J . CHEM. SOC. DALTON TRANS.
1995
Synthesis, Crystal Structure and Catalytic Activities of
[Ru"'(bbpc)(PPh,)CI] [H,bbpc
=
I
,2-bis(4-fert-butyl-
pyrid i ne-2-carboxamido) -4,5=dich
loro benzene]
t
Po-Hung K o , ~ Tai-Yuen Chen,b Jin Zhu,a Kin-Fai Cheng,a Shie-Ming Pengb and Chi
-
M
ing Che"vanba Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong
Department of Chemistry, National Taiwan University, Taipei, Taiwan
The compound H,bbpc [I ,2-bis(4-tert-butylpyridine-2-carboxamido)-4,5-dichlorobenzene] , which has good solubility in organic solvents, has been prepared. Reaction of [Ru( PPh,),CI,] with H,bbpc in ethanol and in the presence of triethylamine gave [Ru"'(bbpc) (PPhJCI], the crystal structure of which has been determined. This complex is an active catalyst for alkene epoxidation by PhlO, cyclopropanation of styrene by ethyl diazoacetate and aziridination of styrene by Phl NO,SC,H,Me-p. Its cyclic voltammogram in dichloromethane showed a reversible Ru"'-Ru" couple at -0.55 V and an oxidation couple at 0.32 V.
The design of new metal catalysts to mimic the oxidative activities of metalloporphyrins has continued to be an active area of research.
'-'
Tetradentate dianionic compounds such as salen [H,salen = N , N'-bis(salicy1idene)ethane- 1,2-diamine] and its derivatives are commonly used for the ~ t u d i e s . ~ Previous works revealed that the diamide, 1,2-bis(pyridine-2-carbox- amido)benzene (H,bpb) and 4,5-dichloro-l,2-bis(pyridine-2- carboxamido)benzene (H,bpc), which are readily prepared from pyridine-2-carboxylic acid and the corresponding diamines,6 form stable complexes with transition-metal ions. The complexes of Mn"' and Cr"' with bpc and bpb were prepared 3 b 9 c and found to be active catalysts for oxidation of alkenes and alkanes. However, our attempts to prepare the related ruthenium complexes have been unsuccessful. We attribute this to the low solubility of the amides and their metal complexes in organic solvents, rendering difficult the complexation reaction and product purification. Introduction of a tert-butyl group on the pyridine ring of H,bpc is likely to circumvent the problem. Herein is described the preparation, crystal structure and catalytic activities of a ruthenium(Ir1) complex of 1,2-bis(4-tert- butylpyridine-2-carboxamido)-4,5-dichlorobenzene (H, bbpc).Experiment
a1Materials. --1,2-Diamino-4,5-dichlorobenzene was obtained from Aldrich and recrystallized from hot ethanol before use. All other chemicals for syntheses were used as received.
The compounds [Ru(PPh,),CI,],' PhIO and PhI= NO,SC,H,Me-p were prepared according to literature methods. Styrene, cyclohexene and cyclooctene for catalytic reactions were purified by repeated fractional distillation. Dichloromethane was washed with concentrated H,S04 and then distilled over CaH, under a nitrogen atmosphere. Tetrabutylammonium tetrafluoroborate for electrochemical studies was dried in vacuum at 100 "C for 1 d.
Instrumentation. -The UV/VIS spectra were obtained on a Perkin-Elmer Lambda 19 spectrophotometer and infrared spectra on a Shimadzu IR-470 spectrophotometer. Gas
t
Supplementury data mailable: see Instructions for Authors, J. Chem. Soc., Dalton Trans., 1995, Issue 1, pp. xxv-xxx.Nun-SI unit employed: pe z 9.27 x 1 0-24 J T-'.
WC'
1 2
chromatography was performed on a Hewlett-Packard 5890 Series I1 gas chromatograph equipped with a flame-ionization detector and a 3396 Series IT integrator. Proton NMR spectra were obtained with a JEOL FX270Q spectrometer using tetramethylsilane as internal standard. Cyclic voltammograms were measured using a Princeton Applied Research (PAR) model 273 potentiostat. The reference electrode was Ag- AgNO, (0.1 mol dm-, in MeCN). All E" values were referenced to ferrocenium-ferrocene.
