pd(PPh3)2Cl2(96 mg, 0.14 mmol) and CuI (26 mg, 0.14 mmol). The solution was stirred at 65◦C for 18 h. After the solution was cooled to room temperature, the solvent was removed by a rotary evaporator. The residue was purified by flash chromatography on silica gel eluted with EA–hexanes, 1 : 3 first, then EA as eluent on a short plug of silica gel. The solvent was removed in vacuum to give a brown powder. The product was washed by methanol and hexanes to yield 2 (160 mg, 31%). Spectroscopic data of 2:1H NMR (CDCl3): d 11.15 (s, 2H, OH), 9.87 (s, 2H, CHO), 7.74 (s, 2H, ArH), 7.66 (dd, 2H, ArH,3J H–H= 5.7 Hz,4JH–H = 2.1 Hz), 7.12 (s, 2H, thiophene), 6.98 (d, 2H, ArH,3J H–H= 8.7 Hz).13C NMR (CDCl3): d 195.9 (CHO), 161.7, 139.5, 136.8, 131.8, 124.3, 120.5, 118.2, 114.4 (Ph), 92.3 (C≡C), 81.7 (C≡C). MS (FAB): m/z 373.3 (M++ 1). Anal. Calc. for C
22H12O4S: C, 70.96; H, 3.25; S, 8.61. Found: C, 71.05; H, 3.33; S, 8.71%.
Compound 3. The mixture of 5-ethynylsalicylaldehyde (100 mg, 0.68 mmol), 1,4-bis-dodecyloxy-2,5-diiodobenzene (159 mg, 0.228 mmol), Pd(PPh3)2Cl2(24 mg, 0.034 mmol) and CuI (6 mg, 0.032 mmol) were dissolved in THF, followed by addition of Et3N (20 mL). The reaction mixture was heated to 70 ◦C overnight. After cooling to room temperature, the solvent was removed by a rotary evaporator. The residue was purified by flash chromatography on silica gel (EA–hexanes, 1 : 3), then EA as elu-ent on a short plug of silica gel. The solvelu-ent was removed and then washed by methanol to yield 3 as a brown powder (134 mg, 80%). Spectroscopic data of 3:1H NMR (CDCl
3): d 11.11 (s, 2H, OH), 9.86 (s, 2H, CHO), 7.73 (s, 2H, ArH), 7.65 (dd, 2H, ArH,3J
H–H= 6.6 Hz,4J H–H = 1.6 Hz), 6.98 (s, 2H, ArH), 6.97 (d, 2H, ArH, 3J H–H= 6.21 Hz), 4.01 (t, 4H, OCH2,3JH–H= 6.5 Hz), 1.87–1.78 (m, 4H, CH2), 1.56–1.46 (m, 4H, CH2), 1.37–1.21 (m, 32H, CH2), 0.85 (t, 6H, CH3,3JH–H= 6.9 Hz).13C NMR (CDCl3): d 196.0 (CHO), 161.4, 153.6, 139.7, 136.7, 1250.5, 118.1, 116.8, 115.4, 113.7 (Ph), 93.0, 85.4 (C≡C), 69.6 (OCH2), 31.8, 29.6, 29.6, 29.6, 29.4, 29.3, 26.0, 22.6 (CH2), 14.0 (CH3). MS (FAB): m/z 735.0 (M++ 1). Anal. Calc. for C48H62O6: C, 78.44; H, 8.50. Found: C, 78.56; H, 8.44%. Monoimine 6b. Compound 5b (200 mg, 0.42 mmol) was treated with 2,3-diaminopyridine (364 mg, 3.34 mmol) and the mixture in THF (20 ml) was stirred at 60◦C. Then MeOH was added slowly into the resulting solution. The mixture was stirred at reflux temperature for 18 hr. Then the solvent was removed under vacuum. The residue was washed with methanol and precipitate was collected by filtration. Yellow organic monoimine 6b (107 mg, 45%) was obtained by extracting from the precipitate by using three lots of 10 mL of CH2Cl2.Spectroscopic data of 6b:1H NMR (CDCl3): d 12.17 (s, 1H, OH), 8.61 (s, 1H, N=CH), 7.87 (dd, 1H, PyrH,3J H–H= 6.0 Hz,4JH–H= 1.7 Hz), 7.69–7.30 (m, 10H, ArH), 7.04 (d, 1H, ArH,3J H–H= 11.1 Hz), 6.81 (dd, 1H, ArH,3JH–H= 7.2 Hz,4J H–H= 2.7 Hz), 6.13 (s, 2H, NH2).13C NMR (CDCl3): d 163.7 (Ph), 160.7 (C=N), 152.7, 150.9, 141.6, 140.3, 139.7, 137.0, 136.7, 135.6, 130.9, 130.4, 127.5, 126.8, 125.9, 122.8, 121.1, 119.9, 119.6, 119.0, 118.1, 117.8, 114.9, 114.2 (Ph), 89.7 (C≡C), 88.0 (C≡C), 55.1, 40.3, 31.4, 29.6, 23.6, 22.5, 13.9 (CH3). MS (FAB):
m/z 570.7 (M++ 1). Anal. Calc. for C
39H43N3O: C, 82.21; H, 7.61; N, 7.37. Found: C, 82.32; H, 7.68; N, 7.44.
