Tuning Excited-State Charge/Proton Transfer Coupled Reaction via the Dipolar
Functionality
Pi-Tai Chou,* Chien-Huang Huang, Shih-Chieh Pu, Yi-Ming Cheng, Yi-Hong Liu, Yu Wang, and Chao-Tsen Chen*
Department of Chemistry, National Taiwan UniVersity, Taipei, Taiwan, R. O. C. ReceiVed: June 1, 2004; In Final Form: June 28, 2004
Based on design and synthesis of I, II, and III, we demonstrate an ingenious approach to fine-tuning the excited-state intramolecular charge transfer (ESICT) coupled excited-state intramolecular proton transfer (ESIPT) reaction via the dipolar functionality of the molecular framework. Both I and II exhibit remarkable dual emission due to the different solvent-polarity environment between ESICT and ESIPT states, while the interplay of two charge-transfer entities in III leads to ESIPT decoupling from the solvent-polarity effect, resulting in a unique proton-transfer tautomer emission. The results make further rational design of the ESICT/ ESIPT coupled systems feasible simply by tuning the net dipolar effect. Accordingly, systematic investigation of the correlation in regards to the difference in dipolar vectors between ESICT and ESIPT versus solvent-polarity induced barriers becomes possible.
Due to its fundamental importance in chemical reactions, the excited-state intramolecular proton transfer (ESIPT) reaction has received much attention.1One important issue of current interest
regarding ESIPT reactions should be ascribed to the ESIPT coupled excited-state intramolecular charge transfer (ESICT). Considering that the Franck-Condon excited state of an ESIPT molecule undergoes a gigantic dipolar change due to the charge separation, the normal and proton-transfer tautomer equilibrium polarizations might be far separated. Since the large dipolar change in solution is normally coupled to solvent polarization effects, the relative energies between normal and tautomer in the excited state are a function of the solvent polarization coordinate. As a result, the long-range polarization interactions may result in a solvent-induced barrier channeling into the proton-transfer reaction.
Seminal studies on molecules exhibiting remarkable solvent polarization dependent ESIPT reaction dynamics should be ascribed to two prototypical systems, namely 4′ -N,N-diethy-lamino-3-hydroxyflavones (I) (see Scheme 1)2a-dand
para-N,N-dimethylamino-methylsalicylate.2e,f For the case of system I,
on one hand, ESICT takes place from the diethylamino nitrogen (electron donor) to the carbonyl oxygen (electron acceptor). On the other hand, similar to its parent molecule 3-hydroxyflavone (3HF),3I also undergoes ESIPT from the hydroxyl proton to
the carbonyl oxygen (see Scheme 1). The difference in solvent-polarity environment between charge transfer (CT) and proton transfer (PT) states leads to a remarkable ESICT/ESIPT coupled reaction. In polar, aprotic solvents, in contrast to a unique PT tautomer emission observed in 3HF,3 dual emissions are
resolved, consisting of CT and PT bands.2a-d Applying the
Marcus electron-transfer model, Swinney and Kelley2cwere able
to describe the overall reaction dynamics as well as to deduce the solvent-induced barrier for I by a combination of solvent polarity and proton-transfer reaction coordinates. In contrast to the ultrafast rate (<50 fs) of ESIPT in 3HF,4the proton tunneling
rate for I in polar solvents is slower than the rate of solvent
relaxation. Thus, a nonadiabatic type of proton transfer takes place essentially after solvent equilibration is established in the excited state.2c Such an intriguing ESICT/ESIPT coupled
property has led to intensive studies on I and its analogues. Most of the approaches focus on the wavelength tunability upon replacing the 2-phenyl ring by other functionalized aromatics,5
the applications in metal ion recognition,6and the use of CT/
PT emission intensity ratio as indicators to probe the empirical solvent polarity.7
Based on the chemical design, we herein demonstrate an ingenious approach to fine-tuning the ESICT/ESIPT coupled reaction via the dipolar orientation of the molecular framework. The synthetic strategy lies in that if the dialkylamine, which acts as an electron donor at the C(4′) position in I, plays a key role in ESICT, then switching the diethylamine from C(4′) to C(7) position, forming II (Scheme 1), may result in a similar ESICT/ESIPT coupled reaction. However, the resulting net dipole moment, the orientation of which is affected by the CT effect, is expected to alter with respect to that of I. Moreover, if 2-phenyl pyrone (region A in Scheme 1) and flavone (region B) chromophores are strongly coupled so that electrons are delocalized throughout the entire molecular framework, it would be intriguing to functionalize 3HF by adding dialkylamine at both the C(4′) and C(7) positions, forming III, of which the net dipole moment may be associated with the interplay between two charge-transfer systems, namely I and II. Thus, switching the dipolar property of the CT state may greatly tune the ESICT/ ESIPT coupling behavior among I-III.
