0022-538X/04/$08.00⫹0 DOI: 10.1128/JVI.78.22.12717–12721.2004 Copyright © 2004, American Society for Microbiology. All Rights Reserved.
Study of Sequence Variation of Dengue Type 3 Virus in Naturally
Infected Mosquitoes and Human Hosts: Implications for
Transmission and Evolution
Su-Ru Lin,
1Szu-Chia Hsieh,
1Yi-Yuan Yueh,
2Ting-Hsiang Lin,
2Day-Yu Chao,
3Wei-June Chen,
4Chwan-Chuen King,
3and Wei-Kung Wang
1,5*
Institute of Microbiology,1Department of Internal Medicine,5College of Medicine, and Institute of Epidemiology,3
College of Public Health, National Taiwan University, and Division of Laboratory Research and Development, Center for Disease Control, Department of Health,2Taipei, and Department of Public Health and
Parasitology, Chang-Guan University, Tao-Yuan,4Taiwan
Received 1 May 2004/Accepted 1 July 2004
Dengue virus is an arbovirus that replicates alternately in the mosquito vector and human host. We investigated sequences of dengue type 3 virus in naturally infected Aedes aegypti mosquitoes and in eight patients from the same outbreak and reported that the extent of sequence variation seen with the mosquitoes was generally lower than that seen with the patients (mean diversity, 0.21 versus 0.38% and 0.09 versus 0.23% for the envelope [E] and capsid [C] genes, respectively). This was further verified with five experimentally infected mosquitoes (mean diversity, 0.09 and 0.10% for the E and C genes, respectively). Examination of the quasispecies structures of the E sequences of the mosquitoes and of the patients revealed that the sequences of the major variants were the same, suggesting that the major variant was transmitted. These findings support our hypothesis that mosquitoes contribute to the evolutionary conservation of dengue virus by maintaining a more homogenous viral population and a dominant variant during transmission.
Dengue viruses are members of the genus Flavivirus of the family Flaviviridae. There are four serotypes of dengue viruses, DEN-1, DEN-2, DEN-3, and DEN-4. Over the past 20 years, epidemics caused by the four dengue viruses have emerged as one of the major public health problems in tropical and sub-tropical regions (4, 6, 18, 30). Dengue virus contains a positive-sense single-stranded RNA genome. Flanked by two nontrans-lated regions, there are three structural genes, the capsid (C),
precursor membrane (PrM), and envelope (E), at the 5⬘
one-fourth and seven nonstructural genes at the 3⬘ three-fourths (4, 14).
Dengue virus is transmitted to human by the bite of an infected mosquito. Aedes aegypti is the principle vector in-volved in the urban transmission cycle (4, 10, 22). After a female mosquito ingests a blood meal from a patient infected with dengue virus, viral replication is initially found in the posterior midgut of the mosquito and then in the proventric-ulus and in other organ systems. Dengue virus appears in the salivary gland after an extrinsic incubation period of 8 to 12 days, when the mosquito becomes capable of transmitting it to another human host (10, 20, 22). Following an incubation period of 3 to 14 days, the infected individuals may be asymp-tomatic or present a mild and self-limited illness, dengue fever (DF), or a severe and potentially life-threatening disease, den-gue hemorrhage fever-denden-gue shock syndrome (DHF-DSS) (4, 30).
The genetic stability of arboviruses that replicate alternately in the vertebrate and arthropod hosts has been well
docu-mented previously (13, 27, 28). In the case of DEN-3 virus, it has been reported that the amino acid similarity of the PrM/E proteins was more than 95% over a 36-year period (13). Pre-viously, it was reported that dengue virus, like other RNA viruses, is present as a population of closely related sequences, the quasispecies, in the human host (2, 3, 8, 16, 24, 25). The extent of sequence variation of dengue virus in the mosquito vector and how the quasispecies structure changes during transmission between human and mosquitoes remain un-known. We hypothesized that mosquitoes may contribute to the genetic stability of dengue virus by maintaining a more homogenous viral population and/or selecting a dominant vari-ant. In this study, we investigated dengue virus sequences de-rived from field-captured A. aegypti mosquitoes in comparison with those obtained from eight patients during a DEN-3 out-break in southern Taiwan in 1998 (9). We analyzed sequence variation of both the E and C genes as well as the quasispecies structures in the mosquitoes and patients. Moreover, sequence variation in five experimentally infected mosquitoes was also examined.
