利用生物資訊方法偵測程序性核糖體移碼之研究
47
0
0
全文
(2) 利用生物資訊方法偵測程序性核糖體移碼之研究 A Bioinformatics Approach of Predicting Programmed Ribosomal Frameshifting. Student:Chia-Jung Wu. 研 究 生:吳家榮 指導教授:盧錦隆. 教授. Advisor:Prof. Chin Lung Lu. 國 立 交 通 大 學 生 物 資 訊 所 碩 士 論 文. A Thesis Submitted to Institute of Bioinformatics College of Biological Science and Technology National Chiao Tung University in partial Fulfillment of the Requirements for the Degree of Master in Biological Science and Technology June 2007 Hsinchu, Taiwan, Republic of China. 中華民國九十六年六月.
(3) 中 文 摘 要 程序性核糖體移碼是一種重新編碼的機制, 藉由這個機制核糖體會在某個特定位置上, 從原 本的零讀碼框切換到 −1 或 +1 的讀碼框 (其中切換到 −1 的讀碼框最為常見), 然後在新 的讀碼框中繼續蛋白質的轉譯。 此機制會導致另一個蛋白質的表現, 而這個蛋白質與未發生 程序性核糖體移碼時所產生的蛋白質不同。 時至今日, 許多生物體, 包含病毒、 細菌和真核 生物等, 已被發現利用程序性核糖體移碼機制來增加其基因表現的多樣性, 或是進行其基因 的調控。 此外, 也有文獻指出, 對利用此機制的病毒, 即便僅僅改變很小的移碼效率就可抑制 其繁殖。 這意味著程序性核糖體移碼位置的發現與辨識, 在為抗病毒藥劑找尋新標的的研究 上, 可能扮演著重要的角色。 在這篇論文中, 我們利用序列模式辨認的方法, 輔之以結構與功 能生物資訊學, 設計出一套更有效的演算法, 可以偵測出基因序列中會發生程序性核糖體移 碼的位置。 根據這個演算法, 我們也已實作出一個名為 PRooF (Programmed Ribosomal Frameshifting 的簡稱) 的網路伺服器, 可供生物學家做線上的分析。 PRooF 的正確性也已. 經過了許多含有一或二個程序性核糖體移碼的基因序列的測試, 而且測試結果也與用現存 最新的工具所得的結果做了比較。 比較結果指出 PRooF 在偵測的敏感度上的確有著大幅度 的改進。 特別的是, PRooF 所偵測出在移碼位置後方的 RNA 二級結構大多數為 H-type pseudoknots 和 bulged helixes。 相較於 simple stem-loops, 以上兩種結構被廣泛認為更能. 促使核糖體移碼的發生。. i.
(4) Abstract Programmed ribosomal frameshifting (PRF) is a recoding mechanism by which the translational ribosome switches from the initial (zero) reading frame to one of the two alternative reading frame (either mostly −1 or +1) at a specific position and continues its translation in the new frame. As a result, the recoding of PRF leads to an expression of an alternative protein, which is different from that produced by standard translation. To date, many organisms, including viruses, bacteria and eukaryotes, have been found to utilize the PRF mechanism for increasing the diversity of gene expression or for gene regulation. In addition, it has been reported that, for viruses that use PRF, even small alterations in their frameshifting efficiencies can inhibit viral propagation, suggesting that the PRF discovery and identification may play a crucial role in identifying new targets for antiviral agents. In this thesis, using a pattern recognition approach aided by structural and functional bioinformatics, we have designed a more effective algorithm to detect −1 and +1 PRF sites in a genomic sequence. This algorithm has also been implemented as a web server, called PRooF (short for Programmed Ribosomal Frameshifting), for online analysis. The accuracy of PRooF was tested with several genomic sequences, each of which has already been known to carry one or two PRF sites, by comparing its testing results with those obtained by the latest existing program. Consequently, the experimental results show that PRooF indeed greatly improves detection sensitivity. In particular, most of the predicted stimulatory RNA structures downstream of slippery sites are H-type pseudoknots and bulged helixes, both of which are widely believed to promote the ribosomal frameshifting events more efficiently than simple stem-loops.. ii.
(5) 誌 謝 感謝我的指導教授盧錦隆老師的耐心指導, 讓我學到許多做研究應有的態度與方法, 並且在 我研究上遭遇瓶頸時, 不厭其煩地提供協助。 也感謝時常鼓勵我的學姊、 同學、 學弟及朋友 們。 最後要感謝家人的栽培與支持, 讓我得以無憂無慮地完成碩士學位。. iii.
(6) Contents. Chinese Abstract. i. Abstract. ii. Acknowledgement. iii. 1 Introduction. 1. 2 Methods. 6. 3 Implementation of PRooF. 10. 3.1. PRooF . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 10. 3.2. Usage of PRooF . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 10. 3.2.1. Input of PRooF . . . . . . . . . . . . . . . . . . . . . . . . . . .. 10. 3.2.2. Output of PRooF . . . . . . . . . . . . . . . . . . . . . . . . . .. 14. 4 Results and Discussion. 17. 5 Conclusions. 30. References. 32. iv.
(7) List of Figures 2.1. The approach pipeline of identifying −1 and +1 PRF sites. . . . . . . .. 7. 3.1. The web interface of PRooF. . . . . . . . . . . . . . . . . . . . . . . . .. 12. 3.2. An output of a detected PRF site of longer protein product. . . . . . .. 16. 3.3. An output of a detected −1 PRF site of shorter protein product. . . . .. 16. v.
(8) List of Tables 4.1. The tested sequences and their −1 PRF numbers . . . . . . . . . . . .. 18. 4.2. The tested sequences and their +1 PRF numbers . . . . . . . . . . . .. 19. 4.3. Summary of the PRooF results for predicting the −1 PRFs of longer product on several sequences from PseudoBase using the slippery sequence X XXY YYZ . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 4.4. 21. Summary of the PRooF results for predicting the −1 PRFs of longer product on several sequences from RECODE using the slippery sequence X XXY YYZ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 4.5. 22. Summary of the PRooF results for predicting the −1 PRFs of longer product on several sequences from PseudoBase and RECODE using the slippery sequence Y YYZ . . . . . . . . . . . . . . . . . . . . . . . . . .. 4.6. 24. Summary of the PRooF results for predicting the −1 PRFs of shorter product on several sequences from RECODE using the slippery sequence X XXY YYZ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 4.7. 25. Summary of the PRooF results for predicting the +1 PRFs on several sequences from RECODE with using the slippery sequence CUU URA C and without detecting downstream RNA structure . . . . . . . . . .. 4.8. 26. Summary of the PRooF results for predicting the +1 PRFs on several sequences from RECODE using the slippery sequence UUU UGA or YCC UGA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . vi. 27.
(9) 4.9. The average sensitivity and specificity of −1 and +1 PRF prediction using PRooF and FSFinder2 . . . . . . . . . . . . . . . . . . . . . . . .. vii. 28.
(10) Chapter 1 Introduction Programmed ribosomal frameshifting (PRF) is a recoding mechanism by which the translational ribosome switches from the initial (zero) reading frame to one of the two alternative reading frame (either −1 or +1) at a specific position and continues its translation in the new frame [1–5]. As a result, the recoding of PRF leads to an expression of an alternative protein, which is different from that produced by standard translation. To date, many organisms, including viruses, bacteria and eukaryotes, have been found to utilize the PRF mechanism for increasing the diversity of gene expression or for gene regulation [4, 6–14]. In addition, it has been reported that, for viruses that use PRF, even small alterations in their frameshifting efficiencies can inhibit viral propagation, which suggests that the PRF sites in viruses may present a potential target for antiviral therapeutics [15, 16]. The PRFs in the −1 direction (−1 PRFs) are the most extensively characterized ones that have often been observed in many RNA viruses and transposons, as well as a few cellular genes. Typically, two cis-acting mRNA signals are critical for −1 PRF to occur. One is a slippery sequence where the −1 PRF event takes place, and the other is a 3’-stimulatory RNA structure, which is separated from the slippery sequence by a short spacer region. The slippery sequence on the mRNA usually is a heptanucleotide 1.
(11) of the general form X XXY YYZ, where the spaces separate codons in the zero frame, with X and Z being any nucleotide and Y being mostly A or U. In some reported cases, however, the slippery sequence can only be presented as Y YYZ, instead of X XXY YYZ. On the other hand, the existence of a 3’-stimulatory RNA structure is important for an efficient −1 PRF, since it forces elongating ribosome to pause over the slippery site such that the ribosome can have a chance to switch from the zero reading frame (X XXY YYZ) to the −1 reading frame (XXX YYY) and then continues its translation in the new frame. The stimulatory RNA structure can be a simple stem-loop in some instances, such as E. coli dnaX [7]. In most cases, however, it is a classical (H-type) pseudoknot, a stem-loop with downstream sequence paired back to the loop [17–21]. A number of studies have indicated that H-type pseudoknots can promote −1 PRF more efficiently than simple stem-loop structures, because a stable stimulatory RNA structure is essential in order to give an efficient −1 PRF and typically H-type pseudoknots are more stable [22, 23]. In fact, the stem-loop previously suspected for −1 PRF in HIV-1 was shown to be a more complex RNA structure, possibly a two-stem structure [24] or a triple-helix one which virtually is a special kind of H-type pseudoknot with an additional stem in its long loop [25]. Recently, Gaudin et al. [26] found the NMR structure of the HIV-1 frameshifting RNA signal to be a long hairpin with an internal 3 nt bulge, which agrees with the structure proposed by Dulude et al. [24] using structure probing and mutagenesis methods. For convenience, this kind of long hairpin with an internal bulge is simply referred to as a bulged helix here. Interestingly, the internal bulge of the bulged helix introduces a bend between the upper and lower helical regions, which is a structural feature often observed in H-type pseudoknots. It has been suggested that the bend conformation of a stimulatory RNA structure is required for an efficient frameshifting to occur, because it causes a specific interaction and recognition between the stimulatory RNA structure and the ribosome [27]. In 2.
