Coupling of Carbomethoxy and Vinylidene Ligand in
Molybdenum Complexes
Jung-Yen Yang, Shou-Ling Huang, Ying-Chih Lin,* Yi-Hong Liu, and Yu Wang
Department of Chemistry, National Taiwan University, Taipei, Taiwan, 106 Republic of China
Received September 13, 1999
Treatment of [Cp(dppe)(CO)ModCdC(Ph)CH
2CHdCH
2]I (2) with MeONa caused
nucleo-philic attack to occur at the terminal CO ligand. This is followed by a coupling reaction of
the resulting carbomethoxy group with CR of the vinylidene ligand accompanied with
coordination of the terminal olefin to afford Cp(dppe)MoC(COOMe)dC(Ph)CH
2CHdCH
2(5).
Similar coupling was observed when [Cp(dppe)(CO)ModCdC(Ph)CH
2Ph]I (3) was treated
with MeONa, but the reaction afforded the neutral allylic complex
Cp(dppe)Mo[CH-(COOMe)C(Ph)CHPh] (6). The structures of complexes 5 and 6 have been determined by
X-ray diffraction analysis.
Introduction
We previously described a new type of deprotonation
reaction of several cationic vinylidene complexes leading
to a rare class of metal complexes containing various
cyclic ligands.
1Using this synthetic strategy, we
suc-cessfully prepared several ruthenium cyclopropenyl
complexes containing various substituents as well as
ruthenium furanyl complexes. For the cyclopropenyl
complex, the cyclization reaction also results in
forma-tion of a chiral carbon center in the three-membered
ring. These features render this cyclization process
potentially useful for organic synthesis.
2Therefore, we
set out to study the chemical reactivity of a molybdenum
vinylidene system of Cp(Ph
2PCH
2CH
2PPh
2)(CO)Mo.
3Such a system was chosen because the presence of donor
phosphine ligands is known to assist the preparation
of cationic metal vinylidene complexes,
4and the
pres-ence of a CO ligand could be utilized to study
carbon-carbon bond formation. Surprisingly, treatment of these
vinylidene complexes containing a terminal CO ligand
with sodium methoxide affords unexpected products via
an unprecedented coupling of a carbomethoxy group
with CR of the vinylidene ligand.
5Coupling reactions
involving CR of a vinylidene ligand on the metal complex
are limited in the literature. Herein we report
prepara-tion of a number of caprepara-tionic molybdenum vinylidene
complexes Cp(Ph
2PCH
2CH
2PPh
2)(CO)ModCdC(Ph)-CH
2R and nucleophilic addition of methoxide to the CO
ligand followed by a coupling reaction leading to
carbon-carbon formation at CR of the vinylidene ligand.
Results and Discussion
Preparation of the Acetylide Complex 1. The
acetylide complex [Mo](CO)CtCPh (1, [Mo] ) (η
5-C
5H
5)(dppe)Mo, dppe ) Ph
2PCH
2CH
2PPh
2) was
pre-pared by two different methods. The reaction of
[Mo]-(CO)Cl with LiCCPh in THF, which gave 1 in reasonable
yield, required handling of an air-sensitive lithium
reagent. We therefore pursued an alternative synthesis,
namely, the reaction of [Mo](CO)Cl with HCtCPh. This
reaction gave
{
[Mo](CO)dCdCHPh
}
Cl, which
under-went thermal decarbonylation to afford
{
[Mo](η
2-HCt
CPh)
}
Cl. Then the η
2-phenylacetylene complex was
treated with CO in the presence of MeONa to give 1.
The tautomerization of alkyne to vinylidene on the
bisdimethylphenylphosphine molybdenum analogue has
been reported before.
3cOur scheme involves more steps
but gives higher overall yield.
The
31P NMR spectrum of 1 exhibits two sets of
doublet resonances at δ 90.4 and 78.7 with J
P-P) 37.2
Hz. In the
1H NMR spectrum, the resonance for the Cp
ligand is a doublet at δ 4.49 with J
H-P) 1.6 Hz,
indicating coupling with only one of the phosphorus
atoms of the dppe. In the
13C NMR spectrum, the signal
for the terminal CO ligand is also a doublet at δ 245.0
with J
C-P) 22.2 Hz, indicating the same coupling
pattern. By the use of selective heteronuclear decoupling
techniques, i.e.,
1H
{
31P
}
and
13C
{
31P
}
NMR spectra, we
are able to determine that both
1H-
31P coupling of the
Cp ligand and
13C-
31P coupling of the CO ligand
originate from the same phosphorus atom displaying the
downfield resonance (at δ 90.4) in the
31P NMR
spec-trum. Coupling between carbon and phosphorus nuclei
of the complex with a piano-stool coordination geometry
(1) (a) Ting, P. C.; Lin, Y. C.; Cheng, M. C.; Wang, Y.
Organome-tallics 1994, 13, 2150. (b) Ting, P. C.; Lin, Y. C.; Lee, G. H.; Cheng, M.
C.; Wang, Y. J. Am. Chem. Soc. 1996, 118, 6443. (c) Chang, C. W.; Ting, P. C.; Lin, Y. C.; Lee, G. H.; Wang, Y. J. Organomet. Chem. 1998,
553, 417. (d) Lo, Y. H.; Lin, Y. C.; Lee, G. H.; Wang, Y. Organometallics 1999, 18, 982.
(2) (a) Kubota, K.; Mori, S.; Nakamura, M.; Nakamura, E. J. Am.
Chem. Soc. 1998, 120, 13334. (b) Muller, P.; Imogai, H. Tetrahedron: Asymmetry 1998, 9, 4419. (c) Plemenkov, V. V.; Rul, S. V.; Gubaidullin,
A. T.; Litvinov, I. A.; Karaseva, I. P.; Nuretdinov, I. A. Russ. J. Org.
Chem. 1998, 34, 971.
