Asiatic acid and maslinic acid protected heart via glycative and
anti-coagulatory activities in diabetic mice
Yi-chih Hunga,b, Hui-ting Yangc, Mei-chin Yinc,d,*
aGraduate Institute of Clinical Medical Science, China Medical University, Taichung City, Taiwan
bDivision of Endocrinology and Metabolism, Department of Internal Medicine, China Medical University Hospital, Taichung City, Taiwan
cDepartment of Nutrition, China Medical University, Taichung City, Taiwan
dDepartment of Health and Nutrition Biotechnology, Asia University, Taichung City, Taiwan
Running title: cardiac protection of asiatic acid and maslinic acid
*To whom correspondence should be addressed: Dr. Mei-chin Yin, Professor, Department of Nutrition, China Medical University, 91, Hsueh-shih Rd., Taichung City, Taiwan
TEL: 886-4-22053366 ext. 7510, FAX: 886-4-22062891, Email: mcyin@ mail.cmu.edu.tw 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Abstract
Cardiac protective effects of asiatic acid (AA) and maslinic acid (MA) in diabetic mice were examined. These triterpenoids at 0.1 or 0.2% of the diet were supplied to diabetic mice for 12 weeks. AA or MA treatments decreased plasma glucose and HbA1c levels, creatine phosphokinase and lactate dehydrogenase activities in diabetic mice (p<0.05). AA or MA intake increased their deposit in heart; retained cardiac glutathione content; and reduced the production of reactive oxygen species, N-(carboxymethyl)-lysine, pentosidine, methylglyoxal, interleukin-6, tumor necrosis factor-alpha and monocyte chemoattractant protein-1 in heart of diabetic mice (p<0.05). AA or MA intake lowered plasma von Willebrand factor and fibrinogen levels, and factor VII activity (p<0.05); also maintained circulating antithrombin-III and protein C activities (p<0.05). AA or MA treatments down-regulated cardiac expression of NADPH oxidase, aldose reductase, nuclear factor kappa B (NF-B) p65 and p-p38; as well as reserved glyoxalase 1 expression (p<0.05). These two compounds only at 0.2% lowered cardiac expression of NF-B p50, p-ERK1/2 and receptor of advanced glycation endproduct (p<0.05). These findings support that the supplement of these triterpenoids could protect heart under diabetic condition via attenuating glycative injury and coagulatory disorders.
Keywords: Diabetes; Glycation; Coagulation; Asiatic acid; Maslinic acid
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Introduction
Coagulatory disorders often occur in diabetic patients with poor glycemic control (Nwose et al. 2009; Lemkes et al. 2010). The increase of coagulation factors such as factor VII (FVII) and fibrinogen; and/or the decrease of anti-coagulation factors such as protein C and antithrombin-III (AT-III) in circulation of diabetic patients caused hypercoagulability, which facilitated the development of cardio-vascular diseases like diabetic cardiomyopathy because heart is vulnerable to diabetes associated thrombosis (Asakawa et al. 2000; Meerarani et al. 2006). Furthermore, excessive production of reactive oxygen species (ROS) under diabetic condition from the activation of NADPH oxidase acts as signaling element in thrombogenic cycle, and favors the progression of local or systemic thrombosis (Herkert et al. 2004; Delmastro and Piganelli 2011). Thus, hemostatic imbalance warrants more attention in order to avoid the occurrence of diabetic cardiomyopathy.
Aldose reductase (AR) is the rate-limiting enzyme of polyol pathway and converts glucose to fructose (Hu et al. 2014). Excessive glucose and fructose facilitate the formation of advanced glycation endproducts (AGEs) under diabetic condition. N-(carboxymethyl)-lysine (CML) and pentosidine are predominant AGEs contributed to the progression of diabetic cardiomyopathy in diabetic patients (Hirata and Kubo 2004; Hu et al. 2013). On the other hand, glyoxalase 1 (GLO-1), the rate-limiting enzyme of glyoxalase system, detoxifies AGE’s precursors such as methylglyoxal to less toxic products like D-lactate (Thornalley 2003). Thus, any agent with the ability to inhibit AR and/or enhance GLO-1 expression may decrease AGEs production and diminish glycative stress. Furthermore, AGEs upregulate their membrane-anchored receptor (RAGE), and AGE-RAGE interaction activates mitogen-activated protein kinase (MAPK) and nuclear factor kappa-B (NF-B) signaling pathways (Barlovic et al. 2010; 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
Xie et al. 2013), which in turn accelerates the production of inflammatory factors like tumor necrosis factor (TNF)-alpha. Thus, the suppression on RAGE, MAPK and NF-B pathways may delay diabetic progression.
Asiatic acid (AA) and maslinic acid (MA) are pentacyclic triterpenes naturally occurring in many vegetables and fruits such as brown mustard (Brassica juncea), centella (Centella
asiatica L.) and olive (Olea europaea L.) (Hashim et al. 2011; Yin et al. 2012). Castellano et al. (2013) indicated that oleanolic acid related pentacyclic triterpenes like MA provided beneficial effects against diabetes through regulating antioxidant enzymes and transduction pathways. Ramachandran et al. (2014) reported that AA improved glycemic control and lipid metabolism in diabetic rats. Thus, AA and MA are potent anti-diabetic agents. However, it remains unknown whether AA or MA could protect heart against diabetic coagulatory and glycative disorders. Our previous study found that dietary intake of AA and MA increased their bioavailability in heart of mice (Yin et al. 2012). Thus, it is highly possible that the presence of these triterpenes in heart could protect this organ against diabetes related damage.