Synthesis of H
,
b bpc .A- t er t-
Bu ty lpyr idine N -oxide. 4- t er t - Butylpyridine (9 g) was dissolved in glacial acetic acid (54 cm3) and 35% H,O, solution (10 cm3) was added dropwise with stirring. The solution was heated at 80 "C for 3 h. Another batch of 35% H,O, solution (8 cm3) was added and the solution was heated for 12 h. It was cooled to room temperature and evaporated to about 25 cm3 under vacuum. Water (25 cm3) was added and the solution adjusted to pH 9 with NaOH solution (40%). The organic product was extracted with dichloromethane (4 x 50 cm3). The dichloromethane extract was dried over MgSO, and evaporated under vacuum to give a pale yellow solid. Yield = 97%. 'H NMR (CDCI,): 6 1.37 (9 H, s, Bu'), 7.26-7.34 (2 H, m, m-H) and 8.15-8.23 (2 H, m, o-H). IR(cm-'): 1680, 858 and 820. m / z = 151.
4-tert-Butyl-2-cyanopyridine. A mixture of 4-tert-butylpyrid- ine N-oxide (9.7 g) in dimethyl sulfate (8.12 g) was heated at 95 "C for 3 h. A brown oily substance was obtained. This was dissolved in water (12 cm3) and a KCN solution (9.5 g KCN in 16 cm3 water) added slowly. The solution was stirred at room temperature for 6 h and then extracted with CHCI, (3 x 50 cm3). The chloroform extract was dried over MgS04 and
2216
J. CHEM. SOC. DALTON TRANS. 1995 evaporated to dryness to give a brown oil. The crude productwas purified on a silica gel column with ethyl acetate-light petroleum (b.p. 60-80 "C) (1 : 5 ) as eluent. The product was obtained as a pale yellow liquid. NMR (CDCl,): 'H, 6 8.38 (1
J = 5.2, 1.9 Hz, H2)and 1.12(9 H, s, Bu'); I3C, 6 161, 150, 133, 125, 124,117,34 and 30. IR (cm-'): 3050,2964,2867,2234 and 1589. m/z = 160.
4-tert-Butylpyridine 2-carboxylic acid. A solution of 4-tert- butyl-2-cyanopyridine in 6 mol dm hydrochloric acid (30 cm3) was refluxed for 24 h. It was then neutralized with a saturated Na,CO, solution. The organic product was extracted with dichloromethane (3 x 50 cm3). The dichloromethane extract was dried over MgS04 and preconcentrated to 10 cm3. Addition of diethyl ether gave a white solid. Yield: 4.8 g. NMR (1 H, s, H3) and 8.26 (1 H, d, J = 5.0 Hz, H'); signal of C 0 2 H too broad to be observed; 13C, 6 168,161,151,149,123,121,34 and 30. IR (cm-'): 3360, 1637 and 1600. nz/z = 179.
1 ,2- Bis( 4- t er t
-
buty lpyr idine-2-car boxam ido)-4,5 -dichlo ro ben- zene (H2 bbpc). A solution of 1,2-diamin0-4,5-dichlorobenzene (0.78 g) and 4-tert-butylpyridine-2-carboxylic acid (1.6 g) inpyridine (20 cm3) was heated up to 100 "C. Triphenyl phosphite (2.76 g) was slowly added and the solution refluxed for 2 h. After refluxing, pyridine was distilled off under vacuum to give a brown oily residue. Upon recrystallization from hot ethanol a pale yellow solid was obtained. 'H NMR (CDCl,): 6 10.23 (2 H, S, NH), 8.42 (2 H, d, J = 5.67, H'), 8.29 (2 H, d, J = 1.38, H3), 8.09 (2 H, s, H4), 7.43 (2 H, dd, J = 5.25, 1.89 Hz, H2) and 1.35 (18 H, s, Bu'). rn/z = 498 (Found: C, 62.35; H, 5.55; N, 11.25. Calc. C, 62.50; H, 5.60; N, 1 1.20%). H, d, J = 4.9, H'), 7.51 (1 H, d, J = 1.5, H3), 7.35 (1 H, dd, (CDCI,): 'H, 6 1.45 (9 H, S, But), 7.15 (1 H, d, J = 5.0, H2), 8.05
Synthesis of [Ru"'(bbpc)(PPh,)Cl] 1.---A solution of H2bbpc (0.2 g), triethylamine (0.4 g) and [Ru(PPh,),CI,] (0.2 g) in ethanol (40 cm3) was refluxed under a slow stream of oxygen gas for 12 h. It was then cooled and filtered. The filtrate was evaporated to dryness and the solid residue extracted with CH,Cl,. The extract was evaporated to dryness and the crude product recrystallized by diffusion of diethyl ether into a dichloromethane solution. Dark green needle-shaped crystals were obtained. Yield = 30% (Found: C, 59.25; H, 4.45; N, 6.30. Calc. C, 58.95, H, 4.60; N, 6.25%) perf = 1 . 9 2 ~ ~ (at room temperature).