(3) (a) Allen, S. R.; Beevor, R. G.; Green, M.; Norman, N. C.; Orpen, A. G.; Williams, I. D. J. Chem. Soc., Dalton Trans. 1985, 435. (b) Beevor, R. G.; Green, M.; Orpen, A. G.; Williams, I. D. J. Chem. Soc.,
Dalton Trans. 1987, 1319. (c) Nickias, P. N.; Seleague, J. P.; Young,
B. A. Organometallics 1988, 7, 2248. (d) Chang, K. H.; Lin, Y. C. Chem.
Commun. 1998, 1441.
(4) (a) Bruce, M. I. Chem. Rev. 1991, 91, 197. (b) Bruce, M. I.; Swincer, A. G. Adv. Organomet. Chem. 1982, 20, 159.
(5) (a) Buil, M. L.; Esteruelas, M. A.; Lo`pez, A. M.; On˜ate, E.
Organometallics 1997, 16, 3169. (b) Bianchini, C.; Casares, J. A.;
Peruzzini, M.; Romerosa, A.; Zanobini, F. J. Am. Chem. Soc. 1996, 118, 4585. (c) Faure, M.; Maurette, L., Donnadieu, B.; Lavigne, G. Angew.
Chem., Int. Ed. Engl. 1999, 34, 518.
10.1021/om9907167 CCC: $19.00 © 2000 American Chemical Society Publication on Web 12/31/1999
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
depends on the orientation.
6In the
1H NMR spectrum
of
{
[Mo](η
2-HCtCPh)
}
Cl, the characteristic triplet
reso-nance at δ 10.02 with J
H-P) 13.8 Hz is assigned to the
terminal CH. The corresponding
1H signal of the
vi-nylidene proton of
{
[Mo](CO)dCdCHPh
}
Cl appears as
a singlet at δ 4.78.
Preparation of Vinylidene Complexes. Treatment
of 1 with BrCH
2R (R ) CHdCH
2, C
6H
5, and CN)
afforded cationic brown-red vinylidene complexes
{
[Mo]-(CO)dCdC(Ph)CH
2R
}
Br (2, R ) CHdCH
2; 3, R ) C
6H
5;
4, R ) CN). All reactions gave the desired products in
70-80% yields. Use of organic iodides gave lower yields.
All these vinylidene complexes are soluble in CHCl
3,
CH
2Cl
2, and CH
3OH but are insoluble in hexane.
Complex 2 is air- and moisture-sensitive but is stable
under nitrogen. The
1H NMR spectrum of 2 displays
multiplet resonances centered at δ 5.00, 4.96 and 2.87,
2.78 assignable to the olefinic and saturated
diaste-reotopic CH
2units, respectively, and the multiplet
resonance at δ 5.62 to the allylic CH unit. The geminal
coupling constant of the diastereotopic methylene
pro-tons is 15.6 Hz, and interestingly, the long-range P-H
coupling with one of the phosphorus atoms (
5J
P-H
) 2.8
and 2.0 Hz) is also observed for these protons. The
1H
NMR spectra of complexes 3 and 4 display the same
features. In the
13C NMR spectrum, the characteristic
doublet of doublet resonance centered at δ 358.4 with
2J
P-C
) 36.0 and 6.5 Hz is assigned to the vinylidene
CR. In the
31P NMR spectrum, two doublet resonances
at δ 71.5 and 67.0 with J
P-P) 37.5 Hz are assigned to
the dppe ligand. In the
31P NMR spectra of 3 and 4, the
31P resonances fall in a similar region with comparable
coupling constants (δ 69.0 and 66.2 with J
P-P) 36.3
Hz for 3 and δ 66.8 and 63.7 with J
P-P) 30.7 Hz for 4).
Other spectroscopic features are consistent with their
formulations.
Reactions of 2 and 3 with MeONa. Having
estab-lished the cyclopropenation reaction of several cationic
vinylidene complexes of ruthenium, we examined the
deprotonation reaction of the molybdenum complex 2
using various bases with an expectation to see similar
results. However, much to our surprise, treatment of 2
with n-Bu
4NOH, DBU, and n-Bu
4NF gave no reaction.
In the reaction of 2 with MeONa the coupling product
[Mo](η
3-C(COOMe)dC(Ph)CH
2
CHdCH
2) (5) (Scheme 1)
was obtained in high yield. Formation of 5 is
rational-ized by nucleophilic addition of MeO
-to the terminal
CO ligand
7giving the carbomethoxy group followed by
a coupling reaction
8of the carboxymethoxy group with
CR of the vinylidene ligand to afford 5 (Scheme 1) in
high yield. The terminal olefin group, acting as a
two-electron donor, fills the vacant site left from the coupling
reaction. It is interesting to note the difference in
spectral characteristics of 2 and 5. Signals of the
terminal vinyl group in the
1H NMR spectrum of 2
appear at δ 5.62 (dCH) and 5.00, 4.96 (dCH
2), while
those of 5 occur at δ 1.78 (dCH) and 2.58, 3.66 (dCH
2).
These assignments are determined by the use of 2D
COSY and HSQC experiments. While most of H-H
(6) (a) Bainbridge, A.; Craig, P. J.; Green, M. J. Chem. Soc. (A) 1968, 2713. (b) Faller, J. W.; Anderson, A. S. J. Am. Chem. Soc. 1970, 92, 5852. (c) Beach, D. L.; Barnett, K. W. J. Organomet. Chem. 1975, 97, C27. (d) Sakaba, H.; Ishida, K.; Horino, H. Chem. Lett. 1995, 1145.