I-III were synthesized by the Claisen condensation.2aWhile
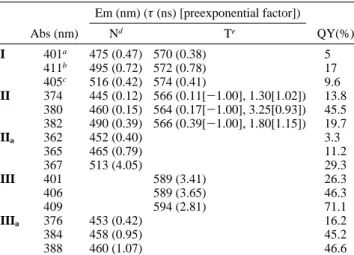
I has been reported, detailed synthetic schemes for II and III as well as their corresponding methoxyl derivatives, IIaand IIIa, respectively, are elaborated in the Supporting Information. Figure 1 shows the emission spectra of I-III in various polar solvents, and detailed photophysical data are listed in Table 1 (see Supporting Information for the absorption spectra). Similar to the steady-state emission behavior of I shown in Figure 1A,2 II exhibits dual emission, in which the peak wavelength of the higher energy band is strongly dependent on the solvent polarity
* Corresponding author. E-mail: [email protected]; [email protected]
6452 J. Phys. Chem. A 2004, 108, 6452-6454
10.1021/jp0476390 CCC: $27.50 © 2004 American Chemical Society Published on Web 07/10/2004
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
and is reasonably ascribed to a CT emission incorporating a charge transfer from C(7)-diethylamine to carbonyl oxygen. Conversely, the lower energy band with an anomalously large Stokes shift of >7000 cm-1reveals negligibly small solvent-polarity dependence and is clearly assigned to a PT tautomer emission. The charge-transfer property in II could be further supported by its methoxyl derivative IIa, in which ESIPT was prohibited and the resulting emission showed similar solvent-polarity dependence with that of the CT band in II (see Table 1). Figure 1C depicts the emission spectra of III in various solvents. In contrast to both I and II, III exhibits predominantly a proton-transfer emission, of which the peak wavelength reveals negligible solvent-polarity dependence. The results indicate that the dipolar change in the excited state for III must be small between normal and PT tautomer species so that ultrafast ESIPT takes place. This viewpoint can be firmly supported by the non-ESIPT analogue IIIa, which exhibits a nearly solvent-polarity
independent, normal Stokes shifted emission (see Table 1 and Figure 1C). The results lead us to propose that, upon excitation, the interplay between two charge-transfer dipole vectors in III yields a net dipole moment nearly the same as that of the PT tautomer. ESIPT is thus decoupled from the solvent polarity effect, resulting in a dominant PT tautomer emission.
A further dynamic approach allows us to establish a precur-sor-successor relationship between CT and PT bands for II. For example, in CH3CN the decay time of 390 ps in II
monitored at the CT band is identical to the rise time of the PT band (see Table 1 and Figure 2). The results are in contrast to I, in which the excited-state equilibrium between CT and PT is established, as supported by their identical population lifetimes (see Table 1 and also ref 2c). For the case of III, the rate of ESIPT is too fast to be resolved by our time-correlated single photon counting system (∼15 ps response time), supporting the solvent-polarity decoupled ESIPT mechanism for III.
Theoretical approaches at an INDO/S-CI level based on AM1 geometry optimized ground state structures have been performed in an attempt to rationalize the experimental results. All singly
SCHEME 1. Structures of Various Systems Studied and the Calculated (see text) Dipole Moments (arrows show
components along the z-x plane) for I, II and III in Singlet normal (S) and Proton Transfer (S′) States (angle specified clockwise to the z-axis)
Figure 1. Emission spectra of (A) I, (B) II, and (C) III (solid line) and IIIa(dash line) in ethyl acetate (0), CH2Cl2(4), and CH3CN (O).
The excitation wavelength is 350 nm.
TABLE 1: Photophysical Properties of I-III in Various Solvents at Room Temperature
Em (nm) (τ (ns) [preexponential factor]) Abs (nm) Nd Te QY(%)f I 401a 475 (0.47) 570 (0.38) 5 411b 495 (0.72) 572 (0.78) 17 405c 516 (0.42) 574 (0.41) 9.6 II 374 445 (0.12) 566 (0.11[-1.00], 1.30[1.02]) 13.8 380 460 (0.15) 564 (0.17[-1.00], 3.25[0.93]) 45.5 382 490 (0.39) 566 (0.39[-1.00], 1.80[1.15]) 19.7 IIa 362 452 (0.40) 3.3 365 465 (0.79) 11.2 367 513 (4.05) 29.3 III 401 589 (3.41) 26.3 406 589 (3.65) 46.3 409 594 (2.81) 71.1 IIIa 376 453 (0.42) 16.2 384 458 (0.95) 45.2 388 460 (1.07) 46.6
aIn ethyl acetate.bIn CH2Cl2.cIn CH3CN.dNormal band.ePT
tautomer.fFor I and II, QY(%)% is the sum of dual emission intensity.
Note that negative preexponential values indicate rise components.