Crystal Structure Determination.-Crystal data. C44H41- Cl,N,O,PRu I , A4 = 896.24, orthorhombic, space group
Prtma, u = 11.939(3), b = 19.599(4), c = 17.709(3)
A,
U =4143.8(15)
A3,
Z = 4,D,
= 1.437 g cm-,, p(Mo-Ka) = 6.3cm-', F(OO0) = 1780, dimensions 0.10 x 0.10 x 0.45 mm. Intensity data were measured on a Nonius CAD-4 diffractometer using the 8-28 scan mode (20,,, = 49.8'). The structure was solved by Patterson and Fourier methods and subsequent refinement by full-matrix least squares using the NRCVAX program. l o The last least-squares cycle was
calculated with 55 atoms, 263 parameters and 2097 reflections ( ! I J > 2011~1) out of 3756 unique reflections, leading to
R
=0.047,
R'
= 0.036 and goodness of fit = 1.58. The weighting scheme was w-'
= a2(F). Table 1 lists the atomic coordinates of the non-hydrogen atoms, Table 2 selected bond distances and angles.Additional material available from the Cambridge Crystallo- graphic Data Centre comprises H-atom coordinates, thermal parameters and remaining bond lengths and angles.
Catalytic Reactions of A1kenes.-All organic substrates were purified by recrystallisation and/or distillation according to the literature methods. Ethyl diazoacetate and tert-butyl hydroper- oxide were used as received. Iodosylbenzene was prepared by hydrolysis of iodosylbenzene diacetate in sodium hydroxide solution. The compound PhI=NO,SC,H,Me-p was prepared by treating iodosylbenzene diacetate with toluene-p-sulfonyl-
amine in potassium hydroxide solution. All manipulations were handled under a nitrogen atmosphere. A typical oxidation reaction was as follows. A mixture of ruthenium catalyst (10 mg), alkene (0.2 g) and PhIO (0.1 g) in CH2C12 (10 cm3) was stirred at room temperature for 6 h. After addition of an internal standard an aliquot was taken for GC and/or 'H NMR analysis. For cyclopropanation, a dichloromethane solution of ethyl diazoacetate (0.2 g in 30 cm3) was added to a
dichloromethane solution mixture of complex 1 (20 mg) and styrene (0.5 g) over a period of 4-5 h. The organic product(s)
was obtained by chromatography of the reaction mixture on a silica gel column with light petroleum (b.p. 40-60 'C)-diethyl ether (9: 1) as eluent. These were analysed by 'H NMR spectroscopy and HPLC. For aziridination of styrene, a mixture of ruthenium catalyst (10 mg), styrene (0.2 g) and PhI= NO,SC,H,Me-p (0.1 g) in CH2Cl, (15 cm3) was stirred for 4-5 h under a nitrogen atmosphere and in the presence of molecular sieves. Chromatography of the reaction mixture on a silica gel column with light petroleum (b.p. 40-60 'C)-diethyl ether (8:2) as eluent gave the organic products which were characterised by 'H NMR spectroscopy.