(7) (a) Nakazawa, H.; Kadoi, Y.; Mizuta, T.; Miyoshi, K.; Yoneda, H. J Organomet. Chem. 1989, 366, 333. (b) Gibson, D. H.; Ong, T. S.; Ye, M.; Franco, J. O.; Owens, K. Organometallics 1988, 7, 2569. (c) Busetto, L. L.; Zanotti, V.; Norfo, L.; Palazzi, A.; Albano, V. G.; Braga, D. Organometallics 1993, 12, 190. (d) Nakazawa, H.; Yamaguchi, M.; Kubo, K.; Miyoshi, K. J Organomet. Chem. 1992, 428, 145. (e) Werner, H.; Hofmann, L.; Zolk, R. Chem. Ber. 1987, 120, 379. (f) Kiel, W. A.; Buhro, W. E.; Gladysz, J. A. Organometallics 1984, 3, 879. (g) Carlos, F.; Barrientos-Penna, A.; Hugo, K. O.; Derek, S. Organometallics 1985, 4, 367. (h) Bao, Q. B.; Rheingold, A. L.; Brill, T. B. Organometallics
1986, 5, 2259.
(8) (a) Wiedemann, R.; Steinert, P.; Gevert, O.; Werner, H. J. Am.
Chem. Soc. 1996, 118, 2495. (b) Braun, T.; Meuer, P.; Werner, H. Organometallics 1996, 15, 4075.
Scheme 1
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
coupling constants in the allylic group are measurable
for 2, only the coupling constant of 17.2 Hz between
mutually trans-protons in the coordinated olefin is
attainable for 5. However, significant upfield shifts
indicate π-coordination of the terminal double bond in
5. Surprisingly, in the
31P NMR spectrum of 5, two
singlet resonances at δ 90.5 and 71.2 with equal
intensity are observed possibly because of a particular
orientation of the dppe ligand.
9In the
13C NMR
spec-trum of 5, resonances of π-coordinated olefinic carbon
atoms appeared at δ 60.3 (dd) and 47.6 (d) with J
C-P)
5.4, 4.0, and 5.9 Hz, respectively. To fully characterize
this product, single crystals of 5 were grown from a
hexane/CH
2Cl
2solution, and the molecular structure
was determined by X-ray diffraction analysis. An ORTEP
drawing is shown in Figure 1, and selected bond
distances and angles are listed in Table 1. The
environ-ment about the metal center corresponds to a structure
of distorted four-legged piano-stool geometry. The
car-bomethoxy group resulting from nucleophilic attack of
MeO
-to the terminal CO ligand is bound to CR. And it
is clear that the internal olefin C4-C5 is in an E
configuration with the phenyl group and the Mo
frag-ment in a trans disposition. The terminal olefin is
oriented such that the CdC bond is contained in the
plane defined by the metal, the center of the Cp ring,
and the trans phosphorus atom. The olefinic ligand
coordinates to the metal center with Mo-C1 )
2.216-(3) Å and Mo-C2 ) 2.2952.216-(3) Å. The Mo-C5 bond length
of 2.248(3) Å is typical of a Mo-C single bond, and the
C4-C5 bond length of 1.338(5) Å typical of a CdC
double bond. The C1-C2 bond length of 1.406(5) Å,
resulting from coordination to the metal center, is
slightly longer that that of the C4-C5 double bond. The
CdC double bond of 1.437(9) Å in the cationic olefin
complex Cp[P
2(Me)
4C
2H
4](CO)Mo(C
2H
3Ph)
10is slightly
longer possibly due to stronger back-bonding to the
antibonding orbital of the olefin.
Treatment of 3 with MeONa similarly causes
nucleo-philic addition of MeO
-to the terminal CO ligand
followed by an analogous coupling reaction and affords
the neutral allylic product [Mo][η
3-CH(COOMe)C(Ph)-CHPh] (6) in high yield. Due to the lack of a terminal
olefin donor group, the vinylidene ligand in 3 transforms
to the η
3-allylic ligand of 6 possibly by coupling followed
by a 1,3-hydrogen shift. The
13C resonance attributed
to the ester carbon appears at δ 183.2, and C-P
coupling constants of three multiplet resonances,
ap-pearing at δ 48.7 (d), 90.3 (t), and 49.2 (d) with J
C-P)
4.5, 3.1, and 2.7 Hz, respectively, clearly indicate the
η
3-allylic bonding mode. Two
31P resonances at δ 71.3
and 72.2 with J
P-P) 36.8 Hz are assigned to the dppe
ligand.
The structure of 6 has also been confirmed by an
X-ray diffraction analysis. An ORTEP drawing is shown
in Figure 2, and selected bond distances and angles are
listed in Table 2. The carbomethoxy group is again
bound to the allylic terminal carbon. The trisubstituted
allylic ligand is in an endo conformation with a
syn-phenyl group and an anti-carbomethoxy group at two
terminal carbon atoms. The two allylic C-C bond
lengths are about equal (C1-C4 ) 1.463(4) Å, C4-C5
) 1.446(4) Å), with the Mo-C1 bond slightly shorter
(9) Morton, M. S.; Lachicotte, R. J.; Vicic, D. A.; Jones, W. D.
Organometallics 1999, 18, 227.
(10) (a) Kegley, S. E.; Bergstrom, D. T.; Crocker, L. S.; Weiss, E. P.; Berndt, W. G.; Rheingold, A. L. Organometallics 1991, 10, 573. (b) Abugideiri, F.; Kelland, M. A.; Poli, R. Organometallics 1992, 11, 1311. (c) Kegley, S. E.; Walter, K. A.; Bergstrom, D. T.; MacFarland, D. K.; Young, B. G.; Rheingold, A. L. Organometallics 1993, 12, 2339.
Figure 1. ORTEP drawing of 5 with thermal ellipsoids
shown at the 30% probability level. For the dppe phenyl groups, only the ipso carbons are shown.
