酵母菌基因體的演化分析研究
78
0
0
全文
(2) 酵母菌基因體的演化分析研究 Evolutionary Analysis of the Yeast Genome. 研 究 生:林勇欣. Student:Yeong-Shin Lin. 指導教授:黃鎮剛. Advisor:Jenn-Kang Hwang. 國 立 交 通 大 學 生 物 科 技 系 博 士 論 文. A Dissertation Submitted to Department of Biological Science and Technology College of Biological Science and Technology National Chiao Tung University in partial Fulfillment of the Requirements for the Degree of PhD in Biological Science and Technology July 2006 Hsinchu, Taiwan, Republic of China. 中華民國九十五年七月.
(3) 酵母菌基因體的演化分析研究 研 究 生:林勇欣. 指導教授:黃鎮剛博士. 國立交通大學 生物科技系 博士班. 摘. 要. Saccharomyces cerevisiae 經由古老的全基因體複製(whole-genome duplication,WGD)產生的重複基因(duplicate genes)中,有許多基因對之間表 現出了較預期低許多的同義分歧(synonymous divergence,KS),有些基因對之 間的序列相似性比該基因和 S. bayanus 的同源基因(orthologue)之間的相似性 更高,或者是和 Kluyveromyces waltii(在 WGD 發生之前分化的物種)的同源基 因相比,擁有較慢的演化速度。這樣的減速演化(decelerated evolution)過去被 歸因於重複基因之間的基因轉換(gene conversion)。在這篇論文的第一部份, 我探討了四個物種中約三百個 WGD 基因對,以及這些基因對在非 WGD 物種中 的同源基因,並因此發現了密碼子使用偏移(codon usage bias)以及蛋白質序列 的保守性是造成重複基因對的減速演化兩個重要的原因。基因轉換只有在巨大的 密碼子使用偏移或是非常保守的蛋白質序列存在的情況下,才能有效地對減速演 化造成影響。我更進一步發現,突變型態的改變,或是 tRNA 編碼基因拷貝數目 (tDNA copy number)的改變,會造成密碼子使用偏移的改變,也因此導致 K. waltii 及 S. cerevisiae 之間同義距離(KS distance)的增加。很有趣地,有些蛋白 質在 WGD 物種輻射狀種化之前表現出很快的演化速率,然而,在輻射狀種化之 後他們的演化速率卻降得很低,甚至不再有變化。這代表功能上的保守性對於重 複基因對的減速演化也有很大的影響。. 接下來,我利用功能性基因體及蛋白質結構資料,探討蛋白質複雜性(蛋 白質次單元種類的數目,protein complexity)對基因的可移除性(dispensability) 及可複製性(duplicability)的影響。結果發現,基因可複製性在異複合體(由兩 i.
(4) 種以上的次單元所構成的複合體,hetero-complexes)及同複合體(單體或是單一 種次單元所構成的複合體,homo-complexes)之間存在明顯的差異。然而,基因 的可移除性則是隨著蛋白質複合體次單元種類數目的增加而逐漸降低。這代表劑 量平衡假說(dosage balance hypothesis)雖然能夠解釋蛋白質複合體的基因可複 製性,卻無法完美的解釋不同的異複合體之間基因可移除性的差異。可能的情況 是當一個異複合體次單元基因被剔除的時候,整個複合體的功能都會受到影響。 因此這個基因被剔除造成的適性(fitness)影響會隨著蛋白質複雜性的升高而增 加。此外我發現具有多功能區(multi-domain)的多肽基因和只具有單一功能區 的多肽基因相比,有較低的可移除性和較高的可複製性。經由 WGD 產生的重複 基因(不含核糖體次單元基因)普遍的比其他的重複基因擁有較高的可移除性。 而屬於同一個複合體的次單元通常傾向有類似的表現量和類似的剔除適性影 響。最後,我估計重複基因對於基因突變頑抗性(genetic robustness against null mutation)的貢獻大約是 9%,比前人估計的要小許多。對酵母菌的基因可移除 性來講,蛋白質的複雜性應該比重複基因的影響來的重大許多。. 最後我所探討的是蛋白質演化速率。最近的研究指出,酵母菌中蛋白質的 演化速率唯一的主要決定因素在於轉譯效率的選擇力(translational selection) 上,這可以由 mRNA 和蛋白質的表現量以及密碼子適應值(codon adaptation index)來表示。本研究則說明蛋白質的結構其實也有舉足輕重的影響。為了要 維持蛋白質的結構穩定,包埋在蛋白質內部或是位於蛋白質交互作用面的殘基 (residue)通常比暴露在蛋白質表面接觸溶劑的殘基面對更強的演化拘束力。經 由淨相關(partial correlation)分析發現,蛋白質中暴露殘基的百分比(Pexposed) 可以解釋的演化速率變異量,可達到轉譯效率的選擇力所能解釋的一半以上。這 個結果和功能性密度(functional density)假說是一致的,也就是說,蛋白質若 擁有較多殘基與特定功能相關(如穩定蛋白質結構或是蛋白交互作用),會因此 而傾向擁有較慢的演化速率。. ii.