Synthesis of dinuclear Zn(II)-salophen complexes of type 1
Complex 7a. Under a nitrogen atmosphere, the mixture of compound 1 (20 mg, 0.075 mmol) and Zn(OAc)2·2H2O (66 mg, 0.30 mmol) dissolved in THF–MeOH (30 mL–5 mL) was stirred at 60◦C for 30 min. Subsequently, 6a (48 mg, 0.15 mmol) dissolved in THF (10 mL) was added to this mixture slowly, and the reaction mixture was heated at 60◦C for 2 day. After cooling to room temperature, the solvent was removed under vacuum and the residue was washed with methanol, hexanes and ether. The precipitate was collected by filtration to yield 7a as orange powder in 70% yield (52 mg). Spectroscopic data of 7a:1H NMR (d
6 -DMSO): d 9.45 (s, 2H, N=CH), 9.13 (s, 2H, N=CH), 8.44 (br, 2H, PyrH), 8.33 (br, 2H, PyrH), 7.70–7.31 (m, 14H, ArH), 7.09 (br, 2H, thiophene), 6.75 (br, 4H, ArH).13C NMR (d
6-DMSO): d 175.4 (Ph), 174.9 (Ph), 164.7 (C=N), 164.4 (C=N), 147.7, 141.2, 139.1, 138.2, 135.9, 132.0, 128.2, 127.8, 125.7, 125.1, 120.7, 120.4, 109.8, 107.9, 95.2 (C≡C), 88.7 (C≡C), 81.3 (C≡C). MALDI-TOF-MS:
m/z 996.38 (M++ 1). Anal. Calc. for C
52H28N6O4S2Zn2·6H2O: C, 56.58; H, 3.65; N, 7.61. Found: C, 56.69; H, 3.68; N, 7.53%.
Complex 7b. Complex 7b as an red powder was similarly prepared by the same procedure as that of complex 7a with 63% yield. Spectroscopic data of 7b:1H NMR (d
8-THF): d 9.61 (s, 2H, N=CH), 9.02 (s, 2H, N=CH), 8.40 (d, 2H, PyrH,3J
H–H = 3.4 Hz), 8.22 (d, 2H, PyrH,3J
H–H = 7.4 Hz), 7.73–7.69 (m, 4H, ArH), 7.58 (s, 4H, ArH), 7.49 (s, 2H, ArH), 7.43–7.35 (m, 10H, ArH), 7.32–7.26 (m, 4H, ArH), 6.82 (d, 4H, ArH,3J
H–H= 9.4 Hz), 2.57 (br, 8H, CH2), 1.73–1.05 (m, 24H, CH2), 0.77 (t, 12H, CH3, 3J H–H = 7.2 Hz), 0.62 (m, 8H, CH2). 13C NMR (d6-DMSO): d 175.5 (Ph), 174.9 (Ph), 164.8 (C=N), 164.5 (C=N), 151.8, 151.6, 151.4, 150.1, 149.8, 149.6, 147.3, 141.5, 140.1, 138.2, 138.1, 135.9, 130.9, 128.1, 127.7, 126.1, 125.6, 125.4, 124.9, 124.3, 124.1, 123.8, 123.6, 120.7, 120.5, 120.4, 120.1, 96.1 (C≡C), 88.8 (C≡C), 81.8 (C≡C), 55.8, 41.1, 32.2, 30.5, 24.6, 23.4, 14.2 (CH3). Anal. Calc. for C94H88N6O4Zn2·6H2O: C, 70.36; H, 6.28; N, 5.24. Found: C, 70.51; H, 6.41; N, 5.13%.
Complex 8a. Complex 8a as a brown powder was similarly prepared by the same procedure as that of complex 7a with 66% yield. Spectroscopic data of 8a:1H NMR (d
8-THF): d 9.58 (s, 2H, N=CH), 8.99 (s, 2H, N=CH), 8.53 (s, 2H, PyrH), 8.21 (d, 2H, PyrH,3J
H–H= 7.9 Hz), 7.64 (s, 2H, ArH), 7.55 (s, 2H, ArH), 7.34 (m, 10H, ArH), 7.17 (s, 4H, thiophene), 6.98 (s, 2H, ArH).13C NMR (d5-Pyridine): d 175.7 (Ph), 174.9 (Ph), 164.7 (C=N), 164.4 (C=N), 151.0, 150.6, 147.7, 142.1, 141.1, 138.6, 138.3, 135.7, 132.0, 128.3, 127.8, 127.8, 125.8, 125.7, 125.2, 125.2, 125.1, 120.7, 120.4, 108.1, 107.9, 95.7 (C≡C), 95.2 (C≡C), 81.4 (C≡C), 81.3 (C≡C). Anal. Calc. for C58H30N6O4S3Zn2·6H2O: C, 57.58; H, 3.50; N, 6.95; S, 7.95. Found: C, 57.76; H, 3.58; N, 7.05; S, 8.05%.