(12) other words, all these findings seem to imply that an H-type pseudoknot or a bulged helix is required as an efficient stimulator of −1 PRF. Nevertheless, in addition to the two cis-acting signals as mentioned above, the spacer between the slippery sequence and the stimulatory RNA structure is also essential for −1 PRF to occur, because presumably its length alters the location of the paused ribosome and hence influences its shifting probability [28]. Moreover, for some bacteria (such as E. coli ), an internal Shine-Dalgarno (SD)-like sequence often can be found upstream of the −1 PRF site [29]. It was reported that the sites +1 PRFs occur less commonly than those of −1 PRFs, although they have been described in some organisms, such as bacteria, yeast and mammals [2]. The most widespread known cellular genes to utilize the recoding of +1 PRF are those prfB genes encoding polypeptide chain release factor 2 (RF2) in E. coli [11] and those ornithine decarboxylase antizyme (oaz ) genes encoding antizyme 1 in mammals [9, 10]. The slippery sequences most commonly found in the prfB and oaz genes are CUU URA C and UUU UGA or YCC UGA, respectively, where R is A or G and Y is C or U. In addition, not all +1 PRF sites, such as in the bacterial prfB genes, have a downstream RNA structure to serve as the frameshifting stimulator. As with similar to the −1 PRF sites in bacteria, however, an upstream SD-like sequence stimulates the efficiency of +1 PRF in the bacterial prfB genes. As mentioned above, the PRF event occurs at a slippery site and causes elongating ribosome to switch from the zero reading frame to the −1 or +1 reading frame. One (the most frequently observed so far) of two consequences is that the PRF event occurs near the end of the zero reading frame and the ribosome switches to translate the new reading frame by extending beyond the terminator of the zero reading frame. As a result, the protein products of such PRFs are longer than those by standard translation. The other consequence is that the PRF event takes place within the zero reading frame 3.
(13) and the ribosome then slips backwards (or forwards) and terminates quickly, because it reaches a stop codon in the new reading frame near the slippery site. Consequently, their protein products are shorter and lack carboxyl-terminal domains as compared to those of the standard translation [3,14]. This notably occurs in a few cases of −1 PRFs as in E. coli dnaX gene [7]. Based on the model described above, several computational approaches, such as pattern recognition [30, 31], statistical analysis [32], machine learning [33] and hidden Markov models [34, 35], have been proposed for prediction of −1 and +1 PRFs in a given genomic sequence. Unfortunately, most of them usually gave too many candidates of false positive in their predictions and even failed to identify the candidates of true positive for some sequences. The cause for the former can be that the adopted model is incomplete (e.g., the 3’-stimulatory RNA structures were not taken into account) or too broad (e.g., the considered stem-loops or pseudoknots were structurally too simple). The reason for the latter can be that the adopted model is too restrained. For example, the parameter settings for slippery sequence pattern and spacer length were too rigid, or only H-type pseudoknots were regarded as 3’-stimulatory RNA structures. In particular, the methods by which they predicted the stem-loops and/or pseudoknots lead to a result that the obtained RNA structures are generally not stable enough to function efficiently as a stimulator of ribosomal frameshifting, which is the common weakness for most programs. In this thesis, we design an algorithm to more accurately detect −1 and +1 PRF sites in a genomic sequence using pattern recognition aided with both structural and functional bioinformatics. We first search for all partially overlapping open reading frames (ORFs) in the given sequence and use the method of pattern recognition to identify all possible slippery sites in the overlapping regions. An approach of functional bioinformatics is then employed to determine whether or not some or all of the 4.
(14) ORFs involved in each possible frameshifting carry a functional protein motif/domain in expressed form. Finally, an efficient heuristic method of structural bioinformatics we developed before [36] is adopted to predict a more accurate and more stable RNA structure downstream of each possible slippery site, where the predicted RNA structures here can be H-type pseudoknots, bulged helixes or simple stem-loops. In addition, we have implemented this algorithm as a web server, called PRooF (short for Programmed Ribosomal Frameshifting) [37], that is open to the public for online analysis. To validate its accuracy, PRooF was tested on several RNA sequences, each of which has already been known to carry one or two −1 or +1 PRF sites, and its testing results were also compared with those obtained by the latest program FSFinder2 (the successor of FSFinder) that was developed by Moon et al. [31, 38]. Consequently, the experimental results reveal that PRooF has high sensitivity and specificity when compared with FSFinder2. Moreover, most of the predicted stimulatory RNA structures downstream of −1 and +1 PRF sites are H-type pseudoknots and bulged helixes, both of which are widely believed to be the stimulators that can promote the PRF events more efficiently than simple stem-loops. The rest of this thesis is organized as follows. In Chapter 2, we describe our method and our implemented program, called PRooF, for detecting −1 and +1 PRF sites more accurately. In Chapter 3, we introduce the PRooF implementation and user interface. In Chapter 4, we demonstrate the applicability of our developed program by testing them on a data set of genomic sequences. Finally, we make some conclusions in Chapter 5.. 5.
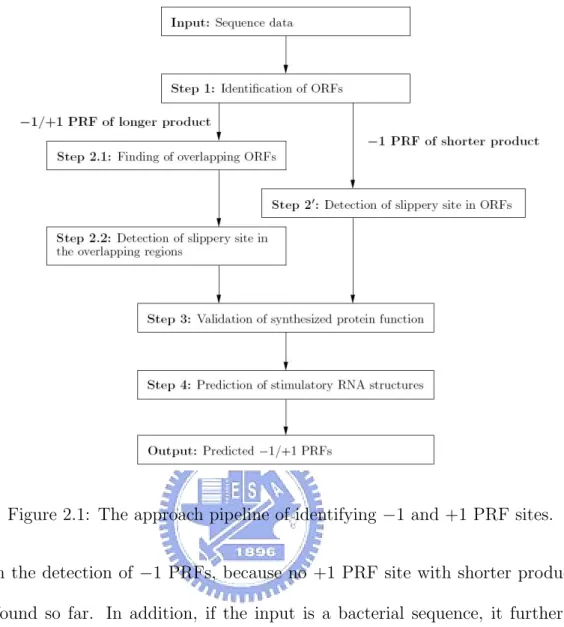
(15) Chapter 2 Methods Figure 2.1 illustrates an approach pipeline of our strategy for predicting −1 and +1 PRF sites in a given genomic sequence. In the first step, all ORFs above a threshold size are identified from an input sequence. By following the convention adopted by Moon et al. [31], the start position of each identified ORF was extended to upstream stop codon from its original start codon. In the second step, two different pathways are designed to deal with all the identified ORFs, primarily depending on the type of PRF protein product. For the PRFs with longer products, the second step first finds all pairs of the partially overlapping ORFs (the zero reading frame as the first and the −1 or +1 reading frame as the second). The second step, then, detects all possible slippery sites in the overlapping regions such that their slippery sequences conform to the default patterns (such as X XXY YYZ or Y YYZ for −1 PRFs, and CUU URA C, UUU UGA or YCC UGA for +1 PRFs) or user-defined patterns. This approach is, however, not suitable or effective for the cases of shorter product, because their second reading frames are either small or non-applicable. Instead, the second step simply searches each identified ORF for its possible slippery sites that possess the required slippery sequences. Notice that the alternative step above for dealing with shorter protein products was implemented 6.
(16) Figure 2.1: The approach pipeline of identifying −1 and +1 PRF sites. only in the detection of −1 PRFs, because no +1 PRF site with shorter product has been found so far. In addition, if the input is a bacterial sequence, it further looks for an internal SD-like sequence about 8–14 nt (for −1 PRF) or 3–5 nt (for +1 PRF) upstream of each slippery site, which can be helpful to reduce the number of false positives. Notice that the distance between the SD-like sequence and the +1 PRF site is usually shorter than that typically seen between the SD-like sequence and the −1 PRF site [2]. The SD-like sequence can be AGGA, AGGG, GAGG, GGAG, GGGA or GGGG in default or any user-defined sequences. The default SD-like sequences used in PRooF are the 4-letter substrings of the widely accepted SD sequences in the standard translation, as well as some variants. The reason is that, for example, the consensus of the SD sequences in E. coli ’s translation is AGGAGG [40]. However, we were able to only find a variant of its substring, which is AGGG, upstream from the slippery site 7.
(17) in E. coli. Next, all ORFs suspected to be involved in frameshifting are further verified in the third step to examine if they bear the potential protein motifs/domains already registered in the InterPro database [41]. InterPro is an integrated documentation resource of protein families, domains and functional sites. The verification procedure proceeds as follows. (1) For the cases of longer product, each of two overlapping ORFs is translated into a protein sequence, which is then examined by InterProScan [42] to look for any existing protein motif/domain registered in InterPro database. InterProScan is a tool that combines various protein signature recognition methods. If InterProScan finds no motif/domain in a translated protein sequence, the corresponding slippery site is not regarded as a candidate associated with PRF. In other words, only those slippery sites whose involved ORFs possess known protein motifs/domains are considered as PRF candidates. (2) For those of shorter product, the full-length ORF is cut into two fragments at the slippery site, 5’-end fragment (left to the slippery site) and 3’-end fragment (right to the slippery site). These two fragments are then translated into protein sequences and are further examined by InterProScan for presence of possible protein motifs/domains. If the motifs/domains exist in both fragments (as defaulted in −1 PRF detection) or in the 3’-end fragment (as defaulted in +1 PRF detection), the corresponding slippery site is then considered as a PRF candidate. Subsequently, by above procedures all the PRF candidates found in this step are passed on to the fourth step for the prediction of their 3’-stimulatory RNA structures. The third step is, in fact, optional in PRooF, because in some cases (e.g., RCD114 and RCD252), as demonstrated in the Results and Discussion section, the motifs/domains of the translated protein sequences may have not yet been registered in the InterPro database. As mentioned before, most of the stimulatory RNA structures currently known are H-type pseudoknots and only a few are bulged helixes or just simple stem-loops. 8.
(18) Recently, we have developed a heuristic approach [36] for efficiently and effectively detecting the H-type pseudoknots in a given RNA sequence by incorporating several existing tools, including RNAMotif [43], PKNOTS [44], NUPACK [45] and pknotsRG [46]. RNAMotif is an RNA structural motif search tool, and PKNOTS, NUPACK and pknotsRG are currently existing tools that can be used to predict RNA secondary structures of minimum free energy and with H-type pseudoknots. Hence, in the fourth step, this heuristic approach is utilized to detect the 3’-stimulatory H-type pseudoknot for the sequence fragment downstream of the slippery site of each PRF candidate. A stable H-type pseudoknot, if found, is taken as the stimulatory RNA structure of the PRF candidate. Otherwise, PRooF continues to use RNAMotif to search the sequence fragment for all possible bulged helixes that conform to a predefined descriptor of bulged helix, and then choose the one with the minimum free energy as the stimulatory RNA structure. If neither a stable H-type pseudoknot nor a bulged helix is found by above procedures, RNAMotif is then used to search for simple stem-loops and designate the one with the minimum free energy as the stimulatory RNA structure. Notice that this step is optional in PRooF, because the +1 PRF sites in some sequences, such as the prfB genes, have no downstream RNA structure as stimulators. Finally, all the qualified −1 or +1 PRF candidates predicted by our algorithm are output along with their predicted 3’-stimulatory RNA structures with corresponding free energies, motifs/domains detected in the protein products, spacer lengths and SD-like sequences.. 9.