(5) Evolutionary Analysis of the Yeast Genome Student: Yeong-Shin Lin. Advisor: Dr. Jenn-Kang Hwang. Department of Biological Science and Technology National Chiao Tung University Abstract Many Saccharomyces cerevisiae duplicate genes that were derived from an ancient whole-genome duplication (WGD) unexpectedly show a small synonymous divergence (KS), a higher sequence similarity to each other than to orthologues in S. bayanus, or slow evolution compared to the orthologue in Kluyveromyces waltii, a non-WGD species. This decelerated evolution was attributed to gene conversion between duplicates. Using ~300 WGD gene pairs in four species and their orthologues in non-WGD species, the first part of my thesis shows that codon usage bias and protein sequence conservation are two important causes for decelerated evolution of duplicate genes, whereas gene conversion is effective only in the presence of strong codon usage bias or protein sequence conservation. Further, I found that change in mutation pattern or in tDNA copy number changed codon usage bias and increased the KS distance between K. waltii and S. cerevisiae. Intriguingly, some proteins showed fast evolution before the radiation of WGD species but little or no sequence divergence between orthologues and paralogues thereafter, indicating that functional conservation after the radiation may also be responsible for decelerated evolution in duplicates. In the second part, I studied the effects of protein complexity (here defined as the number of subunit types in a protein) on gene dispensability and gene duplicability using functional genomic and protein structural data. I found that the major distinction for gene duplicability in protein complexity is between iii.
(6) hetero-complexes, each of which includes at least two different types of subunits (polypeptides), and homo-complexes, which include monomers and complexes that consist of only subunits of one polypeptide type. However, gene dispensability decreases only gradually as the number of subunit types in a protein complex increases. These observations suggest that the dosage balance hypothesis can explain gene duplicability of complex proteins well, but cannot completely explain the difference in dispensabilities between hetero-complex subunits. It is likely that knocking out a gene coding for a hetero-complex subunit would disrupt the function of the whole complex, so that the deletion effect on fitness would increase with protein complexity. I also found that multi-domain polypeptide genes are less dispensable but more duplicable than single domain polypeptide genes. Duplicate genes derived from the whole genome duplication event in yeast are more dispensable (except for ribosomal protein genes) than other duplicate genes. Further, I found that subunits of the same protein complex tend to have similar expression levels and similar effects of gene deletion on fitness. Finally, I estimated that in yeast the contribution of duplicate genes to genetic robustness against null mutation is ~ 9%, smaller than previously estimated. In yeast, protein complexity may serve as a better indicator of gene dispensability than do duplicate genes. The last part is a study related to protein evolutionary rate. Recently, translational selection, including mRNA expression, protein abundance, and codon adaptation index, has been suggested as the single dominant determinant of protein evolutionary rate in yeast. This study shows that protein structure is an important determinant as well. Buried residues, which are responsible for maintaining protein structure or located on a stable interaction surface, are under stronger constraints than solvent-exposed residues. Partial correlation analysis shows that the variance of evolutionary rate explained by the proportion of exposed residues (Pexposed) can reach more than half of that explained by translational selection. This result suggests that proteins with many residues involved in specific functions (e.g. maintaining structure iv.
(7) or protein interaction) may evolve more slowly, which is consistent with the “functional density” hypothesis.. v.