Results and Discussion
Previous studies from this laboratory showed that the deprotonated forms of H,bpb and H,bpc are good (3 donors,
which readily form complexes with transition-metal ions. The [Mn(bpc)X] complexes are efficient oxidative catalysts for hydroxylation of C-H bonds and epoxidation of C=C bonds by PhIO. 3 b However, attempts to prepare ruthenium complexes
with these two amides were unsuccessful. In most cases an amorphous unidentified green solid was obtained. In fact, high- valent ruthenium complexes with chelating amide ligands are sparse. An example is [RurVL'(PPh,)(py)] [H,L' = 1,2- bis( 3,5-dichloro-2-hydroxybenzamido)ethane, py = pyridine] previously reported by Che et ~ 1 . ~ ' In this work a soluble derivative of H2bpc was prepared by introducing a tert-butyl group on the pyridine ring. The complex, [Ru"'( bbpc)(PPh,)Cl] 1 was prepared in a similar manner as that for [Ru"'(salen)- (PPh,)Cl] 4c*' and [ R U ' ~ L ' ( P P ~ , ) ( ~ ~ ) ] , ~ ~ involving the reac-
tion of [Ru(PPh,),Cl,] with the appropriate chelating amide in ethanol and in the presence of triethylamine and molecular oxygen. In the absence of triethylamine the reaction did not proceed. The presence of oxygen is essential to the formation of the ruthenium(1rr) complex. Under anaerobic conditions a diamagnetic red solid was isolated. This was found to react with oxygen in an acetone-methanol solution and in the presence of LiCl to give 1. We tentatively formulated this red solid as [Ru"(bbpc)(PPh,),]. The IR spectrum of 1 shows the absence of v(N-H) at 3296 cm
'
and a lower v(C=O) frequency than that of the free amide, indicating that the co-ordinated amide is in the deprotonated form. The measured perf of 1 . 9 2 ~ ~ is characteristic of a low-spin ruthenium(Ir1) complex. Its UV/VISspectrum in dichloromethane shows an intense absorption band at 791 nm, presumably due to P-+Ru'I' charge transfer. Similar bands in the range 600-800 nm have been observed for [ R U ' ~ L ' ( P P ~ , ) ( ~ ~ ) ] 2 h and [Ru"'(salen)(PPh,)Cl]
.
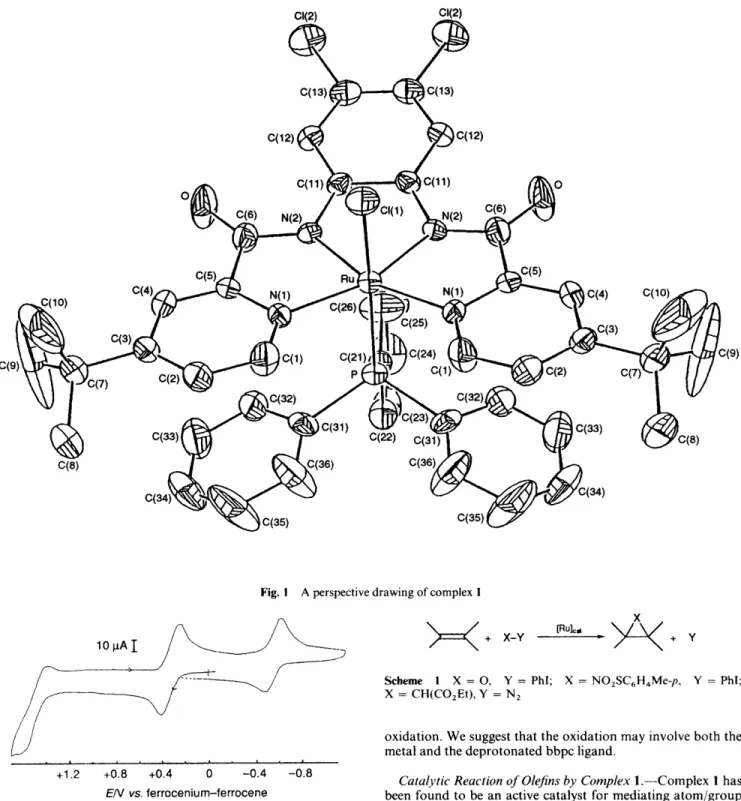
4cThe structure of complex 1 has been characterised by X-ray analysis and a perspective drawing is shown in Fig. 1. The ruthenium atom lies at the centre of the diamide ligand. The two axial ligands, PPh, and Cl, are truns to each other with a P-Ru-Cl angle of 168.38(11)". The Ru-N(2)-C(6) and Ru-N(2)-C( 11) angles are 1 1 8.9(5) and 113.0(4)' respectiveIy. These are close to 120°, which is the expected value for a sp2- hybridised amide nitrogen. The Ru-N(amide) distances of 1.980(6) and 2.088(6) 8, are comparable to those of 1.987(5) and 2.001(5) 8, in [ R u L ' ( P P ~ , ) ( ~ ~ ) ] . ~ ' The Ru-P distance of 2.38 l(3)
A
is significantly longer than that of 2.244(5) 8, in [Ru"- Lz( PPh ,)(OH2)] [H2L2 = N , N'-bis( 3,5-dichlorosalicylidene)- 1,2-diphenylethane- 1,2-diamine]. The shorter Ru-P bond is probably due to the Ru"-+PR, x-back-bonding interaction.1995
Fig. I A perspective drawing of complex I
U
+1.2 +0.8 +0.4 0 -0.4 -0.8
E N vs. ferrocenium-ferrocene
Fig. 2 Cyclic voltammogram of complex 1 in CH,C12 with 0.1 mol
dm tetrabutylammonium hexafluorophosphate as supporting electro- lyte. Scan rate: 100 mV s I . Working electrode: glassy carbon
The cyclic voltammogram of complex 1 in CH,CI, is shown in Fig. 2. There are three reversible/quasi-reversible couples.