The major purpose of our present study was to investigate the anti-glycative effects of AA and MA at various doses in heart of diabetic mice. The influence of these compounds upon protein expression of AR, GLO-1, NADPH oxidase, RAGE and MAPK in heart of diabetic mice was evaluated. Furthermore, the impact of these compounds upon coagulatory and anti-coagulatory factors was also examined. These results will enhance our understanding regarding the application of these triterpenes against diabetic cardiomyopathy.
Materials and Methods
Chemicals 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
Asiatic acid (AA, 98%) and maslinic acid (MA, 97.5%) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA).
Animals and diets
Male Balb/cA mice, 3-week old, were obtained from National Laboratory Animal Center (National Science Council, Taipei City, Taiwan). To induce diabetes, mice with body weight at 23.7 1.2 g were treated with a single i.v. dose (50 mg/kg) of streptozotocin dissolved in citrate buffer (pH 4.5) into the tail vein of mice after 12 hr fast. Blood glucose level was monitored on day 10 from the tail vein after 12 hr fast by using a one-touch blood glucose meter (Lifescan Inc., Milpitas, CA, USA). Mice with fasting blood glucose level 200 mg/dl were used for this study. Animal experiments were performed in accordance with protocols approved by the Animal Care Research Committee of China Medical University (102-36-N).
Experimental design
After diabetes was induced, mice were divided into five groups (10 mice per group): diabetic mice with 0, 0.1% AA, 0.2% AA, 0.1% MA, 0.2% MA. In addition, one group of non-diabetic mice was divided into three sub-groups, in which 0 (control), 0.2% AA or 0.2% MA was supplied. All mice had free access to feed and water at all times. Consumed water volume and feed were recorded. Body weight and plasma glucose level were measured weekly. After 12 weeks supplementation, mice were fasted for 12 hr, and killed with carbon dioxide. Blood and heart from each mouse were collected and weighted. Protein concentration of heart homogenate was determined by a commercial assay kit (Pierce Biotechnology Inc., Rockford, IL, USA) with bovine serum albumin as a standard. Blood analyses 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
Plasma glucose level (mg/dl) was measured by a glucose kit (Sigma Chemical Co., St. Louis, MO, USA). Plasma HbA1c level was measured by a DCA 2000 analyzer (Bayer-Sankyo, Tokyo, Japan). Plasma insulin level (g/l) was determined by an insulin radioimmunoassay kit (Linco Research Inc., St. Charles, MO, USA). Plasma activities of lactate dehydrogenase (LDH), creatine phosphokinase (CPK), alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured by assay kits purchased from Randox Laboratories Ltd. (Crumlin, UK).
Determination of cardiac AA or MA content
The content of target compound in heart was analyzed by the method described in Gerbeth et al. (2011). Cardiac homogenate, 100 μl, was mixed with glycyrrhetinic acid as an internal standard (10 μl of 2.0 μg/ml methanol solution), and followed by extracting with 1 ml ethyl acetate and centrifuging at 3500 xg for 10 min at 4°C. After evaporated by nitrogen, the residue was reconstituted in 100 μl of methanol and water, the mobile phase of HPLC. Identification and quantification was processed by an HPLC-MS system (Agilent Corp, Waldbronn, Germany), in which Agilent 1100 series HPLC equipped with a BDS RP-C18 column (100 mm × 4 mm, 3 μm, Thermo Electron, Bellafonte, PA, USA), and a diode array and a fluorescence detector were applied. An ion-trap mass spectrometer equipped with an electro-spray ionization source was coupled with HPLC, and a negative single ion mode was used for analysis. The limit of detection was 0.1 g/g tissue. The relative standard deviations of precision and accuracy for test compound were 3.7 and 4.1%, respectively.
Measurement of coagulation and anti-coagulation factors
Blood samples were anti-coagulated using sodium citrate. vWF antigen level was measured by a rabbit anti-rat vWF polyclonal antibody (Dako, Glostrup, Denmark), and vWF 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
level was expressed as relative percentage of normal pooled plasma. FVII activity was determined by a kit bought from Chromogenix Co. (Lexington, MA, USA), which was positively correlated to the production of p-nitroaniline, and the absorbance at 405 nm was assayed. Plasma fibrinogen level (g/l) was measured using a commercial kit (Iatroset Fbg, Iatron Laboratory, Tokyo, Japan). PAI-1 activity (kU/l) was assayed by a kit purchased from Trinity Biotech plc. (Wicklow, Ireland). The activity of AT-III and protein C in plasma was measured by AT-III and protein C kits (Sigma Chemical Co., St. Louis, MO, USA), respectively. The activity of AT-III and protein C was expressed as a percentage of standard plasma.