(8) Acknowledgment 感謝黃鎮剛老師這五年來,在學業上的指導及生活上的照顧,讓我有機會 進入結構生物學的殿堂,學習這些有趣新穎的題目。在跨領域的研究中,更要感 謝黃老師耐心的指導、支持以及包容。不論是對學術研究工作的熱忱,或是待人 處事的諄諄教悔,老師都是值得學習的榜樣。在芝加哥一年的生活,則非常感謝 李文雄老師的關照,並開拓了我的視野。97 年夏天認識李老師之後,陸陸續續 受到老師很多的協助,把我當成自己的學生一樣的照顧。李老師對後輩的提攜總 是不遺餘力,令人感動。當然,還要感謝曾晴賢老師多年來的關心指導,帶領我 走入學術研究的道路。如果說,我現在對研究工作稱得上有一些熱情、堅持與自 信,可以說都是在清華的那段歲月所奠定的基礎。我還要感謝楊昀良老師、何信 瑩老師、丁照棣老師及黃憲達老師在口試的時候所提供的許多寶貴的建議,以及 楊進木老師和彭慧玲老師在論文初期所給予的協助與指導。來自不同領域的意見 總是能讓我有機會訓練自己從不同的角度思考,獲益良多。. 從傳統生物學跨足到生物資訊,若不是鎮熊、景盛、存操、啟德屢次傾囊 相授,並將工作站維護的友善而順暢,憑我這只懂皮毛、勉強堪用的電腦知識, 事倍功半可能也不足以形容我的窘狀。另一方面,雖然離開清華到了不同的實驗 室,和玉萍的討論總是能激發出許多有趣的想法。研究的路途一路走來,並不覺 得孤單。還要感謝雅雯、立青、雲輝、Ay、Joshua、Jake 在我待在芝加哥那段期 間生活的照應及研究討論。特別是雅雯像家人般的噓寒問暖,協助日常生活大小 事務,讓我雖然身處異鄉,仍倍覺溫暖。思民學長帶我入門,也為我立下典範, 即使研究的領域不再相同,仍永遠會是我追隨學習的對象。志豪、玉菁、蔚倫, 因為你們的努力,才讓我淺薄的想法有實現的機會。未曾謀面的顏聖紘學長指點 我許多分類學的概念,這是網路世代才有的機緣。其他黃鎮剛實驗室、李文雄實 驗室、曾晴賢實驗室的夥伴,以及我的大學同學們,雖然無法一一將你們的名字 列出來,但是當我挫折沮喪時,有你們的加油打氣,遇到困難時,相互扶持,這 份情誼,不會忘記。. vi.
(9) 最後要感謝上琪及家人的支持與體諒。我知道在親友面前很難解釋這十年 來,我究竟是在做什麼。因為你們,我才能安心的把自己關進這象牙塔裡。你們 永遠是我最好的避風港。. vii.
(10) Contents 中文摘要. …………………………………………………………. i. Abstract. …………………………………………………………. iii. Acknowledgment …………………………………………………………. vi. Contents. …………………………………………………………. viii. Abbreviations. …………………………………………………………. ix. Chapter 1. General Introduction ..…………………………………. 1. Chapter 2. Codon usage bias versus gene conversion in the evolution of yeast duplicate genes. Chapter 3. Chapter 4. Introduction ……………………………………………. 6. Materials and Methods…………………………………. 6. Results and Discussion ..………………………………. 9. Protein complexity, gene duplicability and gene dispensability in the yeast genome Introduction ……………………………………………. 17. Materials and Methods…………………………………. 19. Results ....………………………………………………. 22. Discussion ..……………………………………………. 28. Protein structure and evolutionary rate Introduction ……………………………………………. 32. Materials and Methods…………………………………. 32. Results and Discussion ..………………………………. 33. References. …………………………………………………………. 37. Tables. …………………………………………………………. 44. Figures. …………………………………………………………. 58. viii.
(11) Abbreviations ACC. solvent accessible surface area. CAI. codon adaptation index. DSSP. database of secondary structure assignments for all PDB entries. KA. nonsynonymous distance. KS. synonymous distance. KS test. Kolmogorov-Smirnov test. MIPS. Munich Information Center for Protein Sequences. ORF. open reading frame. PDB. Protein Data Bank. Pexposed. proportion of solvent exposed residues in one protein. PSSM. position-specific scoring matrices. RelACC. relative solvent accessibility. SGD. Saccharomyces Genome Database. SVM. supporting vector machine. tDNA. transfer RNA coding gene. WGD. whole genome duplication. WGD genes. duplicated gene pairs derived from WGD. YDPM. Yeast Deletion Project and Proteomics of Mitochondria Database. ix.