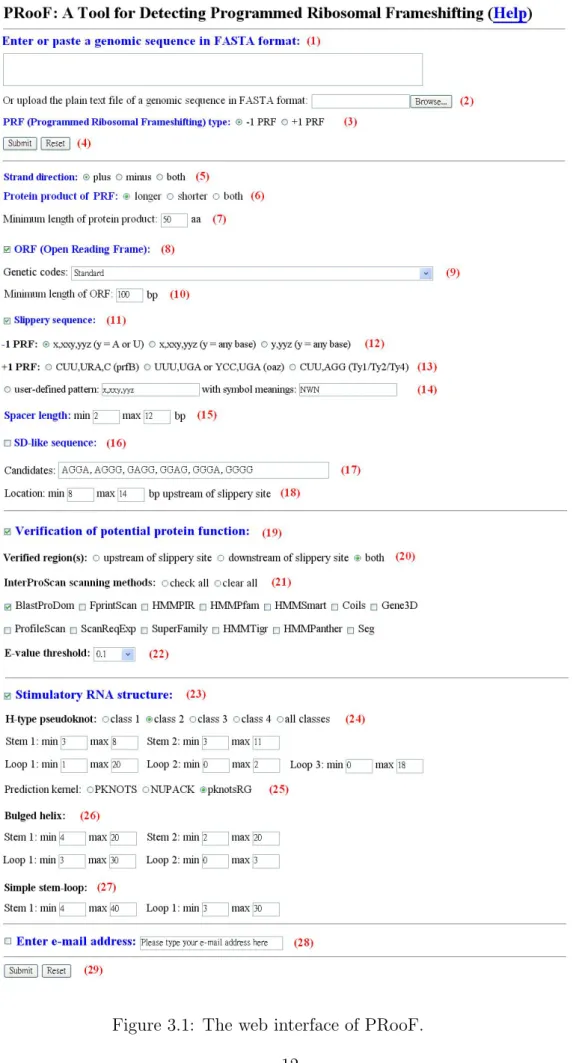
(19) Chapter 3 Implementation of PRooF In this chapter, we shall introduce the implementation of PRooF, as well as its web interface, and then describe how to use it in details.. 3.1. PRooF. The kernal of PRooF (short for Programmed Ribosomal Frameshifting) was implemented by C and its web interface by PHP and HTML. PRooF (http://bioalgorithm.life.nctu.edu.tw/PROOF/) can be easily accessed via a simple web interface (see Figure 3.1).. 3.2. Usage of PRooF. In this section, we shall describe the usage of PRooF step by step and the output of PRooF.. 3.2.1. Input of PRooF. 1. Enter or paste a genomic sequence in FASTA format (1), or simply upload a plain text file of a genomic sequence in FASTA format (2).. 10.
(20) 2. Choose the type of PRF (3), which can be either −1 PRF (default) or +1 PRF. 3. Just click ”Submit” button (4) if users would like to run PRooF with default parameters; otherwise, users continue with the following parameter settings. 4. Choose the strand direction of input sequence (5), which can be either plus (default), minus or both. 5. If the type of PRF is −1, then choose the type of protein product (6), which can be either longer (default), shorter or both. Note that we consider only longer product here if the type of PRF is +1. If needed, further specify the ”minimum length of protein product” (whose default is 50 aa/amino acids) (7), so that only those PRF candidates whose lengths of their protein products are above this threshold will be further considered. 6. Determine whether to search for ORFs in the input sequence (8) or not. If so (default), users need to further select a genetic code (whose default is the standard code) (9) and also specify the ”minimum length” (whose default is 100 bp in −1 PRF and 30 bp in +1 PRF) (10) to identify all ORFs above this threshold length. 7. Determine whether to search for slippery sequences in the input sequence (11) or not. If so (default), users can simply choose one of the pre-defined slippery sequences, which can be ”X,XXY,YYZ with Y being A or U” (default), ”X,XXY,YYZ with Y being any base”, or ”Y,YYZ with Y being any base” for the detection of −1 PRF (12) and ”CUU,URA,C” (default), ”UUU,UGA or YCC,UGA”, or ”CUU,AGG” for the detection of +1 PRF (13). Alternatively, users can choose ”user-defined pattern” (14) by defining their own slippery sequences. (Note that commas in slippery sequence separate codons in the zero frame.) For example, if users would like to define an heptanucleotide as the slip11.
(21) Figure 3.1: The web interface of PRooF. 12.
(22) pery sequence in the form of XXY,YYY,Z with X being any base, Y being A or U, and Z being A, C or U, they can first enter ”XXY,YYY,Z” in the field of the ”user-defined pattern” and then enter ”NWH” in the field of the ”with symbol meanings”, which means that the first used symbol ”X” is ”N”, the second used symbol ”Y” is ”W”, and the third used symbol ”Z” is ”H”, where N, W and H are the IUPAC codes for nucleotides. It should be noticed that currently the length of ”user-defined pattern” is limited between 4 and 7 nucleotides. 8. Specify the spacer length (15) (that ranges from 2 to 12 bp in default). 9. Determine whehter to use SD-like sequence or not (16). If so, users need to further specify the candidates of SD-like sequence (17) (that are AGGA, AGGG, GAGG, GGAG, GGGA and GGGG in default) and the location of the SD-like sequence (18) (whose default ranges from 8 to 14 bp for the detection of −1 PRF and from 3 to 5 for the detection of +1 PRF upstream of slippery site). Notice that users can modify (such as add and delete) the default candidates of SD-like sequences. 10. Determine whether to carry out the verification of protential protein function or not (19). If so (default), users need to further specify which regions around the slippery site should be verified. It can be either the region upstream of slippery site, the region downstream of slippery site, or both (20). In addition, users need to choose the scanning method(s) of InterProScan (21) (multiple choice) to verify the functional protein motifs/domains of the identified ORFs involved in frameshifting, where the default is BlastProDom. Users can also specify the threshold of the E-value (whose default is 0.1) for reporting matches against InterPro database (22).. 13.
(23) 11. Determine whether to search for stimulatory RNA structures downstream of slippery sites or not (23). If so (default), users need to choose the class of H-type pseudoknots (24), which can be ”class 1”, ”class 2”, ”class 3”, ”class 4” or ”all classes (default)”, to act as stimulatory RNA structures. Then the pre-defined size ranges of structural motifs (e.g., two stems and three loops) in the selected class of H-type pseudoknots will be immediately shown below if the selected class is 1, 2, 3, or 4 (all these defaults are manually modifiable). In addition, users can choose the kernel program (25) used to predict the specified H-type pseudoknots (the default is pknotsRG). Notice that if PRooF cannot find the H-type pseudoknots specified by users, it continues to search for possible bulged helixes or simple stem-loops as stimulators of frameshifting. Hence, if necessary, users also can modify the default size ranges of the structural motifs in bulged helixes (26) and/or simple stem-loops (27). 12. Check email box and simultaneously enter an email address (28), if users would like to run PRooF in a batch way. In this way, users will be notified of the output via email when the submitted job is finished. This email check is optional but recommended if the sequences users enter/upload are large-scale. 13. Click ”Submit” button to run PRooF (29).. 3.2.2. Output of PRooF. In the output page, PRooF will show all the detected PRF sites in the input sequence along with their detailed information (refer to Figures 3.2 and 3.3 for examples), including • the strand direction of input sequence, • the slippery sequence along with its position (start nucleotide, end nucleotide), 14.
(24) • the spacer length (nt/bp), • the stimulatory RNA structure along with its type (H-type pseudoknot, bulged helix or simple stem-loop), sequence range (start nucleotide, end nucleotide), sequence content, base pairings (in bucket view) and minimum free energy (kcal/mol), • the SD-like sequence (as shown in Figure 3.3) along with its position (upstream of slippery site), • the type of protein product (either longer or shorter), and • the detected motifs/domains in the fused protein product by using InterProScan, including the one upstream of slippery site and the one downstream of slippery site.. 15.
(25) Figure 3.2: An output of a detected PRF site of longer protein product.. Figure 3.3: An output of a detected −1 PRF site of shorter protein product.. 16.
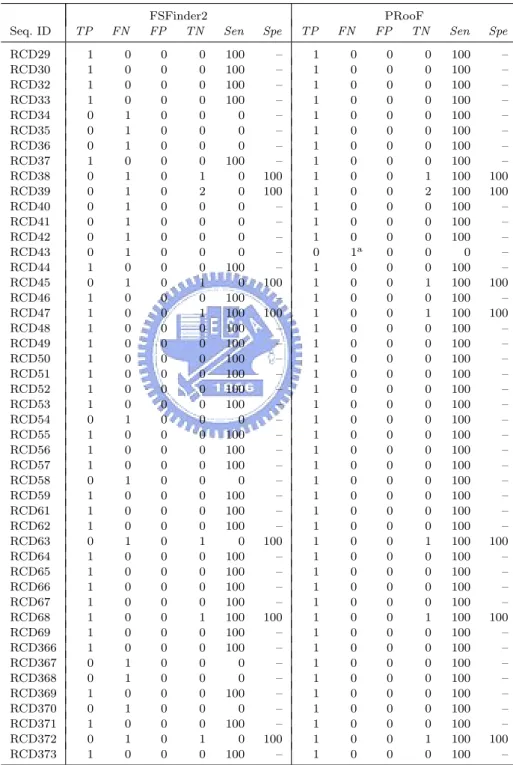
(26) Chapter 4 Results and Discussion We have implemented PRooF based on the algorithm whose details was described in Chapter 3, for the prediction of −1 and +1 PRF sites in a given sequence. The kernel of PRooF was written in C and its web server, available for online analysis at [37], was implemented in PHP. To evaluate its function and correctness, our PRooF was tested with a number of genomic sequences with one or two known PRF sites from many different species. And, its experimental results were compared with those obtained by the latest program FSFinder2 [31, 38]. To reduce the number of false positives, FSFinder2 seems to consider only two pairs of the partially overlapping ORFs whose zero reading frames are the largest two in length, because Moon et al. [31] reported that these two pairs had the highest probability to contain −1 and +1 PRF sites. However, currently there seems to be no biological evidence to support their observation. On the contrary, here we utilized InterProScan to screen out the partially overlapping ORFs whose protein sequences contain no functional motifs/domains. As demonstrated later in our experiments, such an approach of functional bioinformatics is very useful to reduce the number of false positives. In our experiments, the tested sequences were taken from the databases PseudoBase [39] and RECODE [12]. PseudoBase collects RNA pseudoknots, some of which 17.