Determination of CML, pentosidine, fructose, methylglyoxal and D-lactate levels
Cardiac tissue at 50 mg was homogenized and digested with proteinase K (1 mg/ml) for 3 hr at 37°C, and reaction was stopped by 2 mmol/l phenylmethylsulfonyl fluoride. CML was immunochemically determined with a competitive ELISA kit (RocheDiagnostics, Penzberg, Germany) using the CML-specific monoclonal antibody 4G9, and calibration with 6-(N-carboxymethylamino)caproic acid. Absorbance at 405 nm (reference 603 nm) was read in a microtiter ELISA plate reader (Bio-Rad, Hercules, CA, USA). Pentosidine level was analyzed by a HPLC equipped with a C18 reverse-phase column and a fluorescence detector (Waters, Tokyo, Japan) with an excitation and emission wavelength at 335 and 385 nm according to the method described by Miyata et al. (1996). Briefly, sample was lyophilized and acid hydrolyzed in 500 µl 6 N HCl for 16 hr at 110°C in screw-cap tubes purged with nitrogen. After neutralization with NaOH, sample was usedfor HPLC measurement. In addition, 50 mg heart was homogenized with phosphate buffer containing U-[13C]-sorbitol as an internal standard. After precipitating protein and centrifugation, the supernatant was lyophilized. The content of 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
fructose in lyophilized sample was determined by liquid chromatography with tandem mass spectrometry, according to the method of Guerrant and Moss (1984). Methylglyoxal level was analyzed by a HPLC method (Chaplen et al. 1996). Cardiac sample was derivatized with perchloric acid and o-phenylenediamine, and followed by purification with solid-phase extraction on AccuBond extraction cartridges. After centrifugation at 5,000 xg for 20 min, supernatant was collected. Quantification of methylglyoxal was processed by a HPLC system equipped with a fluorescence detector and a RP-C18 column (5 m particle size, 250 x 4 mm i.d.). D-lactate level was determined by an enzymatic assay kit purchased from Eton
Bioscience (San Diego, CA, USA). Cardiac tissue at 25 mg was homogenized, and D-lactate was extracted by ethanol. After centrifugation at 10,000 xg for 10 min, supernatant was used for D-lactate measurement.
Measurement of ROS, glutathione (GSH), interleukin (IL)-6, TNF-alpha and monocyte chemoattractant protein (MCP)-1 levels
Cardiac tissue was homogenized with ice-cold phosphate buffer containing 0.05% Tween 20 and 1 mM EDTA. Intracellular ROS level was determined using an oxidation sensitive dye, 2',7'-dichlorofluorescein diacetate (DCFH-DA). Briefly, 500 l homogenate was mixed with 500 l of 2 mg/ml DCFH-DA for 30 min at 37°C. Fluorescence was measured at 488 nm excitation and 525 nm emission by a fluorescence plate reader. Results are expressed as relative fluorescence unit (RFU) per mg protein. GSH concentration was measured by a commercial colorimetric glutathione assay kit (OxisResearch, Portland, OR, USA). Levels of IL-6 and TNF-alpha were quantified using ELISA kits (R&D Systems, Minneapolis, MN, USA). MCP-1 level was assayed by a cytoscreen immunoassay kit (BioSource International, Camarillo, CA, USA).
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
Western blot analysis
Cardiac tissue, 40 mg, was homogenized in buffer containing 0.5% Triton X-100 and protease-inhibitor cocktail (1:1000, Sigma-Aldrich Chemical Co., St. Louis, MO, USA). This homogenate was further mixed with buffer (60 mmol/l Tris-HCl, 2% SDS and 2% β-mercaptoethanol, pH 7.2), and boiled for 5 min. Sample at 40 μg protein was applied to 10% SDS-polyacrylamide gel electrophoresis, and transferred to a nitrocellulose membrane (Millipore, Bedford, MA, USA) for 1 hr. After blocking with a solution containing 5% nonfat milk for 1 hr, membrane was incubated with RAGE (1:500), AR, SDH, anti-GLO-1, anti-GLO-2, anti-p47phox, anti-gp91phox, anti-NF-κB p65, anti-NF-κB p50 (1:1000) or anti-MAPK (1:2000) monoclonal antibody (Boehringer-Mannheim, Indianapolis, IN, USA) at 4ºC overnight, and followed by reacting with horseradish peroxidase-conjugated antibody for 3.5 hr at room temperature. Glyceraldehyde-3-phosphatedehydrogenase (GAPDH) was used as a loading control. The detected bands were quantified by an image analyzer (ATTO, Tokyo, Japan) and normalized against GAPDH.
Statistical analysis
All data were expressed as mean±standard deviation (SD). Statistical analysis was done using one-way analysis of variance, and post-hoc comparisons were carried out using Dunnett's t-test. Statistical significance is defined as p < 0.05.
Results
Effects of AA or MA upon diabetic characteristic.
For normal mice, AA or MA intake at 0.2% increased their deposit in heart, but did not affect any measurements. As shown in Table 1, diabetes lowered body weight, increased feed intake 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
and water intake (p<0.05); however, AA or MA intake at two doses improved body weight loss and lowered feed intake; only at 0.2% decreased water intake (p<0.05). AA or MA intake at 0.2% increased cardiac content of these compounds in diabetic mice. Diabetes decreased plasma insulin level, increased plasma glucose and HbA1c levels, and CPK and LDH activities (Table 2, p<0.05). AA or MA intake at both doses lowered plasma glucose and HbA1c levels, CPK and LDH activities (p<0.05); but plasma insulin level was restored by these compounds only at 0.2% (p<0.05). AA or MA intake did not affect ALT and AST levels (p>0.05).