(12) Chapter 1 General Introduction For decades, Saccharomyces cerevisiae, also called budding yeast or baker’s yeast, has been one of the best model organisms for genetics, cellular mechanisms and physiological studies. This unicellular organism, unlike more complex eukaryotes, can be grown on various laboratory conditions, which is important for functional genomics analyses. Moreover, many of the substantial cellular functions are highly conserved from yeast to mammals. In 1996, S. cerevisiae became the first completely sequenced eukaryote (Goffeau et al., 1996). Comparative studies with the following sequenced eukaryotic genomes therefore ushered in the “post-genomic era”. Here in this dissertation I used yeast genomic data to study a number of interesting evolutionary problems. One important biological evolutionary mechanism is duplication, which provides extra genetic material that substantially can be remodelled into “novel” gene products. Lynch and Conery (2000) estimated gene duplication rate as one duplication per gene per 100 million years using three completely sequenced eukaryote genomes. Gao and Innan (2004) suggested that this rate should be two orders of magnitude lower because there are many extensive concerted evolution via gene conversion between duplicated genes. Using comparative genomics data from yeast species, Wong, Butler and Wolfe (2002) showed that almost the entire S. cerevisiae genome lies in duplicated sister regions, which suggests that the entire genome became duplicated at some point, followed by rearrangement and gene loss. When the genomes of Kluyveromyces waltii and Ashbya gossypii were completely sequenced (Dietrich et al., 2004; Kellis, Birren and Lander, 2004), the whole-genome duplication (WGD) event was finally confirmed and the ancient gene order was clearly identified. About 10% of the WGD genes have been preserved after massive gene loss. One. 1.
(13) member of each duplicate pairs often has evolved rapidly into a novel gene with a derived function (Wagner, 2002; Kellis, Birren and Lander, 2004). While a majority of yeasts cannot grow in the absence of oxygen (aerobic yeasts), a majority species of the Saccharomyces complex can survive without any oxygen (Pronk, Steensma and van Dijken, 1996; Moller, Olsson and Piskur, 2001). The Saccharomyces sensu stricto yeasts, including S. bayanus, S. cariocanus, S. cerevisiae, S. kudriavzevii, S. mikatae and S. paradoxus, represent an isolated and well-supported monophyletic group with overall phenotypic similarity (Kurtzman and Robnett, 2003). Kwast et al. (2002) showed that some of the segmental duplicated genes have remodelled their expression to become dependent on the presence/absence of oxygen and glucose. The number of shared regulatory motifs in the duplicates decreases with evolutionary times, whereas the total number of regulatory motifs remains unchanged (Papp, Pal and Hurst, 2003a). The ribosomal proteins module can switch from employing one cis-element into another through the formation of redundant intermediate promoters harbouring both cis-elements in a tightly coupled configuration (Tanay, Regev and Shamir, 2005). The loss of a specific cis-regulatory element from dozens of genes following the apparent WGD event is connected to the change in gene expression for mitochondrial and cytoplasmic ribosomal proteins, and the emergence of the capacity for rapid anaerobic growth for these Saccharomyces complex yeasts (Ihmels et al., 2005). These studies suggested that WGD apparently provided new genes or regulatory elements, which were the basis for major remodelling of metabolism, including the development of an efficient glucose repression pathway and oxygen independence, in Saccharomyces complex. Duplicate genes have also been used to explain the genetic robustness against mutations through functional compensation (e.g., Nowak et al., 1997; Gu et al., 2003; Conant and Wagner, 2004; Kafri, Bar-Even and Pilpel, 2005). Most S. cerevisiae genes are nonessential under laboratory conditions (Winzeler et al., 1999; Glaever et 2.
(14) al., 2002; Steinmetz et al., 2002). Based on the dosage balance hypothesis (Veitia, 2002; Veitia, 2003), i.e., similar dosage among subunits in a protein complex is preferred, Papp, Pal and Hurst (2003b) and Yang, Lusk and Li (2003) have shown that protein complexity is an important determinant of gene duplicability. While based on the dosage theory (Kondrashov and Koonin, 2004), duplication is preferred for highly expressed genes. Deutschbauer et al. (2005) showed that the primary mechanism of haploinsufficiency, which is defined as a dominant phenotype in diploid organisms that are heterozygous for a loss-of-function allele, is due to insufficient protein production. He and Zhang (2005) suggested that duplicate genes have longer protein sequences, more functional domains, and more cis-regulatory motifs than singleton genes. They also proposed that non-important (dispensable) genes have higher probability to duplicate (He and Zhang, 2006), although other studies showed that duplicate genes are usually conserved, i.e., with remote orthologues (Davis and Petrov, 2004; Jordan, Wolf and Koonin, 2004). Dispensable genes (Hirsh and Fraser, 2001; Yang, Gu and Li, 2003; Wall et al., 2005; Zhang and He, 2005) or proteins with more interactions (Fraser et al., 2002) have also been proposed to evolve slowly. However, highly expressed proteins also evolve slowly (Pal, Papp and Hurst, 2001; Rocha and Danchin, 2004; Wall et al., 2005), and usually tend to be indispensable; meanwhile, their interactions may have higher chance to be identified (Bloom and Adami, 2003; Pal, Papp and Hurst, 2003). Recently, Drummond, Raval and Wilke (2006) proposed that translational selection, including mRNA expression, protein abundance, and codon adaptation index, is the single dominant determinant of protein evolutionary rate. In the following chapters, I first studied how gene conversion and codon usage bias affect the decelerated evolution of WGD genes. Then I collected protein complex data to analyze its relationships with gene dispensability and gene duplicability, and. 3.