Complex 8b. Complex 8b as an orange powder was similarly prepared by the same procedure as that of complex 7a with 62% yield. Spectroscopic data of 8b:1H NMR (d
8-THF): d 9.57 (s, 2H, N=CH), 9.00 (s, 2H, N=CH), 8.37 (d, 2H, PyrH,3J
H–H= 3.4 Hz), 8.20 (d, 2H, PyrH,3J
H–H= 8.3 Hz), 7.71–7.69 (m, 4H, ArH), 7.65 (s, 2H, ArH), 7.58 (s, 2H, ArH), 7.48 (s, 2H, ArH), 7.43–7.35 (m, 10H, ArH), 7.32–7.26 (m, 4H, ArH), 7.08 (s, 2H, thiophene),
6.79 (d, 4H, ArH, 3J H–H = 8.7 Hz), 2.03 (t, 8H, CH2,3JH–H = 7.9 Hz), 1.12–1.05 (m, 24H, CH2), 0.76 (t, 12H, CH3, 3JH–H = 7.1 Hz), 0.61 (m, 8H, CH2). 13C NMR (d8-THF): d 175.8 (Ph), 174.8 (Ph), 164.4 (C=N), 164.3 (C=N), 151.6, 151.5, 151.3, 150.1, 149.9, 149.7, 147.5, 141.7, 140.4, 138.2, 138.1, 135.9, 131.6, 130.9, 128.1, 127.7, 126.2, 125.6, 125.4, 124.9, 124.5, 124.3, 123.8, 123.7, 123.6, 120.7, 120.5, 120.4, 120.3, 95.7 (C≡C), 91.6 (C≡C), 88.6 (C≡C), 80.5 (C≡C), 55.9, 41.3, 32.5, 30.7, 24.7, 23.5, 14.3 (CH3). Anal. Calc. for C100H90N6O4SZn2·6H2O: C, 70.21; H, 6.01; N, 4.91; S, 1.87. Found: C, 70.36; H, 6.06; N, 4.81; S, 1.93%.
Complex 9a. Complex 9a as an orange solid was similarly prepared by the same procedure as that of complex 7a with 65% yield. Spectroscopic data of 9a:1H NMR (d
6-DMSO): d 9.43 (s, 2H, N=CH), 9.13 (s, 2H, N=CH), 8.43 (d, 2H, PyrH,3J H–H = 3.45 Hz), 8.33 (d, 2H, PyrH,3J H–H= 7.9 Hz), 7.71 (s, 2H, ArH), 7.66 (s, 2H, ArH), 7.58 (d, 2H, thiophene,3J H–H= 5.2 Hz), 7.50 (m, 2H, PyrH), 7.39 (m, 4H, ArH), 7.31 (d, 2H, thiophene,3J
H–H = 3.3 Hz), 7.09 (t, 2H, thiophene, 3J H–H = 3.9 Hz), 7.04 (s, 2H, CH), 6.78 (d, 2H, ArH, 3J H–H = 7.9 Hz), 6.76 (d, 2H, ArH, 3J H–H = 7.2 Hz), 4.01 (t, 4H, OCH2,3JH–H = 5.5 Hz), 1.80–1.74 (m, 4H, CH2), 1.51 (m, 4H, CH3), 1.36–1.11 (m, 32H, CH2), 0.76 (t, 6H, CH3, 3JH–H = 6.9 Hz). 13C NMR (d6-DMSO): d 173.1 (Ph), 172.8 (Ph), 163.8 (C=N), 162.9 (C=N), 149.4, 146.7, 140.1, 140.0, 137.6, 136.6, 134.2, 131.3, 127.7, 127.6, 124.9, 124.1, 123.3, 122.9, 119.6, 119.0, 105.7, 93.7, 79.9 (C≡C), 79.2 (C≡C), 78.9 (C≡C), 78.6 (C≡C), 67.5 (OCH2), 29.6, 29.5, 23.9, 29.1, 28.9, 28.8, 26.4, 25.1, 22.7, 19.3 (CH2), 14.1 (CH3). Anal. Calc. for C84H80N6O6S2Zn2·6H2O: C, 64.16; H, 5.90; N, 5.34; S, 4.08. Found: C, 64.26; H, 5.92; N, 5.19, S, 4.16%.
Complex 9b. Complex 9b as an orange solid was similarly prepared by the same procedure as that of complex 7a with 67% yield. Spectroscopic data of 9b:1H NMR (d
8-THF): d 9.62 (s, 2H, N=CH), 9.02 (s, 2H, N=CH), 8.40 (d, 2H, PyrH,3J
H–H= 3.40 Hz), 8.23 (d, 2H, PyrH,3J
H–H= 8.0 Hz), 7.72–7.69 (m, 4H, ArH), 7.62 (s, 2H, ArH), 7.59 (s, 2H, ArH), 7.49 (s, 2H, ArH), 7.43–7.37 (m, 10H, ArH), 7.32–7.28 (m, 4H, ArH), 7.00 (s, 2H, CH), 6.85 (d, 4H, ArH,3J H–H= 5.04 Hz), 4.05 (t, 4H, OCH2,3JH–H= 6.3 Hz), 2.03 (t, 8H, CH2,3JH–H= 8.0 Hz), 1.95 (m, 4H, CH2), 1.62 (m, 4H, CH2), 1.36–1.06 (m, 32H, CH2), 0.87 (t, 6H, CH3,3JH–H= 7.0 Hz), 0.77 (t, 12H, CH3,3JH–H= 7.2 Hz), 0.62 (m, 8H, CH2).13C NMR (d8-THF): d 173.1 (Ph), 172.8 (Ph), 164.0 (C=N), 163.7 (C=N), 154.5, 151.7, 151.6, 151.5, 147.4, 141.7, 141.7, 141.2, 140.5, 138.6, 138.0, 135.5, 130.9, 128.1, 127.7, 126.2, 125.4, 125.4, 124.5, 123.8, 123.6, 123.5, 120.7, 120.5, 120.4, 120.1, 117.5, 109.4, 108.8, 95.9 (C≡C), 91.1 (C≡C), 88.6 (C≡C), 84.8 (C≡C), 70.2 (OCH2), 55.9, 49.9, 41.3, 32.9, 32.5, 30.8, 30.7, 30.6, 30.5, 30.3, 27.2, 25.8, 25.3, 23.5, 23.4, 14.5 (CH2), 14.3 (CH3). Anal. Calc. for C126H140N6O6Zn2·6H2O: C, 72.99; H, 7.39; N, 4.05. Found: C, 73.11; H, 7.26; N, 4.11%.