Effects of AA or MA upon coagulatory and anti-coagulatory factors.
Diabetes caused the increase in plasma level or activity of vWF, FVII, fibrinogen and PAI-1 (Table 3, p<0.05). AA or MA intake at both doses decreased vWF level, and at 0.2% lowered fibrinogen level and FVII activity (p<0.05); but failed to alter PAI-1 activity (p>0.05). Diabetes reduced plasma AT-III and protein C activities (p<0.05). AA or MA intake retained AT-III activity, and only at 0.2% maintained protein C activity (p<0.05).
Effects of AA or MA upon oxidative and glycative factors.
Diabetes lowered GSH content; and increased ROS level in heart (Table 4, p<0.05); AA or MA intake at both doses retained cardiac GSH content and reduced cardiac ROS level (p<0.05). Diabetes also increased CML, pentosidine, fructose and methylglyoxal levels, as well as decreased D-lactate level in heart (p<0.05). AA or MA intake at test doses lowered cardiac levels of CML, pentosidine, fructose and methylglyoxal; and raised D-lactate generation (p<0.05). As shown in Fig. 1, diabetes up-regulated cardiac expression of p47phox, gp91phox, AR, SDH and RAGE; and down-regulated cardiac expression of GLO-1 and GLO-2 (p<0.05). AA or MA intake at two doses down-regulated p47phox and AR expression, and only at 0.2% reduced gp91phox and RAGE expression (p<0.05). AA or MA intake at test doses also reserved 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
GLO-1 expression (p<0.05). Cardiac SDH and GLO-2 expression were not affected by either AA or MA (p>0.05).
Effects of AA or MA upon NF-B and MAPK expression.
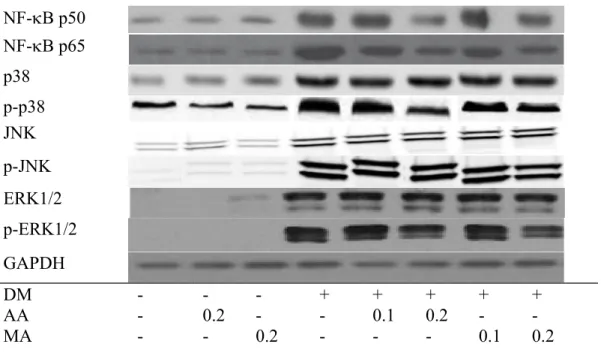
Diabetes increased cardiac release of MCP-1, IL-6 and TNF-alpha (Table 5, p<0.05). AA or MA intake at both doses decreased these cytokines in heart (p<0.05). Diabetes enhanced cardiac expression of NF-B and MAPK (Fig. 2, p<0.05). AA or MA intake at two doses down-regulated NF-B p65 and p-p38 expression (p<0.05). These two compounds only at 0.2% lowered NF-B p50 and p-ERK1/2 expression in heart (p<0.05).
Discussion
Our present study found that the intake of AA or MA resulted in their deposit in heart, benefited glycemic control and protected heart against diabetes induced hyper-coagulatory and glycative stress. Moreover, AA and MA could regulate cardiac NADPH oxidase, polyol, NF-B and MAPK pathways, which in turn alleviated oxidative and inflammatory injury in heart of diabetic mice. These findings suggest that these two triterpenoids could provide multiple protections for heart against diabetes.
Hypercoagulability is an important contributor for the development of vascular complications for diabetic patients (Yamada et al. 2000). vWF is involved in platelet adhesion and aggregation (Vischer 2006). FVII is the first enzyme in blood coagulation system for triggering the clotting cascade (Eigenbrot and Kirchhofer 2002). Fibrinogen is a precursor in fibrin formation and a cofactor in platelet aggregation; and PAI-1 is the primary physiologic inhibitor of fibrinolysis (Urano et al. 2000). Thus, the elevated circulating level or activity of vWF, FVII, fibrinogen and PAI-1 in those diabetic mice revealed a predominance of 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
coagulation, which obviously favored the progression of thrombosis. However, our data indicated that AA or MA supplement at 0.1 and/or 0.2% lowered vWF and fibrinogen levels, and FVII activity, which contributed to attenuate coagulatory disorder. On the other hand, AT-III and protein C are anti-coagulation factors because AT-AT-III inhibits the activity of several proteases in the coagulation cascade, and protein C inactivates coagulation factors such as FVII (Asakawa et al. 2000). We found that AA or MA treatments at 0.2% retained AT-III and protein C activities, which in turn benefited fibrinolysis. Apparently, the anti-coagulatory effects of these compounds could be partially ascribed to they enhance thrombolysis. These findings indicated that AA or MA could reduce the risk of diabetes associated atherogenesis and thrombosis via regulating both coagulatory and anti-coagulatory factors.
It is known that hyperglycemia raises oxidative stress, which subsequently enhances the release of vWF and fibrinogen (ElGendy and Abbas, 2014). Furthermore, ROS could stimulate platelet hyperactivity and facilitate coagulation (Ceriello et al. 1995; Meerarani et al. 2006). Our data revealed that AA or MA could exhibit anti-oxidative activities, via suppressing protein expression of p47phox and gp91phox, subunits of NADPH oxidase, to decrease ROS generation and spare GSH content in heart of diabetic mice. These results supported that AA and MA through their anti-oxidative actions diminished cardiac oxidative injury, which consequently mitigated oxidative stress in circulation and reversed hemostatic imbalance.