(15) also reveal how protein complexity and protein structure may determine protein evolutionary rate.. 4.
(16) Chapter 2 Codon usage bias versus gene conversion in the evolution of yeast duplicate genes. 5.
(17) Introduction Gene conversion has been extensively studied in yeast (Petes and Hill, 1988; Petes, 2001). Recently, Kellis, Birren and Lander (2004) identified 60 gene pairs in Saccharomyces cerevisiae that were derived from an ancient whole-genome duplication (WGD) but showed a small sequence divergence. They suggested that these genes have undergone gene conversion for three reasons. First, in 90% of the cases, both paralogues show decelerated evolution (at least 50% slower than the orthologue in Kluyveromyces waltii). Second, nucleotides at fourfold degenerate codon positions for these genes are highly conserved. Third, in about half of the cases, the two paralogues in S. cerevisiae are closer in sequence to each other than either is to its syntenic orthologue in S. bayanus. Similarly, Gao and Innan (2004) attributed the small synonymous divergence (KS) between ancient duplicated genes in yeast to gene conversion. However, most WGD gene pairs with decelerated evolution (Kellis, Birren and Lander, 2004) have an extremely strong codon-usage bias (Fig. 1). Codon-usage bias is known to increase with gene expression level (Coghlan and Wolfe, 2000; Akashi, 2001) and can slow down synonymous divergence between duplicate genes (Pal, Papp and Hurst, 2001). Therefore, I am interested to investigate whether codon usage bias rather than gene conversion is more important for the decelerated evolution. Materials and Methods Sequence data. I used the whole genome duplication (WGD) gene pairs in S. cerevisiae and their orthologues in K. waltii (Kellis, Birren and Lander, 2004) and Ashbya gossypii (Dietrich et al., 2004), and included their syntenic orthologues from three other species, S. bayanus, S. mikatae and S. paradoxus (Kellis et al., 2003). All sequences were aligned using the amino acid sequences with CLUSTAL W 1.83 (Thompson, Higgins and Gibson, 1994) and their corresponding DNA sequences were therefore used. The synonymous nucleotide divergence (KS) values were estimated 6.
(18) using PAML 3.14 (Yang, 1997). Codon adaptation index (CAI) values (Sharp and Li, 1987), each of which indicates the strength of codon usage bias, were obtained from MIPS (Mewes et al., 2002) for S. cerevisiae genes. Identification of gene conversion events. Numerous methods for gene conversion identification have been developed, but these methods are either not suitable or not powerful enough for the present analysis. For example, S. Sawyer’s method uses measures of the distribution of identical synonymous sites between sequence pairs to identify candidate regions of conversion (Sawyer, 1989). This method assumes a neutral evolutionary process for synonymous sites and may therefore not be suitable for yeast genes in which codon usage bias affects synonymous substitution. More importantly, it does not use any outgroup for reference, so it is in general less powerful than phylogeny-based methods. Other methods, such as those of Jakobsen and coworkers (1996; 1997), rely on the examination of site-by-site phylogenies and the phylogeny for each site in a multiple alignment of paralogues and orthologues is tested for its support of conversion. Although these methods are similar to what proposed in this study, they suffer when there are multiple substitutions at individual sites (Drouin et al., 1999). This may again be a problem in my analysis as I am examining the ancient duplicates retained from the whole genome duplication in yeast in which multiple substitutions are common. Therefore, I have developed a related algorithm for conversion identification. I used WGD orthologues in the 4 genomes, S. cerevisiae, S. bayanus, S. mikatae and S. paradoxus. At nucleotide position i, let Di = the number of nucleotide differences between the two nucleotides in paralogous gene 1 and gene 2 in species 1 (the species under study), and Bji = the number of nucleotide differences in gene j (j = 1, 2) between species 1 and its orthologue in species 2. Let Bi = (B1i + B2i) / 2. Sequences with gaps longer than 50% of the alignment were removed. For a gene under study, species with only one (or no) paralogue available are also removed. Gaps are all removed. For S. cerevisiae, S. paradoxus, or S. mikatae, Bi is calculated 7.