During the outbreak, attempts were made to capture mos-quitoes in pools from different districts for virus isolation. Each
pool of mosquitoes was macerated in 100l of minimal
essen-tial medium (Invitrogen, San Diego, Calif.), triturated on ice, centrifuged, and filtered through a 0.22-m-pore-size filter, and an aliquot of the filtrate was inoculated into C6/36 cells and monitored by an indirect immunofluorescent assay for 7 to 10 days. One pool, which was derived from 22 female A. aegypti mosquitoes captured at the central district of Tainan City, the major area of the outbreak, yielded DEN-3 virus. To study dengue viral sequences in the naturally infected mosquito, the remaining filtrate was subjected to viral RNA isolation and
* Corresponding author. Mailing address: Institute of Microbiology, College of Medicine, National Taiwan University, No. 1 Sec. 1 Jen-Ai Rd., Taipei, Taiwan. Phone: 2312-3456, ext. 8286. Fax: 886-2-2391-5293. E-mail: [email protected].
reverse transcription-PCR (RT-PCR) to amplify a 430-nucle-otide region covering the domain III of the E gene as described previously (17, 21, 25) (Fig. 1). Each PCR product was cloned to the TA cloning vector, PCRII-TOPO (Invitrogen), and 21 clones derived from two separate PCRs were completely se-quenced, aligned, and analyzed as described previously (11, 25).
There were 17 nucleotide substitutions, of which 9 were silent and 8 were nonsilent. The mean diversity, which is the number of substitutions divided by the total number of nucle-otides sequenced, was 0.21% (Table 1) (25, 32). The deduced amino acid sequences of the 21 clones were also aligned. Within the 131-amino-acid region analyzed, there were 13 clones with identical sequences and eight amino acid substitu-tions in other 8 clones (Fig. 2A). Pairwise comparison of amino acid sequences of individual clones revealed that the pairwise p-distance ranged from 0 to 1.53% (mean, 0.58%). We next examined another gene, the C gene, through RT-PCR of a 360-nucleotide region covering the entire C gene (Fig. 1) (26). Among the 21 clones examined, there were six nucleotide substitutions out of 6,678 nucleotides sequenced, correspond-ing to a mean diversity of 0.09% (Table 1). Pairwise compar-ison of amino acid sequences revealed a mean p-distance of 0.27% (range, 0 to 2.83%). These findings indicated that
DEN-3 virus is present as quasispecies in the mosquitoes, al-beit as a relative homogenous population.
To compare the extent of sequence variation in mosquitoes with that in human hosts, dengue viral RNA derived from acute plasma samples of eight confirmed DEN-3 patients (in-cluding four DF and four DHF cases according to the WHO definition) (7, 12, 30), who live in the same district or in nearby districts, was subjected to RT-PCR and clonal sequencing. For the E gene, ten to fifteen clones from each patient were se-quenced and analyzed. The mean diversity ranged from 0.15 to 0.59%, and the overall mean diversity was 0.38%, which was higher than that determined for the mosquitoes (Table 1). Pairwise comparison of amino acid sequences revealed a more homogenous population in the mosquitoes than in the patients (Fig. 3A and C). For the C gene, ten clones from each patient were analyzed as reported previously (26). The mean diversity ranged from 0.13 to 0.41%, and the overall mean diversity was 0.23%, which, in agreement with the results observed for the E gene, was higher than that in the mosquitoes (Table 1). Taken together, these findings indicated that the extent of sequence variation in the naturally infected mosquitoes was generally lower than that in dengue patients.