It is reported that the accumulation of HbA1c, CML and pentosidine in circulation and heart impairs cardiac functions (Schalkwijk et al. 2004; Hu et al. 2013). The enhanced activity and expression of aldose reductase promoted AGEs formation and caused heart failure under diabetic condition (Kaneko et al. 2005; Son et al. 2012). In this present study, AA or MA intake markedly decreased protein expression of this enzyme in heart of diabetic mice, which 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
subsequently reduced fructose flux and AGEs production. These findings suggest that these triterpenoids could abate cardiac glycative injury by inhibiting aldose reductase. On the other hand, GLO-1 is responsible for the detoxification of methylglyoxal to D-lactate (Thornalley 2003; Rabbani and Thornalley 2011). Our data revealed that AA or MA could reserve cardiac expression of GLO-1, decrease methylglyoxal level and increase D-lactate formation. Thus, the observed lower cardiac CML and pentosidine levels in AA or MA-treated diabetic mice could be partially ascribed to these compounds enhance glyoxalase pathway. These findings suggest that these two triterpenoids are potent GLO-1 enhancers.
The activation of NF-B and MAPK pathways by ROS and/or AGE-RAGE interaction exacerbates the progression of inflammation, endothelial dysfunctions and hypercoagulation in heart and/or other organs through accelerating the formation of glycative, oxidant, inflammatory and even coagulant factors (Wang et al. 2006; Lorenzo et al. 2011; Fukami et al. 2014). It is reported that the increased release of inflammatory cytokines and chemoattractants promoted the occurrence of diabetic heart failure (Drimal et al. 2008). Our data revealed that AA or MA treatments already decreased cardiac production of ROS, AGEs and RAGE, which in turn diminished the interaction of AGEs and RAGE, and led to less activation of NF-B and MAPK molecules. Consequently, it was reasonable to observe the limited expression of NF-B p50, p65, p-p38 and p-ERK1/2 in heart of diabetic mice with AA or MA treatments. Meanwhile, the decreased release of IL-6, TNF-alpha and MCP-1 in heart of those diabetic mice also agreed that NF-B and MAPK pathways have been suppressed. Since cardiac oxidative, inflammatory and glycative injury in diabetic mice has been ameliorated, the improvement in cardiac functions, evidenced by the changes in CPK and LDH activities, could be explained. On the other hand, it is possible that these agents decreased the interaction
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
among glycative, inflammatory and oxidative factors in circulation and heart, which subsequently alleviated cardiac damage. The other possibility is that the intake of these triterpenoids also led to their presence in other organs, and protected those tissues to reduce systemic oxidative, inflammatory and glycative responses, which contributed to mitigate cardiac injury. The improvement in glycemic control, body weight, feed and water intake in these AA or MA treated diabetic mice seemingly agreed that the overall oxidative, glycative and inflammatory stresses have been attenuated.
AA and MA are triterpenoids naturally occurring in many plant foods. The intake of these agents at 0.2% for mice is approximately equal to 28 g/day for a 70-kg adult. Our previous study (Yin et al. 2012) and present study indicated that dietary intake of these triterpenoids increased their bioavailability in organs including heart and kidney; and did not cause any toxic sign. Thus, these triterpenoids might be safe and feasible for application. However, we found that diabetic mice had lower AA or MA deposit in hearts when compared with normal mice. Both AA and MA are lipophilic compounds; thus, they might enhance metabolic burden under diabetic condition, and lead to lower cardiac bioavailability. Thus, the supplement of these agents for diabetic subjects should consider their metabolic capability.
In summary, dietary intake of asiatic acid or maslinic acid at 0.2% markedly declined NADPH oxidase, polyol, RAGE, NF-B and MAPK pathways in heart of diabetic mice, which in turn decreased cardiac levels of ROS, AGEs and inflammatory cytokines. Furthermore, these compounds mitigated hemostatic disorders via lowering vWF, FVII and fibrinogen levels or activity, as well as retaining protein C and AT-III activities in circulation. These findings support that the supplement of these triterpenoids or foods rich in these compounds may protect heart under diabetic condition to attenuate glycative injury and coagulatory disorders.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
Conflict of interest statement
There was no Conflict of Interest regarding this manuscript.
Acknowledgement
This study was partially supported by a grant from Ministry of Science and Technology, Taipei City, Taiwan (NSC 102-2313-B-039 -002 -MY3).
1 2 3 4 5 6 7
Table 1
Body weight (BW, g/mouse), water intake (WI, ml/mouse/day), feed intake (FI, g/mouse/day), heart weight (HW, mg/mouse) and cardiac content (g/g) of AA or MA in normal or diabetic mice treated with AA or MA at 0, 0.1 or 0.2% for 12 weeks. Values are mean SD, n=10.