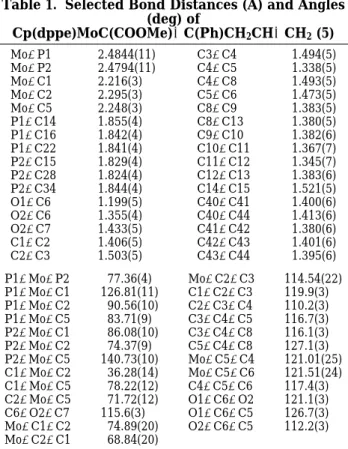
Table 1. Selected Bond Distances (Å) and Angles (deg) of Cp(dppe)MoC(COOMe)dC(Ph)CH2CHdCH2(5) Mo-P1 2.4844(11) C3-C4 1.494(5) Mo-P2 2.4794(11) C4-C5 1.338(5) Mo-C1 2.216(3) C4-C8 1.493(5) Mo-C2 2.295(3) C5-C6 1.473(5) Mo-C5 2.248(3) C8-C9 1.383(5) P1-C14 1.855(4) C8-C13 1.380(5) P1-C16 1.842(4) C9-C10 1.382(6) P1-C22 1.841(4) C10-C11 1.367(7) P2-C15 1.829(4) C11-C12 1.345(7) P2-C28 1.824(4) C12-C13 1.383(6) P2-C34 1.844(4) C14-C15 1.521(5) O1-C6 1.199(5) C40-C41 1.400(6) O2-C6 1.355(4) C40-C44 1.413(6) O2-C7 1.433(5) C41-C42 1.380(6) C1-C2 1.406(5) C42-C43 1.401(6) C2-C3 1.503(5) C43-C44 1.395(6) P1-Mo-P2 77.36(4) Mo-C2-C3 114.54(22) P1-Mo-C1 126.81(11) C1-C2-C3 119.9(3) P1-Mo-C2 90.56(10) C2-C3-C4 110.2(3) P1-Mo-C5 83.71(9) C3-C4-C5 116.7(3) P2-Mo-C1 86.08(10) C3-C4-C8 116.1(3) P2-Mo-C2 74.37(9) C5-C4-C8 127.1(3) P2-Mo-C5 140.73(10) Mo-C5-C4 121.01(25) C1-Mo-C2 36.28(14) Mo-C5-C6 121.51(24) C1-Mo-C5 78.22(12) C4-C5-C6 117.4(3) C2-Mo-C5 71.72(12) O1-C6-O2 121.1(3) C6-O2-C7 115.6(3) O1-C6-C5 126.7(3) Mo-C1-C2 74.89(20) O2-C6-C5 112.2(3) Mo-C2-C1 68.84(20)
Figure 2. ORTEP drawing of 6 with thermal ellipsoids
shown at the 30% probability level. For the dppe phenyl groups, only the ipso carbons are shown.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
than the other two Mo-C(allylic) bonds (Mo-C1 )
2.259(3) Å, Mo-C4 ) 2.328(3) Å, and Mo-C5 )
2.324-(3) Å). The different configuration of the carbomethoxy
group relative to the neighboring phenyl group (cis in 5
and trans in 6) could possibly be attributed to the
additional hydrogen shift process in the formation of 6.
Treatment of 4 with MeONa in MeOH gave, in
moderate yield, an unstable green complex, which
decomposed at room temperature in 2 h in CDCl
3. This
solid product was isolated by precipitation from the CH
2-Cl
2solution of the product via addition of ether. The
FAB mass spectrum of this complex displays peaks that
could be attributed to the cyclopropenyl complex
[Mo]-(CO)CdC(Ph)CHCN (7). In the IR spectrum of 7, the
absorption at 1846 cm
-1is assigned to the vibrational
stretching of the terminal CO ligand, indicating that
there is no nucleophilic addition. And in the
31P NMR
spectrum, chemical shifts of two singlet resonances at
δ 105.4 and 103.9 differ significantly from the range (δ
90.5 and 71.2) observed for 5 and 6. Unfortunately, we
can obtain spectroscopic data only for this green
com-pound, and there is no established data for a
molybde-num cyclopropenyl complex for comparison. This is
somewhat surprising since in the ruthenium system
1the presence of an electron-withdrawing CN substituent
seemed to stabilize a number of cyclopropenyl
com-plexes. In this Mo vinylidene system with a terminal
CO ligand, the CN group provides no similar effect. In
our attempts to carry out addition reactions of 4 using
nucleophilic reagents other than MeO
-, we do not
observe any coupling products as in the reactions of 2
and 3.
In summary, we report high-yield preparation of three
cationic molybednum vinylidene complexes
{
[Mo](CO)d
CdC(Ph)CH
2R
}
Br (2, R ) CHdCH
2; 3, R ) C
6H
5; 4, R
) CN). In the presence of MeONa, the molybdenum
vinylidene complexes 2 and 3, each containing a
termi-nal carbonyl ligand, undergo nucleophilic addition at the
CO ligand followed by a C-C coupling reaction at CR
of the vinylidene ligand to give addition products. The
site preference of this coupling reaction is possibly due
to the relatively strong bond of RudCR and proximity
of the two groups on the ruthenium metal center. For
the analogous CN-substituted vinylidene complex 4, the
same reaction yielded an unstable cyclopropenyl
com-plex through deprotonation.
Experimental Section
General Procedures. All manipulations were performed under nitrogen using vacuum-line, drybox, and standard Schlenk techniques. CH2Cl2 was distilled from CaH2 and diethyl ether and THF from Na/diphenylketyl. All other solvents and reagents were of reagent grade and were used as received. NMR spectra were recorded on Bruker AM-300WB and DMX-500 FT-NMR spectrometers at room temperature (unless states otherwise) and are reported in units of δ with residual protons in the solvents as a standard (CDCl3, δ 7.24; C2D6O, δ 2.04). FAB mass spectra were recorded on a JEOL SX-102A spectrometer. Complex [Mo](CO)Cl ([Mo] ) (η5 -C5H5)(dppe)Mo, dppe ) Ph2PCH2CH2PPh2) was prepared according to the methods reported in the literature.11 Elemen-tal analyses and X-ray diffraction studies were carried out at the Regional Center of Analytical Instrumentation located at the National Taiwan University.