Complex 9c. Complex 9c as a dark red powder was similarly prepared by the same procedure as that of complex 7a with 63% yield. Spectroscopic data of 9c: 1H NMR (d
8-THF): d 9.60 (s, 2H, N=CH), 8.95 (s, 2H, N=CH), 8.37 (d, 2H, PyrH,3J H–H = 4.4 Hz), 8.20 (d, 2H, PyrH,3J H–H= 8.9 Hz), 7.60 (s, 2H, ArH), 7.38–7.33 (m, 6H, ArH), 7.20 (d, 2H, PyrH,3J H–H= 8.7 Hz), 6.99 (s, 2H, ArH), 6.80 (d, 2H, ArH,3J H–H= 8.9 Hz), 6.74 (d, 2H, ArH, 3J H–H= 8.7 Hz), 4.05 (t, 4H, OCH2,3JH–H= 5.8 Hz), 2.37 (t, 4H, C≡CCH2,3JH–H= 7.0 Hz), 1.88–1.26 (m, 72H, CH2), 0.89–0.85
(m, 12H, CH3).13C NMR (d5-Pyridine): d 175.5 (Ph), 174.3 (Ph), 164.6 (C=N), 164.4 (C=N), 154.5, 151.0, 150.6, 147.5, 141.69, 140.5, 139.0, 138.9, 135.4, 125.8, 125.4, 125.1, 123.5, 117.8, 115.3, 109.8, 109.2, 96.9 (C≡C), 88.3 (C≡C), 85.6 (C≡C), 82.0 (C≡C), 70.2 (OCH2), 32.5 (C≡CCH2), 32.5, 30.8, 30.7, 30.6, 30.5, 30.4, 30.3, 30.2, 30.0, 29.9, 29.7, 27.2, 27.0, 25.8, 25.3, 23.5, 23.4, 20.2, 14.5 (CH2), 14.3 (CH3). FAB-MS: m/z 1579.9 (M+). Anal. Calc. for C96H116N6O6Zn2·6H2O: C, 68.27; H, 7.64; N, 4.98. Found: C, 68.45; H, 7.53; N, 4.89%.
Complex 10a. Complex 10a as an orange powder was similarly prepared by the same procedure as that of complex 7a with 60% yield. Spectroscopic data of 10a:1H NMR (d
6-DMSO): d 9.40 (s, 2H, N=CH), 9.11 (s, 2H, N=CH), 8.40 (d, 2H, PyrH,3J H–H = 1.2 Hz), 8.29 (d, 2H, PyrH,3J H–H= 7.2 Hz), 7.69 (d, 2H, ArH, 4J H–H= 2.3 Hz), 7.58 (d, 2H, ArH,4JH–H= 4.1 Hz), 7.43 (dd, 2H, thiophene,3J H–H= 4.9 Hz,4JH–H= 3.5 Hz), 7.37 (dd, 2H, ArH, 3J H–H= 6.5 Hz,4JH–H = 2.4 Hz), 7.31–7.30 (m, 4H, ArH), 7.22 (d, 2H, ArH,3J H–H = 8.8 Hz), 7.08 (dd, 2H, thiophene,3JH–H= 3.7 Hz), 6.74 (d, 2H, ArH,3J H–H= 8.9 Hz), 6.70 (d, 2H, ArH, 3J H–H= 8.7 H), 3.70 (s, 2H, CH).13C NMR (d6-DMSO): d 172.7 (Ph), 172.3 (Ph), 163.6 (C=N), 162.9 (C=N), 149.8, 146.6, 140.0, 136.6, 136.5, 135.6, 133.9, 131.2, 127.7, 127.6, 126.5, 124.6, 124.2, 124.0, 123.6, 122.9, 122.7, 119.6, 118.2, 105.6, 93.8 (C≡C), 79.9 (C≡C), 41.2 (CH2). Anal. Calc. for C51H30N6O4S2Zn2·6H2O: C, 56.00; H, 3.87; N, 7.68; S, 5.86. Found: C, 56.18; H, 3.82; N, 7.57; S, 5.98%.