Normal
AA, 0.2 MA, 0.2
DM
AA, 0.1 AA, 0.2 MA, 0.1 MA, 0.2
BW 30.62.0d 29.71.5d 30.22.2d 12.80.9a 14.90.5b 18.61.0c 15.30.7b 19.21.3c WI 2.30.5a 2.50.6a 2.20.4a 6.11.0c 5.40.8c 4.00.4b 5.50.8c 3.70.7b FI 2.20.7a 2.00.8a 2.30.5a 5.81.2d 4.60.6c 3.40.5b 4.30.7c 3.20.8b HW 1497a 1515a 1478a 1524a 1469a 1506a 14810a 1455a AA -*,a 1.340.11c -a -a -a 0.880.08b -a -a MA -a -a 1.290.07c -a -a -a -a 0.760.05b
*Means too low to be detected.
a-dMeans in a row without a common letter differ, p<0.05. 1 2 3 4 5 6
Table 2
Plasma level of glucose (mmol/l), HbA1c (%), insulin (nmol/l), CPK (IU/l), LDH (IU/l), ALT (U/l) and AST (U/l) in normal or diabetic mice treated with AA or MA at 0, 0.1 or 0.2% for 12 weeks. Values are mean SD, n=10.
Normal
AA, 0.2 MA, 0.2
DM
AA, 0.1 AA, 0.2 MA, 0.1 MA, 0.2
Glucose 9.30.3a 9.10.5a 9.40.7a 28.22.3d 23.01.5c 17.10.9b 21.71.1c 16.00.8b HbA1c 2.50.4a 2.70.5a 2.40.3a 12.11.2d 9.20.9c 6.50.7b 9.61.0c 7.00.6b Insulin 14.10.7c 14.70.6c 14.30.5c 5.30.4a 6.20.7a 7.40.8b 6.00.6a 7.70.3b CPK 56.72.0a 54.21.7a 57.02.3a 202.213.8d 164.710.8c 113.59.6b 156.35.3c 109.67.1b LDH 32.51.8a 33.41.2a 34.11.5a 166.411.0d 129.58.5c 90.36.7b 132.810.4c 96.87.2b ALT 263a 285a 274a 303a 254a 273a 293a 282a AST 334a 352a 353a 362a 342a 354a 332a 343a
a-dMeans in a row without a common letter differ, p<0.05. 1
2 3
Table 3
Plasma level of vWF (%) and fibrinogen (g/l); and activity of FVII activity (%), PAI-1 (kU/l), AT-III (%) and protein C (%) in normal or diabetic mice treated with AA or MA at 0, 0.1 or 0.2% for 12 weeks. Values are mean SD, n=10.
Normal
AA, 0.2 MA, 0.2
DM
AA, 0.1 AA, 0.2 MA, 0.1 MA, 0.2
vWF 100a 984a 973a 41123f 35216e 2679c 31810d 19812b Fibrinogen 2.380.11a 2.250.13a 2.230.10a 5.080.26c 4.950.19c 3.910.15b 5.020.21c 4.040.13b FVII 100a 963a 992a 31319c 30412c 22515b 3017c 20810b PAI-1 7.10.6a 7.30.5a 6.90.7a 20.21.3b 19.51.0b 19.00.8b 19.20.7b 19.11.2b AT-III 14210d 1397d 1448d 682a 835b 1059c 804b 1077c Protein C 985c 958c 976c 544a 605a 793b 592a 774b
a-fMeans in a row without a common letter differ, p<0.05. 1
2 3
Table 4
Cardiac level of GSH (nmol/mg protein), ROS (RFU/mg protein), CML (pmol/mg protein), pentosidine (pmol/mg protein), fructose (nmol/mg protein), methylglyoxal (nmol/mg protein) and D-lactate (nmol/mg protein) in normal or diabetic mice treated with AA or MA at 0, 0.1 or 0.2% for 12 weeks. Values are mean SD, n=10.
Normal
AA, 0.2 MA, 0.2
DM
AA, 0.1 AA, 0.2 MA, 0.1 MA, 0.2
GSH 20.31.1d 21.01.3d 20.60.8d 8.80.7a 11.50.9b 15.21.2c 12.00.6b 14.71.0c ROS 0.240.08a 0.270.05a 0.250.04a 1.250.14d 0.900.11c 0.630.07b 0.950.12c 0.590.06b CML 8.20.8a 8.01.0a 7.81.3a 79.44.2f 66.82.6e 53.23.1c 60.32.9d 45.13.3b Pentosidine 0.210.04a 0.190.5a 0.220.6a 1.420.16d 1.150.11c 0.780.09b 1.070.13c 0.800.10b Fructose 17.11.4a 16.81.2a 17.31.5a 90.14.7d 72.33.5c 46.22.1b 69.52.9c 43.03.1b Methylglyoxal 0.680.04a 0.650.05a 0.630.03a 6.630.28d 4.510.16c 2.060.13b 4.390.09c 2.170.21b D-lactate 79.22.5d 80.51.9d 81.12.7d 49.51.3a 57.31.7b 66.72.0c 59.21.5b 68.11.8c a-fMeans in a row without a common letter differ, p<0.05.