Preparation of [Mo](CO)CtCPh (1). To a sample of [Mo]-(CO)Cl (0.22 g, 0.35 mmol) dissolved in 30 mL of THF at room temperature was added a THF solution of lithium phenyl-acetylide (1.0 M, 0.40 mL) via a syringe. The resulting mixture was heated under reflux for 1 h. After removal of all volatile substances in vacuo, 20 mL of Et2O was added to the residue under nitrogen, and the extract was filtered. The solvent of the filtrate was removed under vacuum to afford the yellow product 1, which was further recrystallized from a 1:1 mixture of hexane/CH2Cl2to afford crystals of 1 (0.14 g, 61% yield). Spectroscopic data for 1: IR (cm-1, CH2Cl2) 2074 (s, νCtC), 1850 (s, νCO);1H NMR (CDCl3) δ 7.95-6.64 (m, 25H, Ph), 4.49 (d, JH-P) 1.6 Hz, 5H, Cp), 2.40, 1.78 (m, 4H, CH2CH2);13C NMR (CDCl3) δ 245.0 (d, JC-P) 22.2 Hz, CO), 141.9-119.2 (Ph), 91.0 (Cp), 31.8 (dd, JC-P) 28.0 Hz, JC-P) 18.6 Hz, PCH2), 28.6 (dd, JC-P) 22.0 Hz, JC-P) 17.3 Hz, PCH2);31P NMR (CDCl3) δ 90.4 (d, JP-P) 37.2 Hz), 78.7 (d, JP-P) 37.2 Hz); MS (FAB, m/z, Mo98) 690 (M+
), 662 (M+- CO). Anal. Calcd for C40H34OP2Mo: C, 69.77; H, 4.98. Found: C, 70.02; H, 4.79. Reaction of [Mo](CO)Cl with HCtCPh. To a sample of [Mo](CO)Cl (0.20 g, 0.32 mmol) dissolved in 50 mL of MeOH at room temperature was added phenylacetylene (0.21 mL, 1.82 mmol) via a syringe. The resulting mixture was heated under reflux for 2.5 h to give a green solution. After removal of all volatile substances in vacuo, 20 mL of CH2Cl2was added to the residue, and the extract was filtered. The solvent of the filtrate was reduced under vacuum to about 5 mL and the solution added to a stirred solution of Et2O to give green precipitates. The solid was collected by filtration and washed with Et2O to give the green product{[Mo](η2-HCtCPh}Cl (0.20 g, 90% yield). Spectroscopic data: 1H NMR (CDCl
3) δ 10.02 (t, JH-P) 13.8 Hz, CH); 7.50-6.82 (m, 25H, Ph), 5.03 (s, 5H, Cp), 2.70 (m, 4H, PCH2CH2P); 31P NMR (CDCl3) δ 79.3 (s); MS (FAB, m/z, Mo98) 663 (M+). Anal. Calcd for C
39H35P2MoCl: C, 67.20; H, 5.06. Found: C, 67.21; H, 5.23.
Reaction of [Mo](CO)Cl with HCtCPh in the Presence of CO. To a sample of [Mo](CO)Cl (0.10 g, 0.16 mmol) dissolved in 30 mL of MeOH at room temperature was added phenyl-acetylene (0.11 mL, 0.92 mmol) via a syringe. The resulting mixture was heated under reflux for 2.5 h to give a green solution. After cooling to room temperature the solution was treated with 1 atm of CO gas and the solution turned yellow. After removal of all volatile substances in vacuo, 20 mL of CH2 -Cl2was added to the residue, and the extract was filtered. The solvent of the filtrate was reduced under vacuum to about 5 mL and the solution added to a stirred solution of hexane to give yellow precipitates, which were collected by filtration and washed with hexane to afford{[Mo](CO)(dCdCHPh}Cl, (0.12
(11) (a) Staerker, K.; Curtis, M. D. Inorg. Chem. 1985, 24, 3006. (b) Lau, Y. Y. Huckabee, W. W.; Gipson, S. L. Inorg. Chim. Acta 1990,
172, 41.
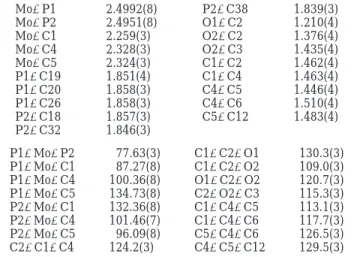
Table 2. Selected Bond Distances (Å) and Angles (deg) of Cp(dppe)MoCH(COOMe)C(Ph)CHPh (6) Mo-P1 2.4992(8) P2-C38 1.839(3) Mo-P2 2.4951(8) O1-C2 1.210(4) Mo-C1 2.259(3) O2-C2 1.376(4) Mo-C4 2.328(3) O2-C3 1.435(4) Mo-C5 2.324(3) C1-C2 1.462(4) P1-C19 1.851(4) C1-C4 1.463(4) P1-C20 1.858(3) C4-C5 1.446(4) P1-C26 1.858(3) C4-C6 1.510(4) P2-C18 1.857(3) C5-C12 1.483(4) P2-C32 1.846(3) P1-Mo-P2 77.63(3) C1-C2-O1 130.3(3) P1-Mo-C1 87.27(8) C1-C2-O2 109.0(3) P1-Mo-C4 100.36(8) O1-C2-O2 120.7(3) P1-Mo-C5 134.73(8) C2-O2-C3 115.3(3) P2-Mo-C1 132.36(8) C1-C4-C5 113.1(3) P2-Mo-C4 101.46(7) C1-C4-C6 117.7(3) P2-Mo-C5 96.09(8) C5-C4-C6 126.5(3) C2-C1-C4 124.2(3) C4-C5-C12 129.5(3)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
g, 87% yield). Spectroscopic data: IR (cm-1, CH
2Cl2) 1909 (s, νCO);1H NMR (CDCl3) δ 7.50-6.82 (m, 25H, Ph), 5.05 (s, 5H, Cp), 4.78 (s, 1H, dCH), 2.70 (m, 4H, PCH2CH2P);31P NMR (CDCl3) δ 58.5 (d, JP-P) 18.0 Hz), 15.5 (d, JP-P) 18.0 Hz); MS (FAB, m/z, Mo98) 691 (M+), 663 (M+- CO). Anal. Calcd for C40H35OP2MoCl: C, 66.26; H, 4.87. Found: C, 66.41; H, 4.98. This reaction is reversible. If the MeOH solution of the product was heated under refluxing, decarbonylation occurred and the η2-acetylide complex was obtained in quantitative yield. The carbonylation reaction in the presence of MeONa (54 mg, 0.10 mmol in 30 mL of MeOH) gave the acetylide complex 1 in 92% yield.