The couple at - 0.55 V us. ferrocenium-ferrocene is tentatively assigned to Ru"'--Ru" since the free amide is electrochemically
inactive in the potential range 0 to - 1.0 V. This E" value is comparable to that of [Ru"'(salen)(PPh,)(O,SC,H,Me-p)] "
which shows a Ru"'-Ru" couple at -0.53 V. The oxidation couple at 0.32 V is comparable to the Ru'~-Ru"' couples of [Ru"'(salen)(PPh,)X] (X = C1 or N3), with E" at 0.38-0.39
V.4' However, the closely related [Os"'(bpc)(PPh,)CI]
complex shows an oxidation couple at 0.26 V, which is just 60 mV less anodic than that for 1. This small difference in
E" values cannot be rationalised by a pure metal-centred
Scheme 1 X = 0, Y = Phi; X = NO,SC,H,Me-p, Y = PhI; X = CH(CO,Et), Y = N2
oxidation. We suggest that the oxidation may involve both the metal and the deprotonated bbpc ligand.
Catalytic Reaction of Olejins by Complex 1.-Complex 1 has been found to be an active catalyst for mediating atom/group transfer to
>
C=C ,:: as in Scheme 1. For catalytic epoxidation the reaction took 4-6 h for completion and yielded either a green or a brown solution. With norbornene (bicyclo- C2.2. Ilhept-2-ene) and cyclooctene, organic epoxides were the dominant products. Allylic oxidation was a major pathway for the oxidation of cyclohexene. The oxidation of cis-stilbene gave a mixture of cis- and trans-stilbene oxide (2,3-diphenyloxirane) in a 4: 1 ratio and some benzaldehyde. For styrene, benz- aldehyde and styrene oxide (phenyioxirane) were obtained in a ratio of 2.4 : 1 . The results of epoxidation are summarised in Table 3. However, unlike the [Mn(bpc)Cl] system,3b 1 is inactive in catalysing alkane hydroxylation by PhIO, even after1 d. This is similar to the behaviour of [Ru(oep)(PPh,)Br]
(H,oep = 2,3,7,8,12,13,17,18-octaethylporphyrin) which is also inactive in alkane hydroxylation by PhI0.I4
The oxidation is proposed to involve dissociation of co- ordinated PPh, as represented by equation ( 1 ) . The
2218
J. CHEM. SOC. DALTON TRANS. 1995Table I
with estimated standard deviations (e.s.d.s) in parentheses
Positional parameters for non-hydrogen atoms in complex 1
X 0.976 39(7) 0.819 42(24) 1.105 5(3) 1.443 58(19) 1.1234(5) 0.917 3(5) 1.071 6(5) 0.840 4(7) 0.81 1 O(7) 0.860 4(6) 0.942 3(6) 0.969 O(6) 1.063 9(7) 0.827 l(7) 0.707 8(9) 0.842 7(16) 0.893 O( 1 1) 1.163 O(6) 1.251 2(6) 1.336 9(6) 0.844 l(9) 0.751 O(10) 0.765 9( 13) 0.868 6( 14) 0.959 8( 12) 0,947 7( 1 1) 0.732 7(6) 0.767 5(7) 0.71 1 5(9) 0.618 5(9) 0.587 6(10) 0.642 O(9) Y Z 0.25 0.122 19(5) 0.25 0.039 24( 17) 0.25 0.224 87( 17) 0.430 3(3) 0.066 6(4) 0.340 5(3) 0.170 O(3) 0.317 l(3) 0.068 7(3) 0.351 O(4) 0.223 7(5) 0.41 5 7(4) 0.249 2(5) 0.472 7(4) 0.219 4(4) 0.461 4(4) 0.165 3(5) 0.396 l(3) 0.142 6(4) 0.384 O(4) 0.087 7(5) 0.543 9(4) 0.243 4(5) 0.547 6(5) 0.265 O(9) 0.593 8(6) 0.181 5(8) 0.566 4(6) 0.305 8(8) 0.287 4(3) 0.032 8(4) 0.330 42(12) -0.079 65(14) 0.321 4(4) - 0.002 2(4) 0.285 4(4) -0.036 7(4) 0.25 - 0.062 7(6) 0.25 - 0.109 7(6) 0.25 -0.187 O(7) 0.25 -0.218 9(7) 0.25 -0.171 4(8) 0.25 - 0.093 4(7) 0.326 7(4) 0.046 9(4) 0.383 7(5) 0.010 5(6) 0.444 8 ( 5 ) 0.016 8(6) 0.450 O ( 5 ) 0.060 O(6) 0.396 3(6) 0.098 8(7) 0.333 O(5) 0.093 4(6) Table 2 e.s.d.s in parentheses