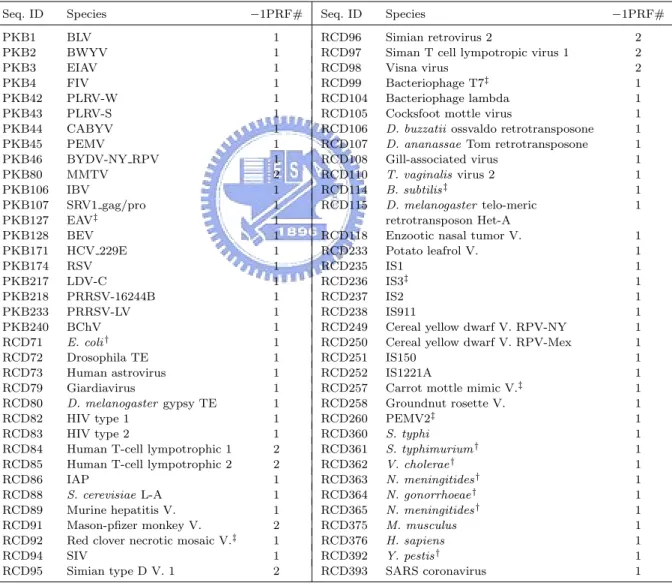
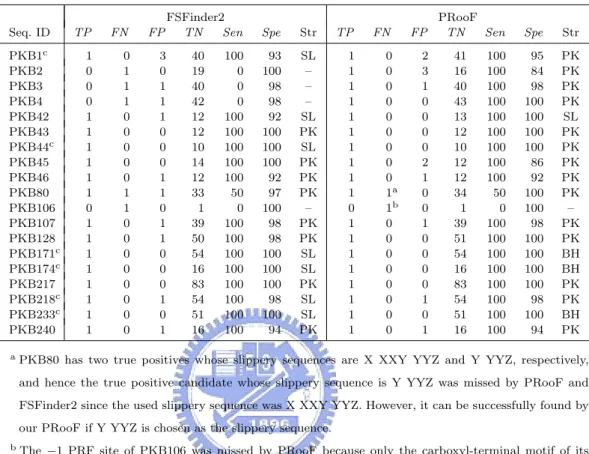
(27) are thought to function as the stimulators of −1 PRFs, and RECODE contains translational recoding events in various biological species, including −1 and +1 PRFs. It should be noted that most of the known PRF sites in these tested sequences are putative, because they have never been proven to be functional and simply just carry the required slippery sequences and downstream RNA secondary structures. Tables 4.1. Table 4.1: The tested sequences and their −1 PRF numbers Seq. ID. Species. PKB1 PKB2 PKB3 PKB4 PKB42 PKB43 PKB44 PKB45 PKB46 PKB80 PKB106 PKB107 PKB127 PKB128 PKB171 PKB174 PKB217 PKB218 PKB233 PKB240 RCD71 RCD72 RCD73 RCD79 RCD80 RCD82 RCD83 RCD84 RCD85 RCD86 RCD88 RCD89 RCD91 RCD92 RCD94 RCD95. BLV BWYV EIAV FIV PLRV-W PLRV-S CABYV PEMV BYDV-NY RPV MMTV IBV SRV1 gag/pro EAV‡ BEV HCV 229E RSV LDV-C PRRSV-16244B PRRSV-LV BChV E. coli † Drosophila TE Human astrovirus Giardiavirus D. melanogaster gypsy TE HIV type 1 HIV type 2 Human T-cell lympotrophic 1 Human T-cell lympotrophic 2 IAP S. cerevisiae L-A Murine hepatitis V. Mason-pfizer monkey V. Red clover necrotic mosaic V.‡ SIV Simian type D V. 1. †. −1PRF#. Seq. ID. Species. 1 1 1 1 1 1 1 1 1 2 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 1 1 1 2 1 1 2. RCD96 RCD97 RCD98 RCD99 RCD104 RCD105 RCD106 RCD107 RCD108 RCD110 RCD114 RCD115. Simian retrovirus 2 Siman T cell lympotropic virus 1 Visna virus Bacteriophage T7‡ Bacteriophage lambda Cocksfoot mottle virus D. buzzatii ossvaldo retrotransposone D. ananassae Tom retrotransposone Gill-associated virus T. vaginalis virus 2 B. subtilis ‡ D. melanogaster telo-meric retrotransposon Het-A Enzootic nasal tumor V. Potato leafrol V. IS1 IS3‡ IS2 IS911 Cereal yellow dwarf V. RPV-NY Cereal yellow dwarf V. RPV-Mex IS150 IS1221A Carrot mottle mimic V.‡ Groundnut rosette V. PEMV2‡ S. typhi S. typhimurium † V. cholerae † N. meningitides † N. gonorrhoeae † N. meningitides † M. musculus H. sapiens Y. pestis † SARS coronavirus. RCD118 RCD233 RCD235 RCD236 RCD237 RCD238 RCD249 RCD250 RCD251 RCD252 RCD257 RCD258 RCD260 RCD360 RCD361 RCD362 RCD363 RCD364 RCD365 RCD375 RCD376 RCD392 RCD393. −1PRF# 2 2 2 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1. Most tested sequences listed in this table have −1 PRFs that produce longer proteins, whereas a few sequences, such as RCD71, RCD360–365 and RCD392, give shorter proteins instead.. ‡. The sequences (PKB127, and RCD92, 99, 114, 236, 257 and 260) possess −1 PRF slippery sequences that conform to the form Y YYZ. Most of the tested sequences, however, have slippery sequences of the general form X XXY YYZ for their −1 PRFs. Notice that in the two −1 PRFs of PKB80, one slippery sequence is X XXY YYZ but the other is Y YYZ.. 18.
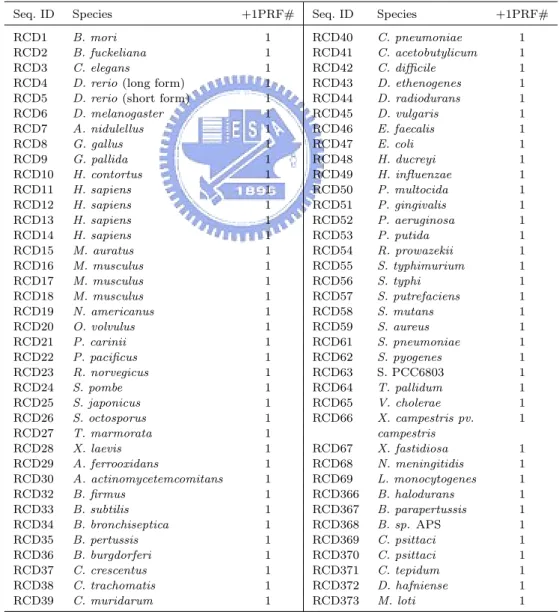
(28) and 4.2 show the information about the sequences we used to predict −1 and +1 PRFs, respectively, and the number of their corresponding PRF sites. For convenience of comparison, here we used the sequence IDs designated by Moon et al. [31], despite the fact that their IDs are inconsistent with those annotated in RECODE. Most sequences listed in Table 4.1 have putative −1 PRFs with longer protein products, whereas only a few sequences, such as RCD71, RCD360–365 and RCD392, have those with shorter. Table 4.2: The tested sequences and their +1 PRF numbers Seq. ID. Species. RCD1 RCD2 RCD3 RCD4 RCD5 RCD6 RCD7 RCD8 RCD9 RCD10 RCD11 RCD12 RCD13 RCD14 RCD15 RCD16 RCD17 RCD18 RCD19 RCD20 RCD21 RCD22 RCD23 RCD24 RCD25 RCD26 RCD27 RCD28 RCD29 RCD30 RCD32 RCD33 RCD34 RCD35 RCD36 RCD37 RCD38 RCD39. B. mori B. fuckeliana C. elegans D. rerio (long form) D. rerio (short form) D. melanogaster A. nidulellus G. gallus G. pallida H. contortus H. sapiens H. sapiens H. sapiens H. sapiens M. auratus M. musculus M. musculus M. musculus N. americanus O. volvulus P. carinii P. pacificus R. norvegicus S. pombe S. japonicus S. octosporus T. marmorata X. laevis A. ferrooxidans A. actinomycetemcomitans B. firmus B. subtilis B. bronchiseptica B. pertussis B. burgdorferi C. crescentus C. trachomatis C. muridarum. +1PRF#. Seq. ID. Species. 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1. RCD40 RCD41 RCD42 RCD43 RCD44 RCD45 RCD46 RCD47 RCD48 RCD49 RCD50 RCD51 RCD52 RCD53 RCD54 RCD55 RCD56 RCD57 RCD58 RCD59 RCD61 RCD62 RCD63 RCD64 RCD65 RCD66. C. pneumoniae C. acetobutylicum C. difficile D. ethenogenes D. radiodurans D. vulgaris E. faecalis E. coli H. ducreyi H. influenzae P. multocida P. gingivalis P. aeruginosa P. putida R. prowazekii S. typhimurium S. typhi S. putrefaciens S. mutans S. aureus S. pneumoniae S. pyogenes S. PCC6803 T. pallidum V. cholerae X. campestris pv. campestris X. fastidiosa N. meningitidis L. monocytogenes B. halodurans B. parapertussis B. sp. APS C. psittaci C. psittaci C. tepidum D. hafniense M. loti. RCD67 RCD68 RCD69 RCD366 RCD367 RCD368 RCD369 RCD370 RCD371 RCD372 RCD373. 19. +1PRF# 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1.