Reaction of 1 with Allyl Bromide. To a sample of 1 (0.60 g, 0.87 mmol) dissolved in 50 mL of CHCl3at room tempera-ture was added excess BrCH2CHdCH2(0.10 mL, 1.16 mmol) via a syringe. The resulting mixture was stirred for 36 h in the dark. The color of the reaction mixture changed from yellow to brown within this period. After removal of all volatile substances in vacuo, 10 mL of CH2Cl2was added to the residue under nitrogen, and the extract was filtered. The solvent of the filtrate was removed under vacuum to afford a brown residue. The residue was redissolved in 5 mL of CH2Cl2and was then added dropwise to a stirred Et2O solution to cause precipitation of brown solid, which was collected by filtration and washed with Et2O under nitrogen and dried under vacuum to afford [[Mo](CO)dCdC(Ph)CH2CHdCH2]Br (2) (0.57 g, 81% yield). Spectroscopic data for 2: IR (cm-1, CH2Cl2) 1931 (s, νCO); 1H NMR (CDCl 3) δ 7.72-6.82 (m, 25H, Ph), 5.62 (m, JH-H) 17.1, 10.1, 6.3, 6.0 Hz, 1H, dCH); 5.20 (s, 5H, Cp), 5.00 (m, JH-H) 17.1, 1.5 Hz, 1H, dCH), 4.96 (m, JH-H) 10.1, 1.5 Hz, 1H, dCH), 3.44, 3.22 (m, 2H, PCH2), 2.87 (ddd, JH-H) 15.6, 6.3 Hz, JH-P) 2.0 Hz, 1H of CH2), 2.78 (ddd, JH-H) 15.6, 6.0 Hz, JP-H) 2.8 Hz, 1H of CH2), 2.42 (m, 2H, PCH2);13C NMR (CDCl3) δ 358.4 (dd, JC-P) 36.0, 6.5 Hz, CR), 227.4 (d, JC-P) 21.8 Hz, CO), 135.0 (CH), 134.3-126.5 (Ph), 116.8 (dCH2), 95.7 (Cp), 33.9 (CH2), 30.1 (dd, JC-P) 27.1, 13.1 Hz, PCH2), 28.9 (dd, JC-P) 28.5, 12.3 Hz, PCH2);31P NMR (CDCl3) δ 71.5 (d, JP-P) 37.5 Hz), 67.0 (d, JP-P) 37.5 Hz); MS (FAB, m/z, Mo98) 731 (M+), 703 (M+- CO). Anal. Calcd for C43H39OP2MoBr: C, 63.79; H, 4.86. Found: C, 64.01; H, 4.97.
Reaction of 1 with Benzyl Bromide. A mixture of 1 (0.30 g, 0.43 mmol) and BrCH2C6H5 (0.11 mL, 0.84 mmol) was dissolved in 40 mL of CHCl3at room temperature, and the resulting mixture was stirred for 36 h in the dark. The color of the reaction mixture changed from yellow to red. After removal of the solvent in vacuo, 10 mL of CH2Cl2was added to the residue under nitrogen, and the extract was filtered. The solvent of the filtrate was reduced in volume to ca. 5 mL. The solution was added dropwise to a stirred Et2O solution to give a brown precipitate. The solid was collected by filtration and washed with 5 mL of Et2O under nitrogen. The solid was dried under vacuum to afford the brown product [[Mo](CO)d CdC(Ph)CH2Ph]Br (3) (0.27 g, 73% yield). Spectroscopic data for 3: IR (cm-1, CH2Cl2) 1931 (s, νCO);1H NMR (CDCl3) δ 7.98-6.73 (m, 30H, Ph), 5.09 (s, 5H, Cp), 3.42 (dd, JH-H) 14.5 Hz, JH-P) 2.0 Hz, 1 H on CH2Ph), 3.27 (dd, JH-H) 14.5 Hz, JH-P ) 3.7 Hz, 1 H on CH2Ph), 3.20 (m, 2H, CH2), 2.38 (m, 2H, CH2); 13C NMR (CDCl 3) δ 356.7 (dd, JC-P) 37.8 Hz, 5.6 Hz, CR), 227.8 (d, JC-P) 20.6 Hz, CO), 134.1-126.8 (Ph), 95.9 (Cp), 36.0 (CH2Ph), 30.3 (dd, JC-P) 28.3, 13.6 Hz, PCH2), 28.5 (dd, JC-P) 28.7, 11.4 Hz, PCH2);31P NMR (CDCl3) δ 69.0 (d, JP-P ) 36.3 Hz), 66.2 (d, JP-P) 36.3 Hz); MS (FAB, m/z, Mo98) 781 (M+), 753 (M+ - CO). Anal. Calcd for C
47H41OP2MoBr: C, 65.67; H, 4.81. Found: C, 65.60; H, 4.99.