Complex 10c. Complex 10c as dark red solid was similarly prepared by the same procedure as that of complex 7a with 63% yield. Spectroscopic data of 10c:1H NMR (d
8-THF): d 9.53 (s, 2H, N=CH), 8.93 (s, 2H, N=CH), 8.33 (s, 2H, PyrH), 8.16 (s, 2H, PyrH), 7.42 (s, 2H, ArH), 7.35–7.14 (m, 8H, ArH), 6.72 (d, 4H, ArH,3J H–H= 9.3 Hz), 3.84 (s, 2H, CH2).13C NMR (d5-Pyridine): d 174.8 (Ph), 174.3 (Ph), 165.0 (C=N), 164.2 (C=N), 151.5, 151.1, 147.4, 140.4, 138.7, 138.1, 136.9, 127.6, 125.5, 125.5, 125.4, 123.2, 120.5, 119.7, 109.7, 88.2 (C≡C), 82.1 (C≡C), 40.5 (CH2), 32.5, 30.3, 30.0, 29.9, 29.9, 29.6, 23.4, 20.2 (CH2), 14.7 (CH3). FAB-MS: m/z 1102.5 (M+). Anal. Calc. for C
63H66N6O4Zn2·6H2O: C, 62.53; H, 6.50; N, 6.94. Found: C, 62.71; H, 6.40; N, 6.99%. Synthesis of dinuclear Zn(II)-salophen complexes of type 2
Complex 11a. Under a nitrogen atmosphere, the compound 5a (60 mg, 0.26 mmol) was first treated with Zn(OAc)2·2H2O (144 mg, 0.66 mmol) and the mixture was stirred in THF– MeOH (30 mL/10 mL) for 30 min at 60◦C. Then the resulting solution of 1,2,4,5-tetraaminobenzene tetrahydrochloride (19 mg, 0.067 mmol) in MeOH (10 mL) was added slowly, and the mixture was stirred for 2 day at 60◦C. Then the solvent of the mixture was removed by vacuum and residue was washed with methanol, hexanes and ether. The precipitate was collected by filtration. The red powder was identified as compound 11a in 65% yield (47 mg). Spectroscopic data of 11a:1H NMR (d
6-DMSO): d 9.13 (s, 4H, N=CH), 8.29 (s, 2H, ArH), 7.72 (d, 4H, ArH,4J H–H = 2.2 Hz), 7.57 (d, 4H, thiophene,3J H–H = 4.1 Hz), 7.40 (dd, 4H, ArH,3J H–H = 8.9 Hz,4JH–H= 2.2 Hz), 7.32 (dd, 4H, thiophene, 3J H–H= 2.5 Hz,4JH–H= 1.1 Hz), 7.09 (dd, 4H, thiophene,3JH–H= 1.5 Hz,3J H–H= 3.6 Hz), 6.76 (d, 4H, ArH,3JH–H= 8.9 Hz).13C NMR (d6-DMSO): d 162.3 (Ph), 152.5 (C=N), 139.9, 138.6, 131.3, 127.7, 127.6, 124.1, 123.9, 122.9, 119.6, 112.1, 93.6, 74.0 (C≡C), 67.6 (C≡C). MS (MALDI-TOF-MS): m/z 1107.0 (M+). Anal. Calc. for C58H30N4O4S4Zn2·6H2O: C, 57.38; H, 3.49; N, 4.62; S, 10.56. Found: C, 57.53; H, 3.48; N, 4.70; S, 10.68%.
Complex 11b. Complex 11b as a red powder was similarly prepared by the synthetic procedure as that of complex 11a with 65% yield. Spectroscopic data of 11b:1H NMR (d
8-THF): d 9.18 (s, 4H, N=CH), 8.35 (s, 2H, ArH), 7.70 (d, 8H, ArH,3J H–H = 7.2 Hz), 7.65 (s, 4H, ArH), 7.51 (s, 4H, ArH), 7.45–7.43 (m, 8H, ArH), 7.37 (d, 4H, ArH,3J H–H= 5.5 Hz), 7.29 (m, 8H, ArH), 6.90 (d, 4H, ArH,3J H–H= 8.8 Hz), 2.04 (t, 16H, CH2,3JH–H= 8.1 Hz), 1.11–1.06 (m, 48H, CH2), 0.74 (t, 24H, CH3,3JH–H= 7.1 Hz), 0.10 (m, 16H, CH2).13C NMR (d8-THF): d 174.8 (Ph), 162.8 (C=N), 151.8, 151.7, 141.8, 141.7, 140.5, 140.2, 138.2, 131.0, 128.3, 127.9, 126.3, 125.5, 123.9, 123.7, 120.8, 120.8, 120.6, 109.1, 104.4, 91.2 (C≡C), 88.8 (C≡C), 56.0, 41.4, 41.4, 32.6, 30.8, 24.8, 23.6 (CH2), 14.5 (CH3). Anal. Calc. for C142H150N4O4Zn2·6H2O: C, 76.98; H, 7.37; N, 2.53. Found: C, 77.15; H, 7.46; N, 2.66%.