(29) products. Moreover, most of the tested sequences bear slippery sequences of the general form X XXY YYZ for −1 PRF, except for a few instances (PKB127, RCD92, 99, 114, 236, 257 and 260) which fit with the shorter form Y YYZ. In Table 4.2, all the tested sequences have +1 PRFs that produce longer proteins. A summary of overall sensitivity and specificity for all the tests is listed in Tables 4.3–4.8, in which we let Sen (Sensitivity) =. 100×T P T P +F N. and Spe (Specificity) =. 100×T N , T N +F P. where TP = true positive (i.e., the number of correctly predicted PRF sites), FN = false negative (i.e., the number of known PRF sites that were not predicted), FP = false positive (i.e., the number of incorrectly predicted PRF sites), and TN = true negative (i.e., the number of predicted non-PRF sites that possess a required slippery sequence but are not annotated as PRF sites in database). The str field denotes the type of the predicted 3’-stimulatory RNA structure, with SL, BH and PK standing for simple stem-loop, bulged helix and H-type pseudoknot, respectively. Unless otherwise specified, all the tests of PRooF and FSFinder2 were run with default parameters. Table 4.3 lists the experimental results of PRooF and FSFinder2 using the PseudoBase sequences whose −1 PRFs result in longer protein products and whose slippery sequences conform to X XXY YYZ. Successfully, our PRooF identified all the −1 PRF sites in this set of tested sequences, except for PKB80 and PKB106. PKB80 actually gave two true positives whose slippery sequences are X XXY YYZ and Y YYZ, respectively. The latter was missed by PRooF, as well as FSFinder2, since the slippery sequence used in the experiment was X XXY YYZ. However, it can be successfully detected by PRooF if Y YYZ is chosen as the slippery sequence. The −1 PRF site in PKB106 was missed by PRooF because only the carboxyl-terminal motif of its protein product is currently registered in the InterPro database. Therefore, if only the region downstream of the slippery site is scanned for potential motifs/domains, then the true −1 PRF site in PKB106 can still be detected by PRooF. In contrast to the result of 20.
(30) Table 4.3: Summary of the PRooF results for predicting the −1 PRFs of longer product on several sequences from PseudoBase using the slippery sequence X XXY YYZ Seq. ID PKB1c PKB2 PKB3 PKB4 PKB42 PKB43 PKB44c PKB45 PKB46 PKB80 PKB106 PKB107 PKB128 PKB171c PKB174c PKB217 PKB218c PKB233c PKB240 a. TP. FN. FP. 1 0 0 0 1 1 1 1 1 1 0 1 1 1 1 1 1 1 1. 0 1 1 1 0 0 0 0 0 1 1 0 0 0 0 0 0 0 0. 3 0 1 1 1 0 0 0 1 1 0 1 1 0 0 0 1 0 1. FSFinder2 TN Sen 40 19 40 42 12 12 10 14 12 33 1 39 50 54 16 83 54 51 16. 100 0 0 0 100 100 100 100 100 50 0 100 100 100 100 100 100 100 100. Spe. Str. TP. 93 100 98 98 92 100 100 100 92 97 100 98 98 100 100 100 98 100 94. SL – – – SL PK SL PK PK PK – PK PK SL SL PK SL SL PK. 1 1 1 1 1 1 1 1 1 1 0 1 1 1 1 1 1 1 1. FN 0 0 0 0 0 0 0 0 0 1a 1b 0 0 0 0 0 0 0 0. FP 2 3 1 0 0 0 0 2 1 0 0 1 0 0 0 0 1 0 1. PRooF TN Sen 41 16 40 43 13 12 10 12 12 34 1 39 51 54 16 83 54 51 16. 100 100 100 100 100 100 100 100 100 50 0 100 100 100 100 100 100 100 100. Spe. Str. 95 84 98 100 100 100 100 86 92 100 100 98 100 100 100 100 98 100 94. PK PK PK PK SL PK PK PK PK PK – PK PK BH BH PK PK BH PK. PKB80 has two true positives whose slippery sequences are X XXY YYZ and Y YYZ, respectively, and hence the true positive candidate whose slippery sequence is Y YYZ was missed by PRooF and FSFinder2 since the used slippery sequence was X XXY YYZ. However, it can be successfully found by our PRooF if Y YYZ is chosen as the slippery sequence.. b. The −1 PRF site of PKB106 was missed by PRooF because only the carboxyl-terminal motif of its protein product is currently registered in the InterPro database. Therefore, if only the region downstream of the slippery site is scanned for potential motifs, then the true −1 PRF site in PKB106 can still be detected by PRooF.. c. In these cases, the stimulatory RNA structures predicted by PRooF are either H-type pseudoknots or bulged helixes, whereas those produced by FSFinder2 are all simple stem-loops.. PRooF, FSFinder2 also failed to find the true −1 PRF sites in PKB2, 3 and 4, whose slippery sequences are in fact X XXY YYZ. For the tested sequences with −1 PRF sites of longer product from RECODE, FSFinder2 failed to identify true −1 PRF sites in RCD91, 96, 104, 107, 110, 115, 237, 238, 251 and 252 as shown in Table 4.4. Our PRooF, however, missed the sites only in three cases of RCD110, 115 and 252. The main reason for the misses in RCD110 and RCD115 is that the Y’s in their slippery sequence X XXY YYZ are C’s or G’s, instead of the defaults A’s or U’s. If the Y used is any base within X XXY YYZ instead, our 21.
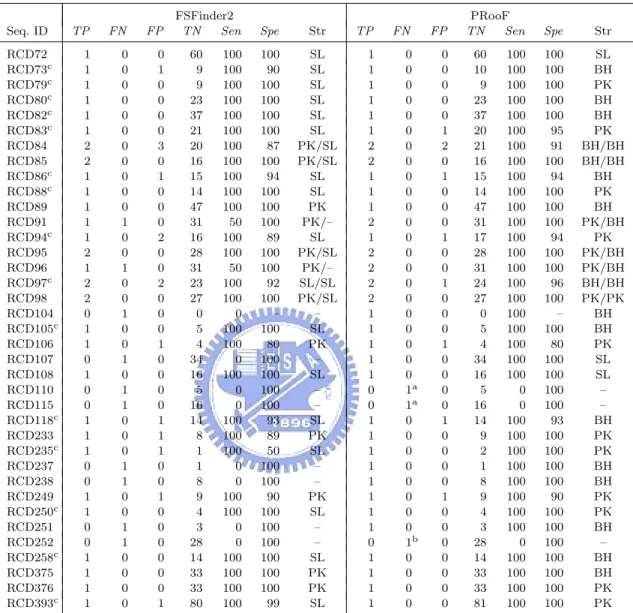
(31) Table 4.4: Summary of the PRooF results for predicting the −1 PRFs of longer product on several sequences from RECODE using the slippery sequence X XXY YYZ Seq. ID RCD72 RCD73c RCD79c RCD80c RCD82c RCD83c RCD84 RCD85 RCD86c RCD88c RCD89 RCD91 RCD94c RCD95 RCD96 RCD97c RCD98 RCD104 RCD105c RCD106 RCD107 RCD108 RCD110 RCD115 RCD118c RCD233 RCD235c RCD237 RCD238 RCD249 RCD250c RCD251 RCD252 RCD258c RCD375 RCD376 RCD393c a. TP. FN. FP. 1 1 1 1 1 1 2 2 1 1 1 1 1 2 1 2 2 0 1 1 0 1 0 0 1 1 1 0 0 1 1 0 0 1 1 1 1. 0 0 0 0 0 0 0 0 0 0 0 1 0 0 1 0 0 1 0 0 1 0 1 1 0 0 0 1 1 0 0 1 1 0 0 0 0. 0 1 0 0 0 0 3 0 1 0 0 0 2 0 0 2 0 0 0 1 0 0 0 0 1 1 1 0 0 1 0 0 0 0 0 0 1. FSFinder2 TN Sen 60 9 9 23 37 21 20 16 15 14 47 31 16 28 31 23 27 0 5 4 34 16 5 16 14 8 1 1 8 9 4 3 28 14 33 33 80. 100 100 100 100 100 100 100 100 100 100 100 50 100 100 50 100 100 0 100 100 0 100 0 0 100 100 100 0 0 100 100 0 0 100 100 100 100. Spe. Str. 100 90 100 100 100 100 87 100 94 100 100 100 89 100 100 92 100 – 100 80 100 100 100 100 93 89 50 100 100 90 100 100 100 100 100 100 99. SL SL SL SL SL SL PK/SL PK/SL SL SL PK PK/– SL PK/SL PK/– SL/SL PK/SL – SL PK – SL – – SL PK SL – – PK SL – – SL PK PK SL. TP 1 1 1 1 1 1 2 2 1 1 1 2 1 2 2 2 2 1 1 1 1 1 0 0 1 1 1 1 1 1 1 1 0 1 1 1 1. FN 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1a 1a 0 0 0 0 0 0 0 0 1b 0 0 0 0. FP 0 0 0 0 0 1 2 0 1 0 0 0 1 0 0 1 0 0 0 1 0 0 0 0 1 0 0 0 0 1 0 0 0 0 0 0 0. PRooF TN Sen 60 10 9 23 37 20 21 16 15 14 47 31 17 28 31 24 27 0 5 4 34 16 5 16 14 9 2 1 8 9 4 3 28 14 33 33 81. 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 0 0 100 100 100 100 100 100 100 100 0 100 100 100 100. Spe. Str. 100 100 100 100 100 95 91 100 94 100 100 100 94 100 100 96 100 – 100 80 100 100 100 100 93 100 100 100 100 90 100 100 100 100 100 100 100. SL BH PK BH BH PK BH/BH BH/BH BH PK BH PK/BH PK PK/BH PK/BH BH/BH PK/PK BH BH PK SL SL – – BH PK PK BH BH PK PK BH – BH BH PK PK. The slippery sites of RCD110 and RCD115 were missed by PRooF (and FSFinder2) since their Y’s in X XXY YYZ are C’s or G’s, instead of the defaults A’s or U’s. Nevertheless, our PRooF, as well as FSFinder2, still can find the slippery site for RCD110 if the Y used within X XXY YYZ is any base instead. As for RCD115, PRooF found an alternative −1 PRF site at around 1269 nt, instead of the reported one in RECODE that starts at 1326 nt, when using X XXY YYZ with Y being any base as the slippery sequence.. b. The candidate of true positive for RCD252 was also missed by our PRooF, because the lengths of the involved ORFs are less than the default minimum length of 100 nt and the motifs/domains of its protein product are not registered in InterPro database. However, it still can be detected by PRooF if the minimum length of ORF is set 40 nt and the verification of protein function is disabled.. c. In these cases, the stimulatory RNA structures predicted by PRooF are either H-type pseudoknots or bulged helixes, whereas those produced by FSFinder2 are all simple stem-loops.. 22.