Reaction of 1 with Bromoacetonitrile. To a sample of 1 (0.61 g, 0.87 mmol) dissolved in 40 mL of CHCl3 at room temperature was added excess BrCH2CN (0.1 mL, 1.44 mmol) via a syringe. The resulting mixture was stirred for 24 h in the dark. The color of the reaction mixture changed from yellow to brown within 12 h. After removal of all volatile substances in vacuo, 10 mL of CH2Cl2was added to the residue
under nitrogen, and the extract was filtered. The solvent of the filtrate was reduced in volume under vacuum to ca. 5 mL. The solution was added dropwise to a stirred Et2O solution to give brown precipitate. The solid was collected by filtration and washed with Et2O under nitrogen. The solid was dried under vacuum to afford [[Mo](CO)dCdC(Ph)CH2CN]Br (4) (0.52 g, 74% yield). Spectroscopic data for 4: IR (cm-1, CH2 -Cl2) 1977 (s, νCO);1H NMR (CDCl3) δ 7.74-6.77 (m, 25H, Ph), 5.21 (s, 5H, Cp), 3.58, 3.29, 2.52, 2.33 (m, 4H, PCH2CH2P), 2.95 (m, JH-H) 18.2 Hz, JH-P) 5.2 Hz, 1H of CH2CN), 2.84 (m, JH-H) 18.2 Hz, JH-P) 5.5 Hz, 1H of CH2CN);31P NMR (CDCl3) δ 66.8, 63.7 (2d, JP-P) 30.7 Hz); MS (FAB, m/z) 730 (M+), 702 (M+ - CO). Anal. Calcd for C42H36NOBrP2Mo (808.5): C, 62.39; H, 4.49; N, 1.73. Found: C, 62.17; H, 4.68; N, 1.76.
Reaction of 2 with NaOMe in Methanol. To a sample of 2 (0.33 g, 0.41 mmol) dissolved in 10 mL of methanol at room temperature was added a methanol solution of sodium meth-oxide (0.15 g, 2.78 mmol) via a cannula. An orange powder precipitated after 1 h, and the resulting solution was stirred for 4 h as the color of the reaction mixture changed from red to yellow. The product was collected by filtration and washed with 20 mL of methanol under nitrogen. The powder was dried under vacuum to afford the product [Mo][η3-C(CO
2 Me)dC(Ph)-CH2CHdCH2] (5) (0.25 g, 80% yield). The powder was further recrystallized from a mixture of hexane/CH2Cl2to afford yellow crystals of 5 for X-ray diffraction analysis. Spectroscopic data for 5: IR (cm-1, CH2Cl2) 1673 (s, νCO);1H NMR (C6D6) δ 8.25-6.71 (m, 25H, Ph), 4.71 (s, 5H, Cp), 3.66 (d, JH-H) 17.2 Hz, 1H, dCH2), 3.41 (s, 3H, OCH3), 2.82 (br, 1H, PCH2), 2.58 (br, 1H, dCH2), 2.52 (br, 1H, PCH2), 2.35 (br, 1H, PCH2), 2.02 (br, 1H, PCH2), 1.97 (br, 1H, CH2), 1.78 (br, 1H, dCH), 1.70 (br, 1H, CH2);13C NMR (C6D6) δ 180.6 (CO2), 144.4-124.7 (Ph), 87.6 (Cp), 60.3 (dd, JC-P) 5.4, 4.0 Hz, dCH), 49.4 (OCH3), 47.6 (d, JC-P) 5.9 Hz, dCH2), 34.6 (CH2), 29.1 (dd, JC-P) 29.9 Hz, JC-P) 15.0 Hz, PCH2), 27.9 (dd, JC-P) 25.8, 14.4 Hz, PCH2);31P NMR (C6D6) δ 90.5 (s), 71.2 (s); MS (FAB, m/z, Mo98) 762 (M+
). Anal. Calcd for C44H42O2P2Mo: C, 69.47; H, 5.57. Found: C, 69.32; H, 5.79.
Reaction of 3 with NaOMe in Methanol. A mixture of 3 (0.40 g, 0.46 mmol) and sodium methoxide (0.20 g, 3.70 mmol) was dissolved in 30 mL of methanol at room temperature. The solution was stirred for 1 h, and a red product precipitated. The product was collected by filtration, washed with methanol under nitrogen, and dried under vacuum to afford the red compound [Mo](η3-CH(CO
2CH3)C(Ph)CHPh) (6) (0.31 g, 83% yield). The solid was further recrystallized from a mixture of Et2O/CH2Cl2 to afford red crystals for diffraction analysis. Spectroscopic data for 6: IR (cm-1, CH2Cl2) 1674 (s, νCO);1H NMR (CD2Cl2) δ 7.98-6.14 (m, 30H, Ph), 4.62 (s, 1H, syn H), 3.88 (d, JH-P) 11.4 Hz, 1H, anti H), 3.73 (s, 5H, Cp), 3.71 (s, 3H, OCH3), 2.68 (m, 1H, PCH2), 2.53 (m, 1H, PCH2), 2.47 (m, 1H, PCH2), 1.90 (m, 1H, PCH2);13C NMR (CD2Cl2) δ 183.2 (d, JC-P) 3.5 Hz, CO2), 149.0-125.0 (Ph), 92.4 (Cp), 90.3 (t, JC-P ) 3.1 Hz, C(Ph)), 50.2 (OCH3), 49.2 (d, JC-P) 2.7 Hz, CHPh), 48.7 (d, JC-P) 4.5 Hz, CHCO2CH3), 33.7 (dd, JC-P) 28.1, 14.3 Hz, PCH2), 24.2 (dd, JC-P) 23.1, 12.9 Hz, PCH2);31P NMR (CD2Cl2) δ 71.3 (d, JP-P) 36.8 Hz), 72.2 (d, JP-P) 36.8 Hz); MS (FAB, m/z, Mo98) 812 (M+
). Anal. Calcd for C48H44O2P2 -Mo: C, 71.11; H, 5.47. Found: C, 71.06; H, 5.59.