Complex 11d. Complex 11d as a red powder was similarly prepared by the synthetic procedure as that of complex 11a with 70% yield. Spectroscopic data of 11d:1H NMR (d
6-DMSO): d 9.11 (s, 4H, N=CH), 8.31 (s, 2H, ArH), 7.65 (d, 4H, ArH,4J H–H= 2.2 Hz), 7.34 (dd, 4H, ArH,3J H–H= 6.3 Hz,4JH–H= 2.3 Hz), 6.71 (d, 4H, ArH,3J H–H= 8.8 Hz), 3.91 (s, 4H, C≡CH).13C NMR (d6 -DMSO): d 172.5 (Ph), 162.4 (C=N), 140.2, 138.6, 136.5, 123.9, 119.3, 109.8, 105.8, 84.0 (C≡C), 77.6 (C≡C). MS (FAB): m/z 777.2 (M+). Anal. Calc. for C
42H22N4O4Zn2·6H2O: C, 56.97; H, 3.87; N, 6.33. Found: C, 57.16; H, 3.75; N, 6.24%.
Complex 11e. Complex 11e as a black powder was similarly prepared by the synthetic procedure as that of complex 11a with 60% yield. Spectroscopic data of 11e:1H NMR (d
8-THF): d 9.12 (s, 4H, N=CH), 8.28 (s, 2H, ArH), 7.54 (br, 4H, ArH), 7.32 (d, 4H, ArH,3J H–H= 8.8 Hz), 7.26 (d, 8H, ArH,3JH–H= 8.6 Hz), 6.82 (br, 4H, ArH), 6.60 (d, 8H, ArH,3J H–H= 8.6 Hz), 3.28 (m, 16H, CH2), 1.59 (m, 16H, CH2), 1.35–1.30 (m, 144H, CH2), 0.91–0.87 (m, 24H, CH3). Anal. Calc. for C162H234N8O4Zn2·6H2O: C, 74.94; H, 9.55; N, 4.32. Found: C, 75.12; H, 9.65; N, 4.42%.
Complex 12. Complex 12 as a brown powder was similarly prepared by the synthetic procedure as that of complex 11a with 98% yield. Spectroscopic data of 12: 1H NMR (d
6-DMSO): d 9.18 (s, 4H, N=CH), 8.36 (s, 2H, ArH), 7.46 (d, 4H, ArH, 3J H–H= 7.8 Hz), 7.28 (t, 4H, ArH,3JH–H= 7.6 Hz), 6.73 (d, 4H, ArH,3J H–H= 8.6 Hz), 6.57 (t, 4H, ArH,3JH–H= 7.3 Hz).13C NMR (d6-DMSO): d 172.5 (Ph), 162.8 (C=N), 138.6, 136.1, 134.5, 123.3, 119.5, 113.1 (Ph). Anal. Calc. for C34H22N4O4Zn2·6H2O: C, 51.73; H, 4.34; N, 7.10. Found: C, 51.81; H, 4.38; N, 7.15%.
Complex 13. Complex 13 as a brown powder was similarly prepared by the synthetic procedure as that of complex 11a with 60% yield. Spectroscopic data of 13: 1H NMR (d
6-acetone): d 9.21 (s, 4H, NCH), 8.46 (s, 2H, ArH), 7.45 (d, 4H, ArH,4J H–H= 2.6 Hz), 7.17 (d, 4H, ArH,4J H–H= 2.6 Hz), 1.32 (s, 36H, CH3).13C NMR (CDCl3): d 172.2 (Ph), 163.2 (C=N), 142.4, 139.6, 134.9, 130.0, 129.3, 119.4, 103.3, 36.2, 34.4, 31.8 (CH3), 31.5 (CH3). MS
(FAB): m/z 1130.19 (M+). Anal. Calc. for C
66H86N4O4Zn2·6H2O: C, 64.02; H, 7.98; N, 4.52. Found: C, 64.17; H, 8.11; N, 4.45%.
1,3,5-Tris[(3-formyl]-4-hydroxyphenyl)ethynyl]benzene (14). A mixture of 1,3,triethynylbenzene (300 mg, 2.00 mmol), 5-bromosalicylaldehyde (1325 mg, 6.59 mmol), Pd(PPh3)2Cl2 (70 mg, 0.010 mmol) and CuI (11 mg, 0.058 mmol) dissolved in 40 mL of Et3N was stirred at 80◦C for 24 hr under nitrogen. After cooling to room temperature, the reaction mixture was poured into a 10% aqueous NH4Cl (50 mL) solution and the product is extracted with CH2Cl2 (2× 60 mL). The combined organic fraction was washed with water, dried over MgSO4. The crude product was purified by flash chromatography using a silica gel packed column by EA/hexanes (2:1) to yield 14 as yellow powder (25 mg, 2.5%): Spectroscopic data of 14:1H NMR (CDCl
3) d 11.14 (s, 3H, OH), 9.89 (s, 3H, CH=O), 7.74 (s, 3H, ArH), 7.64 (d, 3H, ArH,3J
H–H= 8.5 Hz), 7.61 (s, 3H, ArH), 7.00 (d, 3H, ArH,3JH–H= 8.5 Hz).13C NMR (CDCl
3): d 192.4 (CHO), 163.0, 139.7, 135.7, 134.7, 134.5, 125.2, 119.0, 114.5, 91.1 (C≡C), 87.9 (C≡C). FAB-MS: m/z 510.5 (M+). Anal. Calc. for C
33H18O6: C, 77.64; H, 3.50. Found: C, 77.71; H, 3.56%.