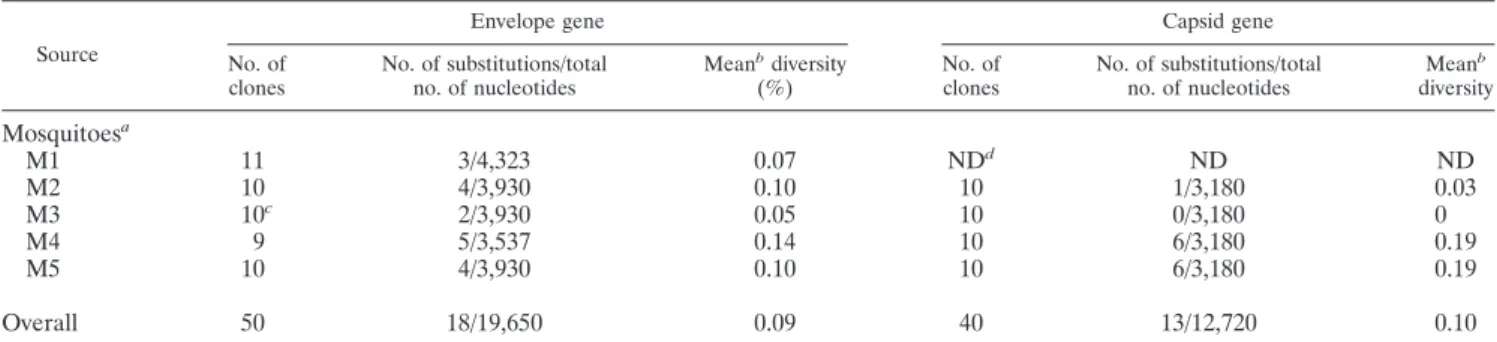
Since homogenates of 22 field-captured mosquitoes were subjected to RNA extraction and sequence analysis, the mean diversity observed may represent the average of the results for two or more mosquitoes rather than the results for a single infected mosquito. To examine the extent of sequence varia-tion in a single mosquito, we infected A. aegypti with 0.17l
(106PFU/ml) of a DEN-3 isolate through intrathoracic
inoc-ulation, identified each infected mosquito by a direct immu-nofluorescent antibody test (1, 5, 23), and used the same pro-tocols for sequence analysis. The DEN-3 virus was isolated from plasma of patient ID23 by use of C6/36 cells, and the titer was determined by a plaque assay of BHK-21 cells (1). The results for five mosquitoes are summarized in Table 2. The overall mean diversity of the E gene, 0.09% (range, 0.05 to
FIG. 1. Schematic diagram of the dengue virus genome and the C and E regions examined in this study. The relative positions of the PCR primers are shown. The sequences of the primers were as de-scribed previously (25, 26). NTR, nontranslated region; C, capsid; PrM, precursor membrane; E, envelope; NS, nonstructural.
TABLE 1. Nucleotide sequence variation of envelope and capsid genes from naturally infected mosquitoes and dengue patients
Source
Envelope gene Capsid gene
No. of clones No. of substitutions/total no. of nucleotides Meancdiversity (%) No. of clones No. of substitutions/total no. of nucleotides Meancdiversity (%) Mosquitoesa 21e 17/8,253 0.21 21 6/6,678 0.09 Patientsb ID7 10 6/3,930 0.15 10 6/3,180 0.19 ID8 11 17/4,323 0.39 10 6/3,180 0.19 ID9 11e 20/4,323 0.46 10 8/3,180 0.25 ID15 10 17/3,930 0.43 10d,f 13/3,180 0.41 ID18 15d 21/5,895 0.36 10 6/3,180 0.19 ID20 11d 15/4,323 0.35 10 4/3,180 0.13 ID23 10 11/3,930 0.28 10 8/3,180 0.25 ID24 10 23/3,930 0.59 10 7/3,180 0.22 Overall 88 130/34,584 0.38 80 58/25,440 0.23
aThe dengue virus sequences in mosquitoes were derived from homogenates of a pool of 22 female A. aegypti mosquitoes.