(19) between the species under study and S. bayanus. For S. bayanus, Bi is calculated as the average of the differences between S. bayanus and the available three species. Under the null hypothesis of no gene conversion, the distance (number of differences) between the two paralogues in a species should be larger than or equal to the distance between orthologues, i.e., Di - Bi ≥ 0, because the duplication event occurred prior to speciation. Dynamic programming is used to select the segment n. ∑ (B. from site m to n that maximizes. i =m. n. Let D =. − Di ) . This segment has N sites, N = n - m + 1.. n. ∑D i =m. i. and B =. i. ∑B i =m. i. . If N ≥ 20, the binomial probability to observe D ≤ B. for a segment of N sites is calculated using the orthologous distance B as the expected distance, i.e., D = B. This is a stringent criterion because the WGD event occurred earlier than speciation events. The estimated probability is N! B k = 0 k!( N − k )! N D. P ( B, D, N ) = ∑. k. B 1 − N. N −k. [1]. However, this segment always has its first and last sites supporting Bi > Di, which may cause an overestimate of the significance. Therefore, I remove the first or n. the last site of the segment, and recalculate B and D as. ∑ Bi and. i = m +1 n −1. ∑ Bi and i =m. n −1. ∑D i =m. i. n. ∑D. i = m +1. i. , or. , and obtain binomial probabilities p1 and p2, respectively. The. higher value of p1 and p2 is used. The segments thus identified with the paralogous distance significantly smaller than the orthologous distance might potentially be derived from gene conversion. However, many possible segments of N sites can be selected from the entire gene sequence, so it is necessary to take this factor into consideration. Therefore, for each segment with a binomial probability p < 0.01 computed from [1], an empirical distribution of B for a segment of length N is constructed using 10,000 bootstrap. 8.
(20) samples from {B1, B2, ..., BL}, where L = alignment length for the gene under consideration. Then, it is possible to determine the significance of D by counting the proportion of samples for which D < B. Segments with a binomial probability p < 0.01 and with an empirical probability < 0.01 are considered candidate gene conversions. Codon usage frequencies and tDNA genes. Relative frequencies of codon usage in. orthologues of WGD genes were calculated for the genomes of K. waltii, A. gossypii, S. cerevisiae, S. bayanus, S. mikatae and S. paradoxus. Two sets of gene pairs were obtained. S. cerevisiae genes with CAI > 0.5 were classified into the highly expressed set and so were their orthologues in other species, whereas genes with CAI < 0.2 were classified into the lowly expressed set. The Chi-square test was used to examine if a codon is favored in highly expressed genes compared with lowly expressed genes. I obtained tDNA genes of S. cerevisiae from MIPS, and used the sequences and genomic BLAST in NCBI to identify orthologues in the other 5 genomes. Results and Discussion. I first use the hypothetical trees in Fig. 2 to explain that a gene conversion event can distort the branch lengths and the topology of the phylogeny of duplicate genes and their orthologues among species. For example, the distance between paralogues α and a is expected to be longer than that between orthologues α and β (Fig. 2A) but the opposite is true in Fig. 2B because of a gene conversion event. To see how often such a situation has occurred in yeast duplicate genes, I studied ~300 WGD gene pairs in S. cerevisiae and their syntenic orthologues from three related species, S. bayanus, S. mikatae and S. paradoxus (Kellis et al., 2003). Because the WGD occurred prior to the radiation of these species, in the absence of gene conversion the synonymous distance (KS) is expected to be larger between S. cerevisiae paralogues than between orthologues in different species. I find that this expectation indeed holds in most cases, with 93.4% of duplicate pairs in S. cerevisiae having a paralogous KS greater than or 9.