Deprotonation of [Cp(dppe)(CO)ModCdC(Ph)CH2 CN]-Br by NaOMe. To a sample of 4 (0.11 g, 0.14 mmol) dissolved in 15 mL of MeOH at room temperature was added 5 mL of a MeOH solution of sodium methoxide (0.05 g, 1.9 mmol) via cannula. The color of the solution changed from red to green immediately. After removal of the solvent in vacuo, 2 mL of CH2Cl2and 20 mL of hexane were added to the residue under nitrogen, and the extract was filtered. The solvent of the filtrate was removed under vacuum to afford a green residue. The residue was redissolved in 2 mL of CH2Cl2. The solution was added dropwise to a stirred Et2O solution to give green
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
precipitates. The solid was collected by filtration, washed with Et2O under nitrogen, and dried under vacuum to afford a green solid, Cp(dppe)(CO)Mo-CdC(Ph)CHCN (7) (0.052 g, 53% yield). Spectroscopic data for 7: IR (cm-1, CH
2Cl2) 1846 (s, νCO); 1H NMR (CDCl
3) δ 7.81-6.91 (m, 25H, Ph), 4.78 (s, 5H, Cp), 3.73, 3.06 (2m, 4H, PCH2CH2P), 2.51 (s, 1H, CHCN);31P NMR (CDCl3) δ 105.4 (s), 103.9 (s); MS (FAB, m/z) 729 (M+), 701 (M+- CO). Anal. Calcd for C
42H35NOP2Mo: C, 69.33; H, 4.85; N, 1.93. Found: C, 69.97; H, 5.35, N, 2.64.
X-ray Structure Determination of 5 and 6. Yellow crystals of 5 suitable for X-ray diffraction study were grown directly from a mixture of hexane/CH2Cl2. A single crystal of dimensions 0.30× 0.35 × 0.40 mm3was glued to a glass fiber and mounted on an Nonius CAD-4 diffractometer. Data were collected and processed, and crystallographic computations were carried out using the NRCC structure determination
package.12 The structure was solved using direct methods13 and was refined on intensities of 4949 reflections to give R ) 0.034, Rw ) 0.037 (I > 2σ(I)). For 6, a single crystal of dimensions 0.20× 0.25 × 0.35 mm3was mounted on a glass fiber with epoxy. Data were collected at room temperature on a Siemens SMART CCD area detector system employing a 3 kW sealed tube X-ray source operating at 1.5 kW. The total data collection was approximately 6 h, yielding 7071 indepen-dent data after integration using SAINT.14Unit cell param-eters were determined from the least-squares refinement of three-dimensional centroids of unique reflections. Data were corrected for absorption with the SADABS program.15 The space group was assigned as P1h, and the structure was solved and refined using direct methods included in the SHELXTL package.16In the final model, non-hydrogen atoms were refined anisotropically, with hydrogen atoms included in idealized locations. The structure was refined to R1 ) 0.0411 and wR2 ) 0.1386 for I > 2σ(I) and to R1 ) 0.0451 and wR2 ) 0.1434 for all data.17Crystal and intensity collection data for 5 and 6 are given in Table 3, and fractional coordinates and thermal parameters are given in the Supporting Information.
Acknowledgment. Financial support from the
Na-tional Science Council, Taiwan, is gratefully
acknowl-edged.
Supporting Information Available: Details of the struc-tural determination for complexes 5 and 6, including crystal and intensity collection data, positional and anisotropic ther-mal parameters, and all of the bond distances and angles. This material is available free of charge via the Internet at http://pubs.acs.org.
OM9907167
(12) Gabe, E. J.; Lee, F. L.; Lepage, Y. In Crystallographic
Comput-ing 3; Sheldrick, G. M., Kruger, C., Goddard, R., Eds.; Clarendon
Press: Oxford, England, 1985; p 167.
(13) Sheldrick, G. M. SHELXS-86, Program for Crystal Structure
Solution; University of Gottingen: Gottingen, Germany, 1986
(14) SAINT (Siemens Area Detector Integration) program; Siemens Analytical X-ray: Madison, WI, 1995.
(15) The SADABS program is based on the method of Blessing; see: Blessing, R. H. Acta Crystallogr., Sect. A 1995, 51, 33.
(16) SHELXTL: Structure Analysis Program, version 5.04; Siemens Industrial Automation Inc.: Madison, WI, 1995.
(17) GOF ) [∑[w(F2
o- F2c)2]/(n - p)]1/2, where n and p denote the number of data and parameters. R1 ) (∑||Fo| - |Fc||)/∑|Fo|, wR2 ) [∑-[w(F2
o- F2c)2]/∑[w(F2o)2]]1/2, where w ) 1/[σ2(F2o) + (aP)2+ bP] and P ) [(max; 0, F2o) + 2F2c]/3.
Table 3. Crystal and Intensity Collection Data for Cp(dppe)MoC(COOMe)dC(Ph)CH2CdCH2(5) and Cp(dppe)MoCH(COOMe)C(Ph)CHPh (6) 5 6 mol formula C45H44O2P2Cl2Mo C48H44O2P2Mo mol wt 845.63 810.71 space group P21/c P1h a, Å 17.998(5) 9.6178(1) b, Å 8.647(3) 10.8305(2) c, Å 26.080(5) 21.3254(3) R, deg 96.911(1) β, deg 100.30(2) 94.388(1) γ, deg 113.258(1) V, Å3 3993.6(19) 2007.22(5) Z 4 2 cryst dimens, mm3 0.30× 0.35 × 0.40 0.20 × 0.25 × 0.35 Mo KR radiation: γ, Å 0.71073
θ range for data
collection 0.55-25.0 0.97-25.0 limiting indices (h, k, l) -21, 21; 0, 10; 0, 30 -12, 12; -14, 14; -28, 28
no. of reflns collected 7015 16793
no. of ind reflns 4949 7071
max. and min. transmn 0.868 and 0.819 0.492/0.408 refinement method full-matrix least-squares on F2
no. of data/restraints/ params 4949/0/470 7071/0/479 GOF 1.55 1.196 final R indices [I > 2σ(I)] 0.034/0.037 0.0411/0.1386 all data 0.0451/0.1434
∆F (in final map), e/Å -0.470, +0.490 -0.846, +0.507
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009