Letters J. Phys. Chem. A, Vol. 108, No. 31, 2004 6453
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009
excited configurations from the 10 highest occupied to the 10 lowest unoccupied molecular orbitals were involved in the computation (see Supporting Information). Dipole moments were calculated for I, II, and III in both ground and excited states, and the dipole vectors along the z-x plane are depicted in Scheme 1. For III, the orientation of the dipole moment is similar between the normal ground (S0, 160°) and excited (S1,
155°) states, while significant differences were obtained for both I and II (see Scheme 1). More importantly, while the dipole vectors of excited CT (S1) and PT (S1′) states are substantially
different for both I and II (see Scheme 1), they are similar in
III (S1, (3.6 D, 155°), S1′(3.9 D, 140°)). Although this approach,
to a certain extent, is qualitative on the magnitude of the dipole moment, according to different dipole orientations, ESICT/ ESIPT coupled reactions are expected for I and II, and solvent polarities should play a key role in describing the reaction dynamics and/or thermodynamics. Conversely, negligible changes in dipolar orientations for III lead to a fast, solvent decoupled ESIPT, consistent with experimental results.
We have thus demonstrated for the first time a prominent dipole orientation tuning ESICT/ESIPT coupled reaction. The preservative net dipolar effect in III leads us to conclude that 2-phenyl pryone and flavone moieties are strongly coupled so CT in III is essentially delocalized in the entire molecule. This makes feasible further rational design of ESICT/ESIPT systems by altering the net dipolar vector. Accordingly, systematic investigation of the correlation in regards to the difference in
dipolar vectors between ESICT and ESIPT as well as the solvent-polarity induced barrier becomes possible, which may be crucial to gain fundamental insights into research fields of current interest regarding, e.g., proton-coupled electron transfer in living systems.8 Unfortunately, for I-III possessing
five-membered ring hydrogen bonds, the intramolecular hydrogen bonding strength is relatively weak. Thus, in protic solvents such as ethanol and water, our preliminary studies have shown that certain percentage of CdO- - -H-O- intramolecular hydrogen bond in compound I-III is ruptured by forming intermolecular hydrogen bond with protic solvents. As a result, excited-state deprotonation instead of ESIPT takes place in these protic solvents. This makes the discussion of the ESICT/ESIPT reaction solely based on the solvent polarity very complicated. Studies on the ESICT/ESIPT coupled reaction, in combination with the excited-state deprotonation, for compounds I-III in protic systems are currently in progress.
Supporting Information Available: Detailed experimental procedures, absorption, emission, time-resolved and X-ray studies. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) For recent reviews, see: (a) Scheiner, S. J. Phys. Chem. A 2000,
104, 5898. (b) Waluk, J. Conformational Aspects of Intra- and Intermo-lecular Excited-State Proton Transfer and Conformational Analysis of Molecules in Excited States; Waluk, J., Ed.; Wiley-VCH: New York, 2000.
(2) (a) Chou, P. T.; Martinez, M. L.; Clements, J. H. J. Phys. Chem.
1993, 97, 2618. (b) Chou, P. T.; Martinez, M. L.; Clements, J. H. Chem. Phys. Lett. 1993, 204, 395. (c) Swinney, T. C.; Kelley, D. F. J. Chem. Phys. 1993, 99, 211. (d) Parsapour, F.; Kelley, D. F. J. Phys. Chem. 1996, 100, 2791. (e) Gormin, D.; Kasha, M. Chem. Phys. Lett. 1988, 153, 574.
(f) Heldt, J.; Gormin, D.; Kasha, M. Chem. Phys. 1989, 136, 321. (3) (a) Sengupta, P.; Kasha, M. Chem. Phys. Lett. 1979, 68, 382. (b) McMorrow, D.; Kasha, M. J. Phys. Chem. 1984, 88, 2235.
(4) Ameer-Beg S.; Ormson, S. M.; Brown, R. G.; Matousek, P.; Towrie, M.; Nibbering, E. T. J.; Foggi, P.; Neuwahl, F. V. R. J. Phys. Chem. A
2001, 105, 3709.
(5) (a) Falkovskaia, E.; Pivovarenko, V. G.; Valle, J. G. Chem. Phys.
Lett. 2002, 352, 415. (b) Klymchenko, A. S.; Pivovarenko, V. G.;
Demchenko, A. P. J. Phys. Chem. A 2003, 107, 4211.
(6) (a) Bublitz, G. U.; Boxer, S. G. Annu. ReV. Phys. Chem. 1997, 48, 213. (b) Klymchenko, A. S.; Demchenko, A. P. J. Am. Chem. Soc. 2002,
124, 12372.
(7) Klymchenko, A. S.; Demchenko, A. P. Phys. Chem. Chem. Phys.
2003, 5, 461.
(8) For example, see: Milligan, J. R.; Aguilera, J. A. Hoang, O.; Ly, A.; Tran, N. Q.; Ward, J. F. J. Am. Chem. Soc. 2004, 126, 1682.
Figure 2. Time-resolved spectra of compound II (CT: monitored at 460 nm; PT: monitored at 640 nm; S: system response function).
6454 J. Phys. Chem. A, Vol. 108, No. 31, 2004 Letters
Downloaded by NATIONAL TAIWAN UNIV on July 27, 2009