(32) PRooF can still identify the slippery site in RCD110. As for RCD115, another −1 PRF site starting at 1269 nt, instead of 1326 nt reported in RECODE, was found by our PRooF when using X XXY YYZ as the slippery sequence with Y being any base. In fact, downstream of 1326 nt, we even detected no simple stem-loop nearby that can serve as a stable RNA structure to stimulate the programmed −1 frameshifting in RCD115. This observation suggests that the −1 PRF site of RCD115 reported in RECODE may be questionable. For RCD252, the failure to identify −1 PRF site by PRooF is caused by the following two reasons. (1) The lengths of the ORFs involved in this frameshifting are less than the default minimum length (i.e., 100 nt) in PRooF. (2) The motifs/domains in the −1 PRF protein product are currently not registered in InterPro database. Consequently, the candidate with this −1 PRF site will be filtered out by PRooF in the step of verifying potential protein function. Therefore, if the minimum length of ORF is set 40 nt and the verification for protein function is disabled, the true −1 PRF site in RCD252 can still, as expected, be successfully detected by PRooF. Table 4.5 lists the experimental results obtained by our PRooF and FSFinder2, for those tested sequences whose −1 PRF slippery sequences conform to Y YYZ, instead of X XXY YYZ. Consequently, PRooF missed the slippery site in RCD114, whereas FSFinder2 missed in RCD99 and 114. PRooF failed to detect the −1 PRF site in RCD114 because the involved ORFs were short and the protein sequence in the region downstream of slippery site contained no motifs/domains currently registered in InterPro database. As expected, it still can be detected by PRooF with the minimum ORF length of 50 nt and with only verifying the protein function of the region upstream from the slippery site. Inevitably, both PRooF and FSFinder2 will generate more false positives by using Y YYZ than X XXY YYZ. But, the numbers of false positives generated by PRooF are still small in all the tested sequences, except for PKB127. In 23.
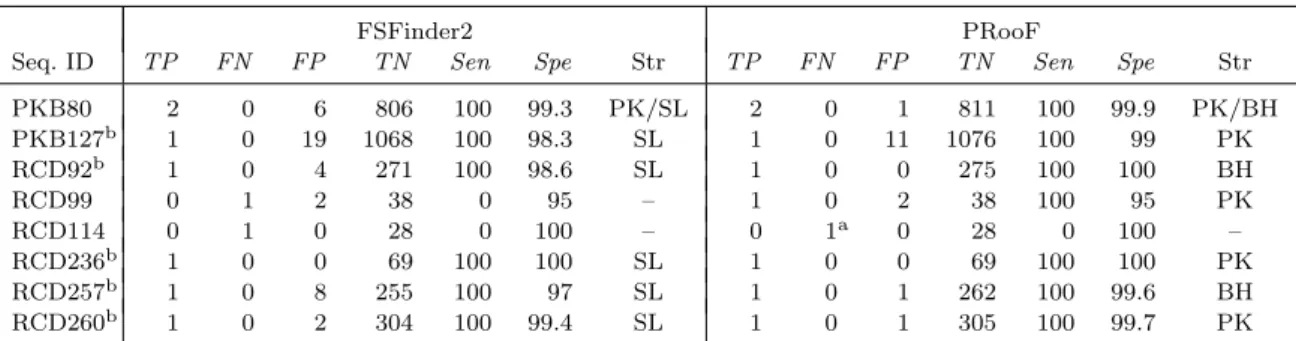
(33) Table 4.5: Summary of the PRooF results for predicting the −1 PRFs of longer product on several sequences from PseudoBase and RECODE using the slippery sequence Y YYZ Seq. ID PKB80 PKB127b RCD92b RCD99 RCD114 RCD236b RCD257b RCD260b a. TP. FN. FP. 2 1 1 0 0 1 1 1. 0 0 0 1 1 0 0 0. 6 19 4 2 0 0 8 2. FSFinder2 TN Sen. Spe. Str. 806 1068 271 38 28 69 255 304. 99.3 98.3 98.6 95 100 100 97 99.4. PK/SL SL SL – – SL SL SL. 100 100 100 0 0 100 100 100. TP 2 1 1 1 0 1 1 1. FN 0 0 0 0 1a 0 0 0. FP 1 11 0 2 0 0 1 1. PRooF TN Sen 811 1076 275 38 28 69 262 305. 100 100 100 100 0 100 100 100. Spe. Str. 99.9 99 100 95 100 100 99.6 99.7. PK/BH PK BH PK – PK BH PK. This true positive of the −1 PRF site in RCD114 can be detected by PRooF if the minimum ORF length is set to 50 nt and only the region upstream of slippery site is scanned for potential motifs/domains.. b. In these cases, the stimulatory RNA structures predicted by PRooF are either H-type pseudoknots or bulged helixes, whereas those produced by FSFinder2 are all simple stem-loops.. the case of PKB127, PRooF totally found nine partially overlapping ORFs, five of which were further screened out for the lack of possible protein motifs/domains. Subsequently, PRooF identified a true positive of −1 PRF site, along with 11 false positives, out of the four remaining overlapping ORFs. Notably, among these 11 false positives, six of them were derived from the same overlapping ORFs and four of them from another same overlapping ORFs. That is, a single overlapping region gave many false positives in the output. According to the −1 PRF model, however, there should be at most one true −1 PRF site in each pair of overlapping ORFs. Furthermore, our results show that a true −1 PRF site is usually accompanied with a 3’-stimulatory RNA structure of lower free energy. Therefore, the number of the false positives in PKB127 can be reduced further if our PRooF continues to filter out those candidates whose predicted RNA structures are of high free energy and those from the same overlapping ORFs. For the sequences with known −1 PRF sites of shorter product, as listed in Table 4.6, PRooF detected all their slippery sites, whereas FSFinder2 failed for the cases 24.
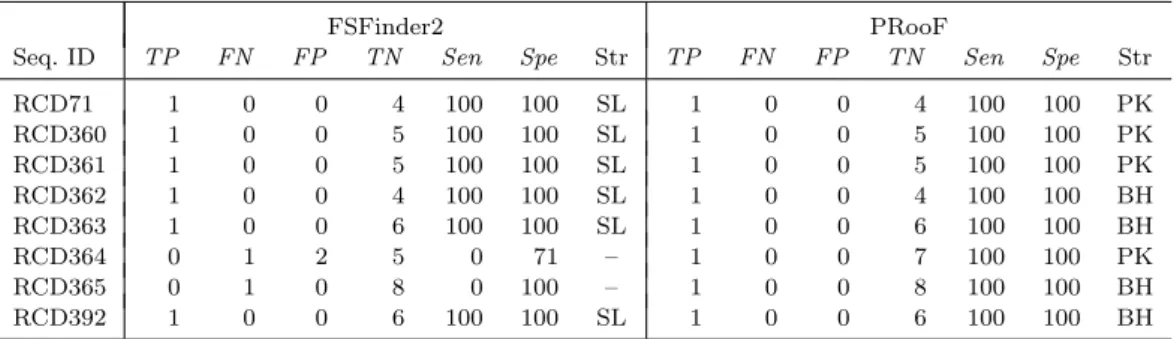
(34) Table 4.6: Summary of the PRooF results for predicting the −1 PRFs of shorter product on several sequences from RECODE using the slippery sequence X XXY YYZ Seq. ID RCD71 RCD360 RCD361 RCD362 RCD363 RCD364 RCD365 RCD392. FSFinder2 TN Sen. TP. FN. FP. 1 1 1 1 1 0 0 1. 0 0 0 0 0 1 1 0. 0 0 0 0 0 2 0 0. 4 5 5 4 6 5 8 6. 100 100 100 100 100 0 0 100. Spe. Str. TP. FN. FP. 100 100 100 100 100 71 100 100. SL SL SL SL SL – – SL. 1 1 1 1 1 1 1 1. 0 0 0 0 0 0 0 0. 0 0 0 0 0 0 0 0. PRooF TN Sen 4 5 5 4 6 7 8 6. 100 100 100 100 100 100 100 100. Spe. Str. 100 100 100 100 100 100 100 100. PK PK PK BH BH PK BH BH. of RCD364 and 365. Moreover, the stimulatory RNA structures detected by PRooF are H-type pseudoknots or bulged helixes, whereas all the RNA structures predicted by FSFinder2 are just simple stem-loops. Actually, such a property can greatly be observed in other experiments as demonstrated in Tables 4.3–4.5. Tables 4.7 and 4.8 presented the experimental results of detecting +1 PRF sites on several sequences from RECODE database. The tested sequences used in Table 4.7 are related to the prfB genes from many bacterial genomes, as mentioned before, whose frameshifting sites have no downstream RNA structures to server as stimulators. Hence, we experimented these sequences with PRooF by selecting CUU URA C (that are most commonly found in the prfB genes) as the slippery sequence, along with detecting their SD-like sequences, but disabling the detection of stimulatory RNA structure. In Table 4.8, the sequences we tested are related to the oaz genes from several eukaryotic genomes whose +1 PRF sites have 3’-stimulatory RNA structures. Therefore, we tested them with PRooF by using UUU UGA or YCC UGA that are common in the oaz genes as the slippery sequence. Consequently, our PRooF had better sensitivity than FSFinder2, because it almost detected the +1 PRF sites on all tested sequences, except for RCD43, and almost predicted H-type pseudoknot or 25.