Synthesis of trinuclear Zn(II)-salophen complexes
Complex 15a. Compound 14 (20 mg, 0.039 mmol) and Zn(OAc)2·2H2O (42 mg, 0.19 mmol) dissolved in pyridine–MeOH (30 mL–5 mL) was stirred at 60◦C for 30 min under nitrogen. Then to the resulting solution was slowly added 6a (38 mg, 0.12 mmol) in THF/pyridine (5 mL/10 mL), and the mixture was heated at 60◦C for 2 days. After cooling to room temperature, the solvent was removed under vacuum and residue was washed with methanol, hexanes and ether. The product was collected by filtration to yield 15a as an orange powder (35 mg, 55%). Spectroscopic data of 15a:1H NMR (d
6-DMSO): d 9.45 (s, 3H, N=CH), 9.13 (s, 3H, N=CH), 8.45 (s, 3H, PyrH), 8.33 (s, 3H, PyrH), 7.77 (s, 3H, ArH), 7.71 (s, 3H, ArH), 7.59–7.31 (m, 18H, ArH), 7.09 (s, 3H, thiophene), 6.76 (d, 6H, ArH,3J H–H = 8.2 Hz).13C NMR (d6 -DMSO): d 173.7 (Ph), 172.7 (Ph), 163.8 (C=N), 162.8 (C=N), 149.5, 149.3, 146.7, 141.1, 140.1, 137.5, 136.6, 134.3, 131.3, 127.7, 127.6, 124.9, 124.4, 124.2, 123.8, 123.4, 122.9, 119.6, 119.1, 106.0, 105.7, 93.7 (C≡C), 91.8 (C≡C), 85.5 (C≡C), 79.9 (C≡C). Anal. Calc. for C87H45N9O6S3Zn3·9H2O: C, 59.14; H, 3.59; N, 7.13; S, 5.44. Found: C, 59.27; H, 3.66; N, 7.25; S, 5.57%.
Complex 15b. Complex 15b as an orange powder was similarly prepared from 6a by the same synthetic procedure as that for complex 15a with 50% yield. Spectroscopic data of 15a:1H NMR (d8-THF): d 9.64 (s, 3H, N=CH), 9.02 (s, 3H, N=CH), 8.44 (d, 3H, PyrH,3J H–H= 3.5 Hz), 8.22 (d, 3H, PyrH,3JH–H= 6.3 Hz), 7.72– 7.28 (m, 39H, ArH), 6.85 (d, 6H, ArH,3J H–H= 8.5 Hz), 2.03 (t, 12H, CH2,3JH–H= 8.1 Hz), 1.15–1.05 (m, 36H, CH2), 0.77 (t, 18H, CH3,3JH–H= 7.2 Hz), 0.62 (m, 12H, CH2).13C NMR (d8-THF): d 173.7 (Ph), 172.8 (Ph) 162.7 (C=N), 162.2 (C=N), 150.1, 149.9, 149.6, 145.9, 140.1, 138.9, 137.1, 136.4, 134.0, 131.3, 129.5, 129.4, 126.6, 126.2, 124.6, 124.3, 123.9, 123.7, 123.1, 122.3, 122.1, 119.2, 119.0, 118.9, 118.9, 118.7, 115.4, 107.3, 90.5 (C≡C), 89.6 (C≡C), 87.1 (C≡C), 82.2 (C≡C). Anal. Calc. for C150H135N9O6Zn3·9H2O: C, 71.55; H, 6.12; N, 5.00. Found: C, 71.71; H, 6.20; N, 5.18%.
Acknowledgements
Financial support provided by the National Science Council of Taiwan is grateful acknowledged. Funding for the instrumentation facility of the department of chemistry of NTU has also been provided in part by the National Science Council of Taiwan. We also thank Dr I-Jui Hsu for helpful comments on the quantum mechanical calculation and Mr Meng-Ju Yang for the lifetime study and measurement.
References
1 W. H. Leung, E. Y. Y. Chan, E. K. F. Chow, I. D. Williams and S. M. Peng, J. Chem. Soc., Dalton Trans., 1996, 1229–1236.
2 D. A. Atwood and M. J. Harvey, Chem. Rev., 2001, 101, 37–52. 3 G. A. Morris, H. Zhou, C. L. Stern and S. T. Nguyen, Inorg. Chem.,
2001, 40, 3222–3227.
4 M. Bandini, P. G. Cozzi and A. Umani-Ronchi, Chem. Commun., 2002, 919–927, and references therein.
5 L. Canali and D. C. Sherrington, Chem. Soc. Rev., 1999, 28, 85–93. 6 N. Y. Ito and T. Katsuki, Bull. Chem. Soc. Jpn., 1999, 72, 603–619. 7 T. Katsuki, Curr. Org. Chem., 2001, 5, 663–678.
8 For reviews: (a) N. Str¨ater, W. N. Lipscomb, T. Klabunde and B. Krebs,
Angew. Chem., Int. Ed. Engl., 1996, 35, 2024–2055; (b) D. E. Wilcox, Chem. Rev., 1996, 96, 2435–2458; (c) H. Steinhagen and G. Helmchen, Angew. Chem., Int. Ed. Engl., 1996, 35, 2339–2442.
9 Representative examples: (a) M. J. Young and J. Chin, J. Am. Chem.
Soc., 1995, 117, 10577–10578; (b) P. Molenveld, S. Kapsabelis, J. F. J.