1 2 3 4
Fig. 1. Cardiac expression of p47phox, gp91phox, AR, SDH, RAGE, GLO-1 and GLO-2 in normal or diabetic mice treated with AA or MA at 0, 0.1 or 0.2% for 12 weeks was detected by western blot analyses. GAPDH was used as loading control. p47phox, gp91phox, AR, SDH, RAGE,
GLO-1 and GLO-2 protien bands were normalized to GAPDH expression. A representative image is shown. Each bar represents the mean SD, n=10. a-dMeans among bars without a common letter differ, p<0.05. p47phox gp91phox AR SDH RAGE GLO-1 GLO-2 GAPDH DM - - - + + + + + AA - 0.2 - - 0.1 0.2 - -MA - - 0.2 - - - 0.1 0.2 1 2 3 4 5 6 7
p47phox/GAPDH gp91phox/GAPDH
1 2
Table 5
Cardiac level (pg/ml) of MCP-1, IL-6 and TNF-alpha in normal or diabetic mice treated with AA or MA at 0, 0.1 or 0.2% for 12 weeks. Values are mean SD, n=10.
Normal
AA, 0.2 MA, 0.2
DM
AA, 0.1 AA, 0.2 MA, 0.1 MA, 0.2
MCP-1 12.21.3a 10.51.2a 11.41.1a 79.54.2d 58.12.6c 41.62.9b 55.93.0c 38.32.5b IL-6 13.40.8a 12.81.5a 11.91.2a 92.13.9d 74.73.0c 47.83.1b 75.21.8c 45.22.0b TNF-alpha 11.61.0a 11.01.4a 10.70.7a 108.65.0d 80.23.6c 52.63.3b 76.12.2c 48.62.7b a-dMeans in a row without a common letter differ, p<0.05.
1 2 3
Fig. 2. Cardiac expression of NF-B and MAPK in normal or diabetic mice treated with AA or MA at 0, 0.1 or 0.2% for 12 weeks was detected by western blot analyses. GAPDH was used as loading control. NF-B and MAPK protien bands were normalized to GAPDH expression. A representative image from each group is shown. Each bar represents the mean SD, n=10.
a-dMeans among bars without a common letter differ, p<0.05.
NF-B p50 NF-B p65 p38 p-p38 JNK p-JNK ERK1/2 p-ERK1/2 GAPDH DM - - - + + + + + AA - 0.2 - - 0.1 0.2 - -MA - - 0.2 - - - 0.1 0.2 1 2 3 4 5 6
1 23
4 5 6
References
Asakawa, H., Tokunaga, K., Kawakami, F., 2000. Elevation of fibrinogen and thrombin-antithrombin III complex levels of type 2 diabetes mellitus patients with retinopathy and nephropathy. J. Diabet. Complicat. 14, 121-126.
Barlovic, D.P., Thomas, M.C., Jandeleit-Dahm, K., 2010. Cardiovascular disease: what's all the AGE/ RAGE about? Cardiovasc. Hematol. Disord. Drug. Targets. 10, 7-15.
Castellano, J.M., Guinda, A., Delgado, T., Rada, M., Cayuela, J.A., 2013. Biochemical basis of the antidiabetic activity of oleanolic acid and related pentacyclic triterpenes. Diabetes 62, 1791-1799.
Ceriello, A., Giacomello, R., Stel, G., Motz, E., Taboga, C., Tonutti, L., Pirisi, M., Falleti, E., Bartoli, E., 1995. Hyperglycemia-induced thrombin formation in diabetes. The possible role of oxidative stress. Diabetes 44, 924-928.
Chaplen, F.W., Fahl, W.E., Cameron, D.C., 1996. Method of determination of free intracellular and extracellular methylglyoxal in animal cells grown in culture. Anal. Biochem. 238, 171-178.
Delmastro, M.M., Piganelli, J.D., 2011. Oxidative stress and redox modulation potential in type 1
diabetes . Clin. Dev. Immunol. 2011, 593863.
Drimal, J., Knezl, V., Navarova, J., Nedelcevova, J., Paulovicova, E., Sotnikova, V.R., Drimal, D., 2008. Role of inflammatory cytokines and chemoattractants in rat model of streptozotocin-induced diabetic heart failure. Endocr. Regul. 42, 129-135.
Eigenbrot, C., Kirchhofer, D., 2002. New insight into how tissue factor allosterically regulates factor VIIa. Trends Cardiovasc. Med. 12, 19-26.
ElGendy, A.A., Abbas, A.M., 2014. Effects of warfarin and L-carnitine on hemostatic function and oxidative stress in streptozotocin-induced diabetic rats. J. Physiol. Biochem. 70, 535-546.
Fukami, K., Yamagishi, S., Okuda, S., 2014. Role of AGEs-RAGE system in cardiovascular disease. Curr. Pharm. Des. 20, 2395-2402.
Gerbeth, K., Meins, J., Kirste, S., Momm, F., Schubert-Zsilavecz, M., Abdel-Tawab, M., 2011. Determination of major boswellic acids in plasma by high-pressure liquid chromatography/mass spectrometry. J. Pharm. Biomed. Anal. 56, 998-1005.
Guerrant, G., Moss, C.W., 1984. Determination of monosaccharides as aldononitrile, O-1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31
methyoxime, alditol, and cyclitol acetate derivatives by gas chromatography. Anal. Chem. 56, 633-638.
Hashim, P., Sidek, H., Helan, M.H., Sabery, A., Palanisamy, U.D., Ilham, M., 2011. Triterpene composition and bioactivities of Centella asiatica. Molecules 16, 1310-1322.
Herkert, O., Djordjevic, T., BelAiba, R.S., Görlach, A., 2004. Insights into the redox control of blood coagulation: role of vascular NADPH oxidase-derived reactive oxygen species in the thrombogenic cycle. Antioxid. Redox. Signal. 6, 765-776.