(21) equal to the KS between orthologues (Fig. 3). This result indicates that only in a small proportion of these WGD duplicate genes has the tree topology been distorted by gene conversion because only when a point is below the line in Fig. 3 would a distortion in topology have occurred. Interestingly, most S. cerevisiae paralogous pairs with a small KS also show a small KS between orthologues and many have a high codon adaptation index value (CAI, a large circle in Fig. 3), a measure of codon usage bias (Sharp and Li, 1987). This analysis suggests that decelerated evolution of S. cerevisiae paralogues is at least in part due to biased codon usage, which serves as an evolutionary constraint (Pal, Papp and Hurst, 2001; Hirsh, Fraser and Wall, 2005). Here are two examples illustrating different effects of gene conversion and codon usage bias on the evolution of duplicate genes. The first one is the gene pair YGR138C / YPR156C indicated by the red arrow in Fig. 3. The small circle indicates that these two genes have a weak codon usage bias (CAI 0.310 / 0.261), which is also reflected in the large KS distance between orthologues. However, contrary to expectation, the KS distance between the two S. cerevisiae paralogues is smaller than those between orthologues (Fig. 3), suggesting that gene conversion has occurred between the two S. cerevisiae paralogues. Indeed, the phylogenetic tree in Fig. 4A shows that the paralogues in each of the first three species are clustered, indicating gene conversions in these species after speciation. The second example is the gene pair YML063W / YLR441C indicated by the green arrow in Fig. 3. The large circle indicates a strong codon usage bias (CAI 0.769 / 0.696), which is reflected by small KS values. The tree topology is as expected (Fig. 4B), so it provides no evidence of gene conversion. Despite this, the tree branches in Fig. 4B are in general much shorter than those in Fig. 4A. Clearly, codon usage bias can slow down sequence evolution in the entire tree, whereas gene conversion can shorten only sequence divergences between paralogues, but not those between syntenic orthologues.. 10.
(22) To pursue the analysis further, I reconsidered the 66 duplicate gene pairs identified by Gao and Innan (2004) to have a small KS between S. cerevisiae paralogues. I found that 57 of them were duplicated before the divergence between S. cerevisiae and S. bayanus and only one of these 57 pairs (YGL147C / YNL067W) is not from WGD (Dietrich et al., 2004; Kellis, Birren and Lander, 2004). In the 57 phylogenies for these 57 pairs, only 8 pairs showed a completely distorted tree topology (suggesting conversion in all lineages) like Fig. 4A, 23 pairs showed a partially distorted topology, while about half of them (26 pairs) showed no topology distortion (Table 1). I note that with the exception of two (YDL131W / YDL182W and YDR312W / YHR066W) all of the 57 pairs have a strong codon usage bias (CAI > 0.5). Therefore, in many of these gene pairs the small KS values between S. cerevisiae paralogues (and between orthologues) might be largely due to strong codon usage bias constraint. The above phylogenetic analysis, however, is not powerful enough for detecting all gene conversion events because conversion events involving only a small DNA region are unlikely to change the tree topology. For this purpose, a statistical method has been developed to detect gene conversion events and has been applied to ~300 WGD duplicate gene pairs in S. cerevisiae, S. paradoxus, S. mikatae and S. bayanus. The main purpose is to see whether gene conversion occurred primarily in high CAI genes. Indeed, Table 2 shows that about half of the genes with CAI ≥ 0.7 have undergone gene conversion events, while only 2% of the genes with CAI < 0.5 have conversions (p < 10 –8 for all species). Apparently, codon usage bias increases the rate of gene conversion by reducing the rate of sequence divergence. In the absence of strong codon usage bias, synonymous divergence between duplicate genes increases with time, and the chance of gene conversion is concomitantly reduced. Another intriguing observation was that for most duplicate gene pairs that show a small protein distance, the divergence between the K. waltii - A. gossypii and 11.
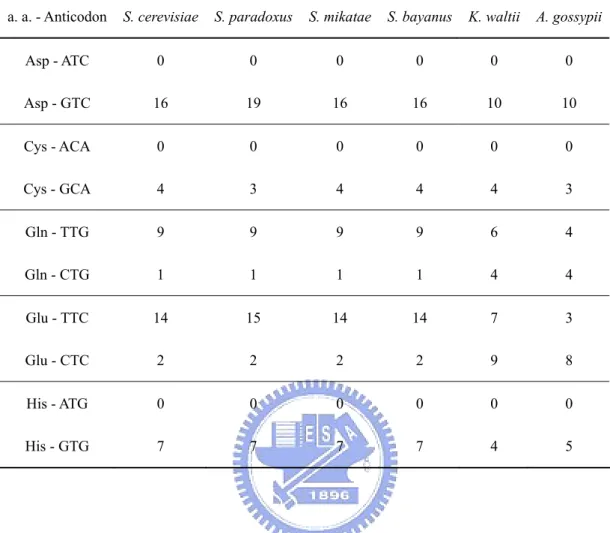
(23) Saccharomyces sensu stricto species lineages is much longer (e.g. Fig. 5). This observation has been taken as evidence of gene conversion in the Saccharomyces species under study (Kellis, Birren and Lander, 2004). However, I notice that in these genes the protein distances are short not only between paralogues in the same species but also between orthologues in different WGD species, indicating that protein sequence conservation rather than gene conversion was the major cause of decelerated evolution. In the period immediately following the WGD event the duplicate proteins had apparently evolved rather rapidly (Fig. 5), likely due to relaxed functional constraints following WGD or the emergence of anaerobic growth, which has been found to be connected with cis-regulatory element evolution (Ihmels et al., 2005) . During this period gene conversion might have played a key role in maintaining the sequence similarity between the two paralogues. However, the rate of evolution had evidently become very slow prior to the radiation of the four Saccharomyces species (Fig. 5) and this largely explains why the sequence divergence is small between not only paralogues but also orthologues. As for synonymous substitutions, previous studies showed that overlooking nucleotide composition differences (Tarrio, Rodriguez-Trelles and Ayala, 2001) or codon-usage patterns (Christianson, 2005) among sequences can mislead phylogenetic reconstruction. An examination of the codon usage patterns reveals that genes in K. waltii and A. gossypii have a stronger preference for G and C at third codon positions than genes in the four Saccharomyces species (Table 3). This may be one reason for the large KS values in highly expressed genes between the K. waltii - A. gossypii lineage and the Saccharomyces lineage. It was proposed that codon-usage bias is generally correlated with overall genome GC content, which is largely determined by mutational processes (Chen et al., 2004). Moreover, in most prokaryotic genomes, codons that are favored in highly expressed genes are well conserved (Rocha, 2004). In this study, the codon 12.