bThe patients included four DF (ID7, ID8, ID9, and ID15) and four DHF (ID18, ID20, ID23, and ID24) cases according to the World Health Organization definition
(30).
cThe mean diversity is the number of substitutions divided by the total number of nucleotides sequenced. dOne clone contained a stop codon.
eOne clone contained a single-nucleotide deletion.
fTwo clones contained single-nucleotide deletions (one each).
0.14%), was slightly lower than that seen with the patients (Table 1 and Table 2). Pairwise comparison of amino acid sequences revealed a more homogenous population in each mosquito than in the patients (Fig. 3B and C). Similarly, the overall mean diversity of the C gene, 0.10% (range, 0 to 0.19%), was slightly lower than that seen with the patients (Table 1 and Table 2). Of note was that the sequence variation observed could not be totally attributed to in vitro artifacts introduced by RT or Taq polymerase. This is because the error frequencies, ranging from 0.02 to 0.05%, introduced by the use of Taq polymerase for 60 cycles of PCR and by the use of RT are lower than the mean diversity observed with most of our samples (25, 26). For two mosquitoes (M2 and M3), the mean diversities for the C gene were 0 and 0.03% (Table 2), sug-gesting a very homogenous population.
A closer examination of the quasispecies structures of the E sequences of the field-captured mosquitoes and the patients revealed that the consensus sequences of 21 clones from the mosquitoes and of multiple clones from each patient were the same (Fig. 2 and data not shown). Moreover, the consensus sequence corresponds to the sequences of the major variants of the quasispecies (Fig. 2), suggesting that the major variant is transmitted between mosquitoes and humans. To further verify this, RNAs derived from the mosquitoes and four of the eight patients were subjected to RT-PCR and direct sequencing, the sequence derived from which is believed to represent the dom-inant sequence. The results revealed that the full-length E sequences of the mosquitoes and of the four patients were the same (data not shown), supporting the notion that dengue viruses with an E sequence of the major variant were trans-mitted.
To our knowledge, this was the first study that directly ex-amined dengue viral sequences from naturally infected mos-quitoes. Based on the analysis of both the E and C genes, we
FIG. 2. Alignment of the deduced amino acid sequences of E proteins of multiple clones from the field-captured mosquitoes (A) and patient ID18 (B). The positions of amino acid residues and the corresponding domains and regions within the E protein as determined on the basis of the TBE virus model (21) are shown on top. A consensus sequence (CON) was generated for each sample. Dashes indicate sequence identity, an asterisk indicates an in-frame stop codon, and an underline indicates a deletion at that position. Names and numbers of individual clones are shown at the left, with the letters A and B indicating two separate PCRs. For simplicity, only one of the clones with identical sequences is shown.
FIG. 3. Amino acid sequence similarity of the E proteins from the field-captured mosquitoes (A), experimentally infected mosquitoes (B), and dengue patients (C). Amino acid sequences of multiple clones from the mosquitoes and patients were subjected to pairwise compar-ison using the program MEGA (19), and the percentages of total comparisons with a given similarity are presented as a histogram for the field-captured (MOS) and experimentally infected (M1 to M5) mosquitoes and for each patient (shown by patient ID number).
reported that the extent of sequence variation was generally lower in the naturally infected mosquitoes than in the human hosts. This was further verified with each of the five experi-mentally infected mosquitoes. A low extent of sequence vari-ation of the nonstructural genes is expected to be seen in the mosquitoes, since a previous study had shown a similar extent of intrahost sequence variation of the structural gene (C) and the nonstructural gene (NS2B) (26). Dengue virus is known to cause a productive and cytolytic infection in the mammalian host and a persistent and noncytolytic infection in the arthro-pod vector (4, 14). A recent study of eastern equine encepha-litis virus, which is an arbovirus, showed that persistent infec-tion in the mosquito cell line was associated with a lower rate of genetic change (28). It is conceivable that persistent infec-tion of dengue virus in mosquitoes would contribute to a lower extent of sequence variation in the mosquitoes than in the patients observed in our study.