Hirata, K., Kubo, K., 2004. Relationship between blood levels of N-carboxymethyl-lysine and pentosidine and the severity of microangiopathy in type 2 diabetes. Endocr. J. 51, 537-544.
Hu, S., He, W., Liu, Z., Xu, H., Ma, G., 2013. The accumulation of the glycoxidation product N(ε)-carboxymethyllysine in cardiac tissues with age, diabetes mellitus and coronary heart disease. Tohoku. J. Exp. Med. 230, 25-32.
Hu, X., Li, S., Yang, G., Liu, H., Boden, G., Li, L., 2014. Efficacy and safety of aldose reductase inhibitor for the treatment of diabetic cardiovascular autonomic neuropathy: systematic review and meta-analysis. PLoS One 9, e87096.
Kaneko, M., Bucciarelli, L., Hwang, Y.C., Lee, L., Yan, S.F., Schmidt, A.M., Ramasamy, R., 2005. Aldose reductase and AGE-RAGE pathways: key players in myocardial ischemic injury. Ann. N. Y. Acad. Sci. 1043, 702-709.
Lemkes, B.A., Hermanides, J., Devries, J.H., Holleman, F., Meijers, J.C., Hoekstra, J.B., 2010.
Hyperglycemia: a prothrombotic factor? J. Thromb. Haemost. 8, 1663-1669.
Lorenzo, O., Picatoste, B., Ares-Carrasco, S., Ramírez, E., Egido, J., Tuñón, J., 2011. Potential role of nuclear factor κB in diabetic cardiomyopathy. Mediators Inflamm. 2011, 652097. Meerarani, P., Badimon, J.J., Zias, E., Fuster, V., Moreno, P.R., 2006. Metabolic syndrome and
diabetic atherothrombosis: implications in vascular complications. Curr. Mol. Med. 6, 501-514.
Miyata, T., Taneda, S., Kawai, R., Ueda, Y., Horiuchi, S., Hara, M., Maeda, K., Monnier, V.M., 1996. Identification of pentosidine as a native structure for advanced glycation end products in 2-microglobulin-containing amyloid fibrils in patients with dialysis-related amyloidosis. Proc. Natl. Acad. Sci. USA93, 2353-2358.
Nwose, E.U., Jelinek, H.F., Richards, R.S., Tinley, P., Kerr, P.G., 2009. Atherothrombosis and
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31
oxidative stress : the connection and correlation in diabetes. Redox. Rep. 14, 55-60.
Rabbani, N., Thornalley, P.J., 2011. Glyoxalase in diabetes , obesity and related disorders .
Semin. Cell Dev. Biol. 22, 309-317.
Ramachandran, V., Saravanan, R., Senthilraja, P., 2014. Antidiabetic and antihyperlipidemic activity of asiatic acid in diabetic rats, role of HMG CoA: in vivo and in silico approaches.
Phytomedicine 21, 225-232.
Schalkwijk, C.G., Baidoshvili, A., Stehouwer, C.D., van Hinsbergh, V.W., Niessen, H.W., 2004. Increased accumulation of the glycoxidation product Nepsilon-(carboxymethyl)lysine in hearts of diabetic patients : generation and characterisation of a monoclonal anti- CML antibody. Biochim. Biophys. Acta. 1636, 82-89.
Son, N.H., Ananthakrishnan, R., Yu, S., Khan, R.S., Jiang, H., Ji, R., Akashi, H., Li, Q., O'Shea, K., Homma, S., Goldberg, I.J., Ramasamy, R., 2012. Cardiomyocyte aldose reductase causes heart failure and impairs recovery from ischemia. PLoS One 7, e46549. Thornalley, P.J., 2003. Glyoxalase I-structure, function and a critical role in the enzymatic
defence against glycation. Biochem. Soc. Trans. 31, 1343-1348.
Urano, T., Ihara, H., Suzuki, Y., Takada, Y., Takada, A., 2000. Coagulation-associated enhancement of fibrinolytic activity via a neutralization of PAI-1 activity. Semin. Thromb. Hemost. 26, 39-42.
Vischer, U.M., 2006. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J. Thromb. Haemost. 4, 1186-1193.
Wang, J., Song, Y., Wang, Q., Kralik, P.M., Epstein, P.N., 2006. Causes and characteristics of diabetic cardiomyopathy. Rev. Diabet. Stud. 3, 108-117.
Xie, J., Méndez, J.D., Méndez-Valenzuela, V., Aguilar-Hernández, M.M., 2013. Cellular signalling of the receptor for advanced glycation end products ( RAGE ). Cell Signal. 25, 2185-2197.
Yamada, T., Sato, A., Nishimori, T., Mitsuhashi, T., Terao, A., Sagai, H., Komatsu, M., Aizawa, T., Hashizume, K., 2000. Importance of hypercoagulability over hyperglycemia for vascular complication in type 2 diabetes. Diabetes Res. Clin. Pract. 49, 23-31.
Yin, M.C., Lin, M.C., Mong, M.C., Lin, C.Y., 2012. Bioavailability, distribution, and antioxidative effects of selected triterpenes in mice. J. Agric. Food Chem. 60, 7697-7701. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31