(35) Table 4.7: Summary of the PRooF results for predicting the +1 PRFs on several sequences from RECODE with using the slippery sequence CUU URA C and without detecting downstream RNA structure Seq. ID RCD29 RCD30 RCD32 RCD33 RCD34 RCD35 RCD36 RCD37 RCD38 RCD39 RCD40 RCD41 RCD42 RCD43 RCD44 RCD45 RCD46 RCD47 RCD48 RCD49 RCD50 RCD51 RCD52 RCD53 RCD54 RCD55 RCD56 RCD57 RCD58 RCD59 RCD61 RCD62 RCD63 RCD64 RCD65 RCD66 RCD67 RCD68 RCD69 RCD366 RCD367 RCD368 RCD369 RCD370 RCD371 RCD372 RCD373 a. TP. FN. 1 1 1 1 0 0 0 1 0 0 0 0 0 0 1 0 1 1 1 1 1 1 1 1 0 1 1 1 0 1 1 1 0 1 1 1 1 1 1 1 0 0 1 0 1 0 1. 0 0 0 0 1 1 1 0 1 1 1 1 1 1 0 1 0 0 0 0 0 0 0 0 1 0 0 0 1 0 0 0 1 0 0 0 0 0 0 0 1 1 0 1 0 1 0. FSFinder2 FP TN 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0. 0 0 0 0 0 0 0 0 1 2 0 0 0 0 0 1 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 1 0 0 0 0 0 0 0 1 0. Sen. Spe. TP. 100 100 100 100 0 0 0 100 0 0 0 0 0 0 100 0 100 100 100 100 100 100 100 100 0 100 100 100 0 100 100 100 0 100 100 100 100 100 100 100 0 0 100 0 100 0 100. – – – – – – – – 100 100 – – – – – 100 – 100 – – – – – – – – – – – – – – 100 – – – – 100 – – – – – – – 100 –. 1 1 1 1 1 1 1 1 1 1 1 1 1 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1. FN 0 0 0 0 0 0 0 0 0 0 0 0 0 1a 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0. PRooF FP TN 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0. 0 0 0 0 0 0 0 0 1 2 0 0 0 0 0 1 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 1 0 0 0 0 0 0 0 1 0. Sen. Spe. 100 100 100 100 100 100 100 100 100 100 100 100 100 0 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100. – – – – – – – – 100 100 – – – – – 100 – 100 – – – – – – – – – – – – – – 100 – – – – 100 – – – – – – – 100 –. For RCD43, its true positive candidate was missed by PRooF with default parameters. However, it can still be found by PRooF if the detection of SD-like sequence is disabled.. 26.
(36) Table 4.8: Summary of the PRooF results for predicting the +1 PRFs on several sequences from RECODE using the slippery sequence UUU UGA or YCC UGA Seq. ID RCD1a RCD2a RCD3 RCD4 RCD5 RCD6a RCD7a RCD8 RCD9 RCD10a RCD11 RCD12a RCD13 RCD14 RCD15 RCD16 RCD17a RCD18a RCD19a RCD20 RCD21 RCD22a RCD23 RCD24 RCD25a RCD26 RCD27a RCD28 a. TP. FN. FP. 1 1 0 1 1 1 1 1 0 1 1 1 0 0 1 1 1 1 1 0 0 1 1 1 1 1 1 1. 0 0 1 0 0 0 0 0 1 0 0 0 1 1 0 0 0 0 0 1 1 0 0 0 0 0 0 0. 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0. FSFinder2 TN Sen. Spe. Str. TP. FN. FP. 1 0 2 1 1 4 0 1 0 0 1 4 0 1 2 2 2 0 1 1 0 0 2 2 0 2 2 2. 100 – 100 100 100 100 – 100 – – 100 100 – 100 100 100 100 – 100 100 – – 100 100 – 100 100 100. SL SL – PK PK SL SL PK – SL PK SL – – PK PK SL SL SL – – SL PK PK SL PK SL PK. 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1. 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0. 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0. 100 100 0 100 100 100 100 100 0 100 100 100 0 0 100 100 100 100 100 0 0 100 100 100 100 100 100 100. PRooF TN Sen 1 0 2 1 1 4 0 1 0 0 1 4 0 1 2 2 2 0 1 1 0 0 2 2 0 2 2 2. 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100 100. Spe. Str. 100 – 100 100 100 100 – 100 – – 100 100 – 100 100 100 100 – 100 100 – – 100 100 – 100 100 100. BH BH BH PK PK BH BH PK BH BH PK PK SL PK PK PK PK BH BH BH BH BH PK BH BH BH PK BH. In these cases, the stimulatory RNA structures predicted by PRooF are either H-type pseudoknots or bulged helixes, whereas those produced by FSFinder2 are all simple stem-loops.. bulged helixes as the stimulatory RNA structures on all sequences, excepted for RCD13. The failure to detect the frameshifting site in RCD43 was due to the fact that there is no any pre-defined SD-like sequence upstream of the slippery site. Hence, we can correctly detect it with PRooF if the detection of SD-like sequence is disabled. Generally speaking, the average sensitivity and specificity of PRooF are both better than those of FSFinder2, as depicted in Tables 4.9. In particular, PRooF greatly improves the sensitivity when compared with FSFinder2. In addition, almost all the stimulatory RNA structures predicted by PRooF are either H-type pseudoknots or 27.
(37) Table 4.9: The average sensitivity and specificity of −1 and +1 PRF prediction using PRooF and FSFinder2 −1 and +1 PRF prediction Average sensitivity Average specificity PRooF. 149×100 149+7. = 96. 4288×100 4288+37. = 99. FSFinder2. 114×100 114+42. = 73. 4255×100 4255+70. = 98. The total TP, FN, TN and FP in Tables 4.3–4.8 of −1 and +1 PRF prediction are 149, 7, 4288 and 37, respectively, for PRooF, and 114, 42, 4255 and 70, respectively, for FSFinder2.. bulged helixes, except those for PKB42 in Table 4.3, RCD72, 107 and 108 in Table 4.4 and RCD13 in Table 4.8. Recall that H-type pseudoknots and bulged helixes both share a similar structural feature of bend conformation, and are structurally more complex and more stable than simple stem-loops. Therefore, they are believed to be more useful and constructive to promote the efficiency of −1 PRFs and some +1 PRFs. As for PKB42 and RCD72, 107, 108 and 13, their stimulators found by PRooF are just simple stem-loops, and neither a stable H-type pseudoknot nor a bulged helix downstream of their slippery sites was detected. In contrast to our PRooF, a great number of the stimulatory RNA structures identified by FSFinder2 are just simple stem-loops, because the algorithm employed by FSFinder2 for the RNA structure prediction first searches for possible stem-loops (without bulges or interior loops) by examining the nucleotides in both directions from every pivot for possible base pairing, and then considers any two simple stem-loops as an H-type pseudoknot if they cross with each other. In addition, it is worth mentioning that some simple stem-loops (such as RCD72 and 108) predicted by FSFinder2 do not seem to be stable RNA structures, since their loops are only 1 nt long, leading to sharp stem-loops. Recall that the stimulatory RNA structure in the −1 PRF of HIV-1 was first thought 28.
(38) to be a simple stem-loop, but it was then proved experimentally to be a bulged helix. Interestingly, the stimulatory RNA structure predicted by PRooF for the −1 PRF of HIV-1 (i.e., RCD82) is indeed a bulged helix, exactly the same as that determined by Gaudin et al. [26] using heteronuclear NMR spectroscopy. However, the one predicted by FSFinder2 is just a simple stem-loop. It should be worthwhile to further determine experimentally the stimulatory RNA structures for −1 and +1 PRF sites in other similar cases where their RNA structures predicted by PRooF are H-type pseudoknots or bulged-helixes, but are just simple stem-loops by FSFinder2 or reported in the literature.. 29.
(39) Chapter 5 Conclusions In this thesis, we studied and designed a bioinformatics approach for automatically detecting −1 and +1 PRF sites in a given genomic sequence. Using the pattern recognition approach incorporated with structural and functional bioinformatics, we have designed a computational approach that is capable of predicting the −1 and +1 PRF sites accurately in an input sequence. Such an approach ensures that each predicted −1 PRF site, as well as some predicted +1 PRF sites, has two cis-acting signals, a slippery sequence and a stimulatory RNA structure, and its produced polypeptide truly carries protein motifs/domains to present related biological functions. Based on this method, we have developed a web server PRooF that is open to the public for online analysis. In addition, we have evaluated the accuracy of PRooF predictions on several genomic sequences with known −1 or +1 PRF sites. Consequently, the testing results showed that PRooF indeed greatly improves sensitivity by comparing its computational results with those by the latest program FSFinder2. Especially, most of the stimulatory RNA structures predicted by PRooF downstream of PRF sites are H-type pseudoknots and bulged helixes, both of which are widely believed to be the stimulators that can more efficiently promote the PRF events than simple stem-loops, whereas those produced by FSFinder2 are mostly simple stem-loops. It is worth mentioning that our PRooF 30.
(40) was implemented in a flexible way that it allows the user to modify all the default parameters such that some exceptional PRF sites can still be detected, as demonstrated in our experiments.. 31.
(41) References [1] Farabaugh, P. J. (1996) Programmed translational frameshifting. Microbiol Rev, 60, 103–134. [2] Farabaugh, P. J. (1996) Programmed translational frameshifting. Annu Rev Genet, 30, 507–528. [3] Baranov, P. V., Gesteland, R. F. & Atkins, J. F. (2002) Recoding: translational bifurcations in gene expression. Gene, 286, 187–201. [4] Namy, O., Rousset, J. P., Napthine, S. & Brierley, I. (2004) Reprogrammed genetic decoding in cellular gene expression. Mol Cell, 13, 157–168. [5] Namy, O., Moran, S. J., Stuart, D, I., Gilbert, R. J. C. & Brierley, I. (2006) A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature, 441, 244–247. [6] Brierley, I. (1995) Ribosomal frameshifting viral RNAs. J Gen Virol, 76, 1885– 1892. [7] Larsen, B., Gesteland, R. F. & Atkins, J. F. (1997) Structural probing and mutagenic analysis of the stem-loop required for Escherichia coli dnaX ribosomal frameshifting: programmed efficiency of 50%. J Mol Biol, 271, 47–60.. 32.