This is also the first report showing that the major variant of the dengue viral quasispecies is transmitted between the mos-quito and its human hosts. This is different from what had been reported in the transmission study of human immunodefi-ciency virus type 1; in both reports, a minor variant was dem-onstrated to be transmitted and a mechanism of selective transmission or selective amplification was proposed (29, 32). Although the infection rate of mosquitoes varies with the titers of the viremia in dengue patients, it has been reported that an amount equal to as much as 104mosquito 50% infectious doses
of virus was transmitted (10, 22, 23). Such an amount would allow the dominant variant to be transmitted. Skin Langerhans cells were recently shown to be the initial targets of dengue virus infection after a bite by a mosquito (31). Our findings suggest that selective transmission or amplification of a minor variant is unlikely to have occurred before dengue virus enters the circulation.
A study of the structural proteins of several serological sub-groups of flaviviruses has shown a high degree of sequence conservation within each subgroup that includes dengue vi-ruses (15). Continuous adaptation of dengue virus between mosquitoes and humans has been suggested to account for the sequence conservation (13). By examining the quasispecies structures, we demonstrated in this study that only the major variant was transmitted between the mosquito and patients.
Moreover, after transmission to the mosquito, which thereafter has a life-long infection, dengue virus tends to maintain a more homogenous population. These facts would contribute to the evolutionary conservation of dengue virus.
It has also been suggested that the genetic stringency im-posed on dengue virus and other arthropod-borne viruses that replicate in both vertebrate and arthropod hosts would con-tribute to the sequence conservation (13, 27, 28). We have used the program MEGA (19) to calculate the differences in the number of synonymous nucleotide substitutions per site (dS) and the differences in the number of nonsynonymous nucleo-tide substitutions per site (dN). The ratio of dS to dN (dS/dN) was thus determined. In the mosquitoes, the mean dS/dN of the E gene was 3.92, a result which was higher than 1, suggest-ing the presence of negative (purifysuggest-ing) selection pressure. Similarly, the mean dS/dN of the E gene was higher than 1 in seven out of the eight dengue patients (range, 1.13 to 4.79), suggesting negative selection in human hosts as well. The neg-ative selection probably results from certain structural or func-tional constraints on the virus, though the nature of the con-straints remains to be determined.
Nucleotide sequence accession numbers. The sequences have been submitted to GenBank; the accession numbers are AY053396, AY053397, AY053399, AY082898, AY082899, AY082901, AY082904, AY082905, AF495893 to AF495896, AF495899, AF495901, AF495904, and AF495905.
We thank Chien-Ming Li at the Chi-Mei Foundation Medical Cen-ter, Shih-Chung Lin at the Kuo General Hospital, and Shih-Ting Ho at the Sin-Lau Christian Hospital for kindly providing clinical samples.
This work was supported by the National Science Council Taiwan (NSC92-2320-B-002-151).
REFERENCES
1. Chen, W. J., H. L. Wei, E. L. Hsu, and E. R. Chen. 1993. Vector competence of Aedes Albopictus and Ae. aegypti (Diptera: Culicidae) to dengue 1 virus on Taiwan: development of the virus in orally and parenterally infected mos-quitoes. J. Med. Entomol. 30:524–530.