Selected bond distances
(A)
and angles (") for complex 1 withRu-P 2.381(3) Ru-N( 1 ) 2.088(6) Ru-CI( 1) 2.384(3) Ru-N(2) 1.980(6) P-Ru-CI(I) 168.38(11) Cl(l)-Ru-N(I) 84.79(16) P-RU-N( 1) 89.1 O( 16) C1( 1 )-Ru-N(2) 89.64( 18) P-Ru-N(2) 99.01(18) Cl(bbpc)Ru"'-PPh,
-
Cl(bbpc)Ru'*' + PPh, (1) Cl(bbpc)Ru"'+ PhIO
---
Cl(bbpc)RuV=O (2) Cl(bbpc)RuV=O+
PPh,-
Cl(bbpc)Ru"'-O=PPh3 (3) [Ru(bbpc)Cl) intermediate is subsequently oxidised by PhIO to give a RuV=O species [equation (2)]. This reacts with PPh, to give [Ru"'(bbpc)(OPPh,)Cl] [equation (3)]. Indeed, treatment of complex 1 with PhIO in dichloromethane and in the absence of alkene for about 3 h gave [Ru"'(bbpc)(OPPh,)Cl] which has been isolated and characterised by a X-ray analysis.*Complex 1 also catalyses cyclopropanation of styrene by ethyl diazoacetate. The combined yield of cis- and trans-ethyl 2-phenylcyclopropanecarboxylate was 98%. The reaction is quite stereospecific as a trans : cis ratio of 6 : 1 was found. Very little coupling product was detected. (However, in the presence of acetonitrile, no cyclopropanation reaction occurred and the
*
Crystal data: A4 = I 196.13, orthorhombic, space group Pnma, a = D , = 1.347 g ~ m - ~ , p(Mo-Ka) = 5.70 cm-', F(000) = 1936. There is a serious disorder of the co-ordinated O=PPh,. The final R and R' values were 0.047 and 0.036 respectively.'*12.623(4), b = 20.461(5), c = 18.122(4) A, U = 4680(2)
A3,
2 = 4,Table 3 Oxidation of alkenes by iodosylbenzene catalysed by com-
plex 1
Substrate Product(s)" Yield (%) (turnover) Norbornene exo-Norbornene oxide
Cyclooctene Cyclooctene oxide Styrene Styrene oxide
Benzaldeh yde Cyclohexene Cyclohexene oxide
Cyclohex-2-en- 1-01
Cyclohex-2-en-1 -one trans-Stilbene oxide Benzaldehyde trans-Stilbene' trans-Stilbene oxide
Benzaldehyde Cyclohexaned Cyclohexanol cis-Stilbene cis-Stilbene oxide
26.0 (1 2) 59.0 ( I 3) 19.0 (4) 45.0 (9) 14.0 (6) 12.0 (5) 25.0 ( I 1) 7.5 (4) 2.0 ( 1 ) 13.0 (7) 7.0 (4) 14.0 (8) < 3 . 0 ( < 1)
a Norbornene = bicyclo[2.2.l]hept-2-ene. Product yield determined by GC and based on the PhIO consumed unless otherwise specified. The yield was determined by 'H NMR spectroscopy with 1,l- diphenylethylene as internal standard. Reaction time over 24 h.
/c'(L)Ru-pPh3
\
+ N&H(COZEt) + PPh3f
CI(L)RU= CH(CO2Et) CI(L)Ru 'Ph Scheme2 L = bbpcunreacted ethyl diazoacetate was recovered even after stirring the reaction mixture for 24 h.) During the reaction, the solution mixture became deep green ( h = 610 nm). Presumably, the complex was first converted into an active species. The cyclopropanation reaction is proposed to occur via Scheme 2.