(42) [8] Mejlhede, N., Atkins, J. F. & Neuhard, J. (1999) Ribosomal −1 frameshifting during decoding of Bacillus subtilis cdd occurs at the sequence CGA AAG. J Bacteriol, 181, 2930–2937. [9] Ivanov, I. P., Gesteland, R. F., & Atkins, J. F. (2000) Antizyme expression: a subversion of triplet decoding, which is remarkably conserved by evolution, is a sensor for an autoregulatory circuit. Nucleic Acids Res, 28, 3185–3196. [10] Ivanov, I. P., Matsufuji, S., Murakami, Y., Gesteland, R. F., & Atkins, J. F. (2000) Conservation of polyamine regulation by translational frameshifting from yeast to mammals. EMBO J, 19, 1907–1917. [11] Baranov, P. V., Gesteland, R. F., & Atkins, J. F. (2002) Release factor 2 frameshifting sites in different bacteria. EMBO Rep, 3, 373–377. [12] Baranov, P. V., Gurvich, O. L., Hammer, A. W., Gesteland, R. F., & Atkins, J. F. (2003) RECODE 2003. Nucleic Acids Res, 31, 87–89. [13] Manktelow, E., Shigemoto, K. & Brierley, I. (2005) Characterization of the frameshift signal of Edr, a mammalian example of programmed −1 ribosomal frameshifting. Nucleic Acids Res, 33, 1553–1563. [14] Wills, N. M., Moore, B., Hammer, A., Gesteland, R. F. & Atkins, J. F. (2006) A functional −1 ribosomal frameshift signal in the human paraneoplastic Ma3 gene. J Biol Chem, 281, 7082–7088. [15] Dinman, J. D., Ruiz-Echevarria, M. J. & Peltz, S. W. (1998) Translating old drugs into new treatments: ribosomal frameshifting as a target for antiviral agents. Trends Biotechnol, 16, 190–196.. 33.
(43) [16] Harger, J. W., Meskauskas, A. & Dinman, J. D. (2002) An ”integrated model” of programmed ribosomal frameshifting. Trends Biochem Sci, 27, 448–454. [17] Brierley, I., Digard, P. & Inglis, S. C. (1989) Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell, 57, 537–547. [18] ten Dam, E. B., Pleij, C. W. & Bosch, L. (1990) RNA pseudoknots: translational frameshifting and readthrough on viral RNAs. Virus Genes, 4, 121–136. [19] Somogyi, P., Jenner, A. J., Brierley, I. & Inglis, S. C. (1993) Ribosomal pausing during translation of an RNA pseudoknot. Mol Cell Biol, 13, 6931–6940. [20] Giedroc, D. P., Theimer, C. A. & Nixon, P. L. (2000) Structure, stability and function of RNA pseudoknots involved in stimulating ribosomal frameshifting. J Mol Biol, 298, 167–185. [21] Lopinski, J. D., Dinman, J. D. & Bruenn, J. A. (2000) Kinetics of ribosomal pausing during programmed −1 translational frameshifting. In Mol Cell Biol, 20, 1095–1103. [22] Plant, E. P., Jacobs, K. L. M., Harger, J. W., Meskauskas, A., Jacobs, J. L., Baxter, J. L., Petrov, A. N. & Dinman, J. D. (2003) The 9-˚ A solution: how mRNA pseudoknots promote efficient programmed −1 ribosomal frameshifting. RNA, 9, 168–174. [23] Plant, E. P. & Dinman, J. D. (2005) Torsional restraint: a new twist on frameshifting pseudoknots. Nucleic Acids Res, 33, 1825–1833.. 34.
(44) [24] Dulude, D., Baril, M. & Brakier-Gingras, L. (2002) Characterization of the frameshift stimulatory signal controlling a programmed −1 ribosomal frameshift in the human immunodeficiency virus type 1. Nucleic Acids Res, 30, 5094–5102. [25] Dinman, J. D., Richter, S., Plant, E. P., Taylor, R. C., Hammell, A. B. & Rana, T. M. (2002) The frameshift signal of HIV-1 involves a potential intramolecular triplex RNA structure. Proc Natl Acad Sci USA, 99, 5331–5336. [26] Gaudin, C., Mazauric, M. H., Tra¨ıkia, M., Guittet, E., Yoshizawa, S. & Fourmy, D. (2005) Structure of the RNA signal essential for translational frameshifting in HIV-1. J Mol Biol, 349, 1024–1035. [27] Chen, X., Kang, H., Shen, L. X., Chamorro, M., Varmus, H. E. & Tinoco, I. (1996) A characteristic bent conformation of RNA pseudoknots promotes −1 frameshifting during translation of retroviral RNA. J Mol Biol, 260, 479–483. [28] Kollmus, H., Honigman, A., Panet, A. & Hauser H. (1994) The sequences of and distance between two cis-acting signals determine the efficiency of ribosomal frameshifting in human immunodeficiency virus type 1 and human T-cell leukemia virus type II in vivo. J Virol, 68, 6087–6091. [29] Weiss, R. B., Dunn, D. M., Dahlberg, A. E., Atkins, J. F. & Gesteland, R. F. (1988) Reading frame switch caused by base-pair formation between the 3’ end of 16S rRNA and the mRNA during elongation of protein synthesis in Escherichia coli. EMBO J, 7, 1503–1507. [30] Hammell, A. B., Taylor, R. C., Peltz, S. W. & Dinman, J. D. (1999) Identification of putative programmed −1 ribosomal frameshift signals in large DNA databases. Genome Res, 9, 417–427.. 35.
(45) [31] Moon, S., Byun, Y., Kim, H. J., Jeong, S. & Han, K. (2004) Predicting genes expressed via −1 and +1 frameshifts. Nucleic Acids Res, 32, 4884–4892. [32] Shah, A. A., Giddings, M. C., Parvaz, J. B., Gesteland, R. F., Atkins, J. F. & Ivanov, I. P. (2002) Computational identification of putative programmed translational frameshift sites. Bioinformatics, 18, 1046–1053. [33] Bekaert, M., Bidou, L., Denise, A., Duchateau-Nguyen, G., Forest, J. P., Froidevaux, C., Hatin, I., Rousset, J. P. & Termier M. (2003) Towards a computational model for −1 eukaryotic frameshifting sites. Bioinformatics, 19, 327–335. [34] Bekaert, M. & Rousset, J. P. (2005) An extended signal involved in eukaryotic −1 frameshifting operates through modification of the E site tRNA. Mol Cell, 17, 61–68. [35] Bekaert, M., Atkins, J. F. & Baranov, P. V. (2006) ARFA: a program for annotating bacterial release factor genes, including prediction of programmed ribosomal frameshifting. Bioinformatics, 22, 2463–2465. [36] Huang, C. H., Lu, C. L. & Chiu, H. T. (2005) A heuristic approach for detecting RNA H-type pseudoknots. Bioinformatics, 21, 3501–3508. [37] Huang, C.. L.. Y.. PRooF:. L.,. Wu,. C.. J.,. Tang,. a. tool. for. detecting. C.. Y.,. programmed. Chiu,. H.. ribosomal. T.. &. Lu,. frameshift-. ing[http://bioalgorithm.life.nctu.edu.tw/PROOF/]. [38] Byun, Y., Moon, S. & Han, K. (2005) Prediction of ribosomal frameshift signals of user-defined models. In Proceedings of the Fifth International Conference on Computational Science (ICCS 2005), Volume 3514 of Lecture Notes in Computer Science. Edited by Sunderam, V. S., van Albada, G. D., Sloot ,P. M. A. & Dongarra, J. J., Springer, Berlin 2005, 948–955. 36.
(46) [39] van Batenburg, F. H., Gultyaev, A. P. & Pleij. C. W. (2001) PseudoBase: structural information on RNA pseudoknots. Nucleic Acids Res, 29, 194–195. [40] Osada, Y., Saito, R. & Tomita M. (1999) Analysis of base-pairing potentials between 16S rRNA and 5’ UTR for translation initiation in various prokaryotes. Bioinformatics, 15, 578–581. [41] Mulder, N. J., Apweiler, R., Attwood, T. K., Bairoch, A., Bateman, A., Binns, D., Bradley, P., Bork, P., Bucher, P., Cerutti, L., Copley, R., Courcelle, E., Das, U., Durbin, R., Fleischmann, W., Gough, J., Haft, D., Harte, N., Hulo, N., Kahn, D., Kanapin, A., Krestyaninova, M., Lonsdale, D., Lopez, R., Letunic, I., Madera, M., Maslen, J., McDowall, J., Mitchell, A., Nikolskaya, A. N., Orchard, S., Pagni, M., Ponting, C. P., Quevillon, E., Selengut, J., Sigrist, C. J. A., Silventoinen, V., Studholme, D. J., Vaughan, R. & Wu, C. H. (2005) InterPro, progress and status in 2005. Nucleic Acids Res, 33, D201–D205. [42] Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R. & Lopez, R. (2005) InterProScan: protein domains identifier. Nucleic Acids Res, 33, W116–W120. [43] Macke, T. J., Ecker, D. J., Gutell, R. R., Gautheret, D., Case, D. A. & Sampath, R. (2001) RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res, 29, 4724–4735. [44] Rivas, E. & Eddy, S. (1999) A dynamic programming algorithm for RNA structure prediction including pseudoknots. J Mol Biol, 285, 2053–2068. [45] Dirks, R. M. & Pierce, N. A. (2003) A partition function algorithm for nucleic acid secondary structure including pseudoknots. J Comput Chem, 24, 1664–1677.. 37.
(47) [46] Reeder, J. & Giegerich, R. (2004) Design, implementation and evaluation of a practical pseudoknot folding algorithm based on thermodynamics. BMC Bioinformatics, 5, 104.. 38.
(48)
數據

+7
Outline
相關文件
From these results, we study fixed point problems for nonlinear mappings, contractive type mappings, Caritsti type mappings, graph contractive type mappings with the Bregman distance
Recently, the paper [36] investigates a family of smoothing functions along with a smoothing-type algorithm to tackle the absolute value equation associated with second-order
The algorithms have potential applications in several ar- eas of biomolecular sequence analysis including locating GC-rich regions in a genomic DNA sequence, post-processing
Start with a STUN header, followed by a STUN payload (which is a series of STUN attributes depending on the message type).
Indicate the type and format of information included in the message body. Content-Length: the length of the message
電機工程學系暨研究所( EE ) 光電工程學研究所(GIPO) 電信工程學研究所(GICE) 電子工程學研究所(GIEE) 資訊工程學系暨研究所(CS IE )
Microphone and 600 ohm line conduits shall be mechanically and electrically connected to receptacle boxes and electrically grounded to the audio system ground point.. Lines in
Generic methods allow type parameters to be used to express dependencies among the types of one or more arguments to a method and/or its return type.. If there isn’t such a