2. Domingo, E. J., J. J. Holland, and P. Ahlquist. 1988. RNA genetics, vol. 3. CRC Press Inc., Boca Raton, Fla.
3. Farci, P., A. Shimoda, A. Coiana, G. Diaz, G. Peddis, J. C. Melpolder, A. Strazzera, D. Y. Chien, S. J. Munoz, A. Balestrieri, R. H. Purcell, and H. J. Alter.2000. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 288:339–344.
4. Gubler, D. J. 1998. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 11:480–496.
TABLE 2. Nucleotide sequence variation of envelope and capsid genes from experimentally infected mosquitoes
Source
Envelope gene Capsid gene
No. of clones No. of substitutions/total no. of nucleotides Meanbdiversity (%) No. of clones No. of substitutions/total no. of nucleotides Meanb diversity Mosquitoesa M1 11 3/4,323 0.07 NDd ND ND M2 10 4/3,930 0.10 10 1/3,180 0.03 M3 10c 2/3,930 0.05 10 0/3,180 0 M4 9 5/3,537 0.14 10 6/3,180 0.19 M5 10 4/3,930 0.10 10 6/3,180 0.19 Overall 50 18/19,650 0.09 40 13/12,720 0.10 a
Dengue virus sequences were derived from each A. aegypti mosquito after intrathoracic injection of dengue virus and confirmation by an immunofluorescent antibody test (1, 23).
b
The mean diversity is the number of substitutions divided by the total number of nucleotides sequenced.
c
One clone contained a single-nucleotide deletion.
d
ND, not done.
5. Gubler, D. J., and L. Rosen. 1976. A simple technique for demonstrating transmission of dengue virus by mosquitoes without the use of vertebrate hosts. Am. J. Trop. Med. Hyg. 25:146–150.
6. Halstead, S. B. 1988. Pathogenesis of dengue: challenges to molecular biol-ogy. Science 239:476–481.
7. Harris, E., G. Roberts, L. Smith, J. Selle, L. D. Kramer, S. Valle, E. San-doval, and A. Balmaseda.l998. Typing of dengue viruses in clinical speci-mens and mosquitoes by single-tube multiplex reverse transcriptase PCR. J. Clin. Microbiol. 36:2634–2639.
8. Holland, J. J., J. C. De La Torre, and D. A. Steinhauer. 1992. RNA virus populations as quasispecies. Curr. Top. Microbiol. Immunol. 176:1–20. 9. King, C. C., Y. C. Wu, D. Y. Chao, T. H. Lin, L. Chow, H. T. Wang, C. C. Ku,
C. L. Kao, K. P. Hwang, S. K. Lam, and D. J. Gubler.2000. Major epidemics of dengue in Taiwan in 1991–2000: related to intensive virus activities in Asia. Dengue bulletin 24:1–10. World Health Organization, Geneva, Swit-zerland.
10. Kuno, G. 1997. Factors influencing the transmission of dengue viruses, p. 61–88. In D. J. Gubler and G. Kuno (ed.), Dengue and dengue hemorrhagic fever. CAB International, London, United Kingdom.
11. Kwok, S., and R. Higuchi. 1989. Avoiding false positives with PCR. Nature 339:237–238.
12. Lanciotti, R. S., C. H. Calisher, D. J. Gubler, G.-J. Chang, and A. V. Vorndam.1992. Rapid detection and typing of dengue viruses from clinical samples by using reverse transcriptase-polymerase chain reaction. J. Clin. Microbiol. 30:545–551.
13. Lanciotti, R. S., J. G. Lewis, D. J. Gubler, and D. W. Trent. 1994. Molecular evolution and epidemiology of dengue-3 viruses. J. Gen. Virol. 75:65–75. 14. Lindenbach, B. D., and C. M. Rice. 2001. Flaviviridae: the viruses and their
replication, p. 991–1041. In D. M. Knipe, P. M. Howley, et al. (ed.), Fields virology, 4th ed. Lippincott Raven, Philadelphia, Pa.