Engbersen and D. N. Reinhoudt, J. Am. Chem. Soc., 1997, 119, 2948– 2949; (c) W. H. Chapman and R. Breslow, J. Am. Chem. Soc., 1995, 117, 5462–5469; (d) J. C. M. Ritter and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 6826–6827; (e) T. Arai, Y. M. A. Yamada, N. Yamamoto, H. Sasai and M. Shibasaki, Chem.–Eur. J., 1996, 2, 1368–1372, and references therein.
10 R. G. Konsler, J. Karl and E. N. Jacobsen, J. Am. Chem. Soc., 1998,
120, 10780–10781.
11 (a) A. C. W. Leung, J. H. Chong, B. O. Patrick and M. J. MacLachlan,
Macromolecules, 2003, 36, 5051–5054; (b) C. Ma, A. Lo, A.
Abdol-maleki and M. J. MacLachlan, Org. Lett., 2004, 6, 3841–3844. 12 (a) S.-S. Sun, C. L. Stern, S. T. Nguyen and J. T. Hupp, J. Am. Chem.
Soc., 2004, 126, 6314–6326; (b) G. A. Morris, H. Zhou, C. L. Stern
and S. T. Nguyen, Inorg. Chem., 2001, 40, 3222–3227; (c) K. E. Splan, A. M. Massari, G. A. Morris, S.-S. Sun, E. Reina, S. T. Nguyen and J. T. Hupp, Eur. J. Inorg. Chem., 2003, 2348–2351.
13 (a) K.-H. Chang, C.-C. Huang, Y.-H. Liu, Y.-H. Hu, P.-T. Chou and Y.-C. Lin, Dalton Trans., 2004, 1731–1738; (b) H.-C. Lin, C.-C. Huang, C.-H. Shi, Y.-H. Liao, C.-C. Chen, Y.-C. Lin and Y.-H. Liu, Dalton
Trans., 2007, 781–791.
14 A. W. Kleij, D. M. Tooke, A. L. Spek and J. N. H. Reek, Eur. J. Inorg.
Chem., 2005, 4626–4634.
15 (a) O. Karthaus, K. Ueda, A. Yamagushi and M. Shimomura,
J. Photochem. Photobiol., A, 1995, 92, 117–120; (b) D. D. Mysyk, I. F.
Perepichka and N. I. Sokolov, J. Chem. Soc., Perkin Trans. 2, 1997, 537–546.
16 (a) I. F. Perepickha, D. D. Mysyk and N. I. Sokolov, in Current Trends
in Polymer Photochemistry, ed. N. S. Allen, M. Edge, I. R. Bellobono
and E. Selli, Ellis Horwood, Chichester, UK, 1995, pp. 318–327; (b) M. Matsui, K. Shibata, H. Muramatsu and H. Nakazumi, J. Mater. Chem., 1996, 6, 1113–118; (c) D. D. Mysyk, I. F. Perepichka, D. F. Perepichka, M. R. Bryce, A. F. Popov, L. M. Goldenberg and A. J. Moore, J. Org.
Chem., 1999, 64, 6937–6950; (d) I. F. Perepichka, A. F. Popov, T. V.
Orekhova, M. R. Bryce, A. M. Andrievskii, A. S. Batsanov, J. A. K. Howard and N. I. Sokolov, J. Org. Chem., 2000, 65, 3053–3063. 17 (a) K.-T. Wong, Y.-Y. Chien, R.-T. Chen, C.-F. Wang, Y.-T. Lin,
H.-H. Chiang, P.-Y. Hsieh, C.-C. Wu, C.-H.-H. Chou and Y. O. Su, J. Am.
Chem. Soc., 2002, 124, 11576–11577; (b) W.-L. Yu, J. Pei, W. Huang
and A. J. Heeger, Adv. Mater., 2000, 12, 828–831; (c) S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. M ¨ullen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946–953.
18 K. Sonogashira, Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, New York, 1991, vol. 3, pp. 521–549.
19 (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975,
16, 4467–4470; (b) T. Hundertmarl, A. F. Littke, S. L. Buchwald and
G. C. Fu, Org. Lett., 2000, 2, 1729–1731.
20 R. I. Kureshy, N. H. Khan, S. H. R. Abdi, S. T. Patel and R. V. Jasra,
Tetrahedron: Asymmetry, 2001, 12, 433–437.
21 M. Nielsen, A. H. Thomsen, T. R. Jensen, H. J. Jakobsen, J. Skibsted and K. V. Gothelf, Eur. J. Org. Chem., 2005, 342–347.
22 (a) C.-C. Kwok, S.-C. Yu, I. H. T. Sham and C.-M. Che, Chem.
Commun., 2004, 2758–2579; (b) C.-M. Che, S.-C. Chan, H.-F. Xiang,
M. C. W. Chan, Y. Liu and Y. Wang, Chem. Commun., 2004, 1484–1485. 23 (a) A. N. Fletcher, Photochem. Photobiol., 1969, 9, 439–444; (b) K.
Nakamaru, Bull. Chem. Soc. Jpn., 1982, 55, 2697–2705.
24 E. Weber, M. Hecker, E. Koepp, W. Orlia, M. Czugler and I. Cs ¨oregh,