Aziridination of styrene by PhI=NO,SC,H,Me-p is also catalysed by complex 1. To our knowledge, this is the first ruthenium complex which catalyses alkene aziridination. The yield of 2-phenyl-N-toluene-p-sulfonylaziridines based on the amount of PhI=NO,SC,H,Me-p consumed is only 15% and a significant amount of sulfonylamine was formed. In addition, benzaldehyde was found to be a major product. During the reaction the brown solution became green. The UV/VIS spectroscopic studies showed that a broad absorption band at 610 nm slowly developed and the original band at 791 nm diminished. The optical spectrum, however is different from that of [Ru"'(bbpc)(PPh,)Cl] and [Ru1"(bbpc)(OPPh3)Cl]. However, attempts to characterize the complex have so far been unsuccessful.
Conclusion
The aim of this study was to develop new ruthenium catalysts for epoxidation, cyclopropanation, and aziridination of alkenes. The approach was to generate high-valent ruthenium 0x0, imido and alkylidene complexes with chelating amide ligands. The complex [Ru(bbpc)(PPh,)Cl], which has good solubility in organic solvents, has been prepared and found to
CLC bonds. Although the reactive intermediate has not been characterised, it is likely that a high-valent ruthenium- heteroatom multiple-bonded species, stabilised by the chelating diamide ligands is involved. The aziridination of styrene highlights the potential of imidoruthenium complexes in nitrogen atom-transfer reactions.
Acknowledgements
We acknowledge support from the Hong Kong Research Grants Council and The Frontier Applied Chemistry Laboratory of The University of Hong Kong. P.-H. K. acknowledges the receipt of a postgraduate studentship
administered by the University of Hong Kong.
References
K. A. Jmgensen, Chem. Rev., 1989,89,431.
( a ) T. J. Collins, Acc. Chem. Rex, 1994, 27, 279; (6) C. M. Che,
W. K. Cheng, W. H. Leung and T. C. W. Mak, J. Chem. Soc.,
Chem. Cummun., 1987,418.
(a)C. M. Che, W. K. ChengandT. C. W. Mak, J. Chem. Soc., Chem. Commun., 1986, 200; (b) C. M. Che and W. K. Cheng, J. Chem. SOC., Chem. Commun., 1986, 1443; ( c ) W. H. Leung, J. X. Ma, V. W. W. Yam, C. M. Che and C. K. Poon, J. Chem. SOC., Dalton
Trans., 1991, 1071; (d) Y. Yang, F. Z. Diederich and J. S. Valentine, J. Am. Chem. Soc., 1991,113,7195.
4 ( a ) K. Srinivasan, P. Michaud and J. K. Kochi, J. Am. Chem. Soc.,
1986, 108, 2309; (6) E. G. Samsel, K. Srinivasan and J. K. Kochi, J. Am. Chem. Soc., 1985,107,7606; ( c ) W. H. Leung and C. M. Che, Inorg. Chem., 1989,28,4619.
5 H. Nishino and J. K. Kochi, horg. Chim. Acta, 1990, 174,93. 6 R. L. Chapman, F. S. Stephens and R. S. Vagg, Innorg. Chim. Acta,
7 T. A. Stephenson and G. Wilkinson, J. Inorg. Nucl. Chem., 1966,28, 8 H. Saltzman and J. G. Sharekin, Org. Synth., 1963,43,60. 9 J. P. Mahy, G. Bedi, P. Battioni and D. Mansuy, J. Chem. Soc., 10 NRCVAX, E. J. Gabe, Y. Le Page, J. P. Charland, F. L. Lee
1 1 K. S. Murray, A. M. van den Bergen and B. 0. West, Aust. J. Chem.,
12 C. M. Che and S. M. Peng, unpublished work.
13 W. K. Cheng, K. Y. Wong, W. F. Tong, T. F. Lai and C. M. Che,
14 T. Leung, B. R. James and D. Dolphin, Inorg. Chim. Acta, 1983,79,
15 T. Y. Chen, Master Thesis, National Taiwan University, 1993. 1981,52, 161.
945.
Perkin Trans. 2, 1988, 1517.
and P. S. White, J. Appl. Crystallogr., 1989, 22, 384. 1978,31,203.
J. Chem. SOC., Dalton Trans., 1992,91. 180.