15. Mandl, C. W., F. X. Heinz, and C. Kunz. 1988. Sequence of the structural proteins of tick-borne encephalitis virus (western subtype) and comparative analysis with other flaviviruses. Virology 166:197–205.
16. Martell, M., J. I. Esteban, J. Ouer, J. Genesca, A. Weiner, R. Esteban, J. Guardia, and J. Gomez. 1992. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J. Virol. 66:3225–3229.
17. Modis, Y., S. Ogata, D Clements, and S. C. Harrison. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313–319. 18. Monath, T. P. 1994. Dengue, the risk to developed and developing countries.
Proc. Natl. Acad. Sci. USA 91:2395–2400.
19. Nei, M., and T. Gojobori. 1986. Simple methods for estimating the numbers
of synonymous & nonsynonymous nucleotide substitutions. Mol. Bio. Evol. 3:418–426.
20. Reiter, P., M. A. Amador, R. A. Anderson, and G. G. Clark. 1995. Short report: dispersal of Aedes aegypti in an urban area after blood feeding as demonstrated by rubidium-marked eggs. Am. J. Trop. Med. Hyg. 52:177– 179.
21. Rey, F. A., F. X. Heinz, C. Mandl, C. Kunz, and S. C. Harrison. 1995. The envelope glycoprotein from tick-borne encephalitis virus at resolution. Na-ture 375:291–298.
22. Rodhain, F., and L. Rosen. 1997. Mosquito vectors and dengue virus-vector relationships. p. 45–60. In D. J. Gubler and G. Kuno (ed.), Dengue and dengue hemorrhagic fever. CAB International, London, United Kingdom. 23. Rosen, L., and D. J. Gubler. 1974. The use of mosquitoes to detect and
propagate dengue viruses. Am. J. Trop. Med. Hyg. 23:1153–1160. 24. Steinhauer, D. A., and J. J. Holland. 1987. Rapid evolution of RNA viruses.
Annu. Rev. Microbiol. 41:409–433.
25. Wang, W.-K., S.-R. Lin, C.-M. Lee, C.-C. King, and S.-C. Chang. 2002. Dengue type 3 virus in plasma is a population of closely related genomes: quasispecies. J. Virol. 76:4662–4665.
26. Wang, W. K., T. L. Sung, C. N. Lee, T. Y. Lin, and C. C. King. 2002. Sequence diversity of the capsid gene and the nonstructural gene NS2B of dengue-3 virus in vivo. Virology 303:181–191.
27. Weaver, S. C., R. Rico-Hesse, and T. W. Scott. 1992. Genetic diversity and slow rates of evolution in new world alphaviruses. Curr. Top. Microbiol. Immunol. 176:99–117.
28. Weaver, S. C., A. C. Brault, W. Kang, and J. J. Holland. 1999. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 73:4316–4326.
29. Wolinsky, S. M., C. M. Wike, B. T. Korber, C. Hutto, W. P. Parks, L. L. Rosenblum, K. J. Kunstman, M. P. Furtado, and J. L. Munoz.1992. Selec-tive transmission of human immunodeficiency virus type 1 variants from mother to infants. Science 255:1134–1137.
30. World Health Organization. 1997. Dengue hemorrhagic fever, diagnosis, treatment and control, 2nd ed. World Health Organization, Geneva, Swit-zerland.
31. Wu, S.-J. L., G. Grouard-Vogel, W. Sun, J. R. Mascola, E. Brachtel, R. Putvatana, M. K. Louder, L. Filgueira, M. A. Marovich, H. K. Wong, A. Blauvelt, G. S. Murphy, M. L. Robb, B. L. Innis, D. L. Birx, C. G. Hayes, and S. S. Frankel.2000. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 6:816–820.
32. Zhu, T., H. Mo, N. Wang, D. S. Nam, Y. Cao, R. A. Koup, and D. D. Ho. 1993. Genotypic and phenotypic characterization of HIV-1 in patients with pri-mary infection. Science 261:1179–1181.