(24) preferences for these yeast species also agree with their genome GC content, i.e., 44% and 52% for K. waltii and A. gossypii, and 38% ~ 40% for the four Saccharomyces species. However, although most favored codons are the same among these species (Table 3), I found a switch of the preferred codon of glutamine (Gln) between CAA and CAG and a switch of the preferred codon of glutamic acid (Glu) between GAA and GAG between S. cerevisiae and A. gossypii. As shown in Table 4, these switches might be due to changes in tDNA gene copy number. For instance, the numbers of tDNA-Glu genes for anticodons TTC and CTC are 14 and 2 in S. cerevisiae but 3 and 8 in A. gossypii, and this may explain why the GAA codon is preferred in S. cerevisiae, whereas GAG is preferred in A. gossypii. Such a difference in codon preference can increase the synonymous distance between species. The tDNA gene phylogeny suggests that the change of gene copy number can be derived from a point mutation at anticodon or from duplication/deletion of tDNA genes in the genome (Fig. 6). Codon-usage bias is a compromise between compositional constraint (genomic GC content) and natural selection acting at the level of translation (Powell and Moriyama, 1997; Musto et al., 1999; Kliman, Irving and Santiago, 2003). If these two forces act in the same direction, for example, a preferred codon ending in G or C in a GC-rich genome, codon-usage bias could be extremely strong for highly expressed genes. On the other hand, the two forces may counteract each other; for example, a preferred codon ending in G or C in an AT-rich genome may only have its frequency slightly higher than 50% for highly expressed genes. This might explain why the high divergence between the K. waltii - A. gossypii and the Saccharomyces sensu stricto species mostly occurred in highly expressed genes. Gao and Innan (2004) estimated the expected length of concerted evolution in S. cerevisiae as 25 million years based on the theory the same group previously proposed (Teshima and Innan, 2004) (f = 9/51; 51 gene pairs shows concerted 13.
(25) evolution at the divergence time between S. cerevisiae and S. bayanus, while 9 gene pairs are still under concerted evolution at the divergence time between S. cerevisiae and S. paradoxus). I selected 18 gene pairs for which the paralogues and orthologues in S. cerevisiae, S. paradoxus and S. bayanus are all available and with CAI ≥ 0.7. Gene conversion was detected in 11 S. cerevisiae gene pairs. When I used S. paradoxus to calculate the orthologous distance instead, 6 gene pairs still have gene conversion events detectable. The expected length of concerted evolution for S. cerevisiae genes with CAI ≥ 0.7 thus estimated is 70 million years (f = 6/11; from S. cerevisiae - S. bayanus divergence to S. cerevisiae - S. paradoxus divergence). Note that this value may be underestimated because these genes are highly constrained and have evolved slowly. Informative sites indicating gene conversion may be too few to make the statistics significant. However, a similar estimate was obtained assuming the duration of concerted evolution started at the WGD event and the WGD occurred 100 million years ago (f = 12/21; from WGD to S. cerevisiae - S. bayanus divergence). Using the same method, I can estimate the expected lengths of concerted evolution for S. cerevisiae genes with CAI between 0.5 and 0.7, and CAI < 0.5 as 20 million years and 10 million years, respectively (f = 4/31 and 4/238; from WGD to S. cerevisiae - S. bayanus divergence). In summary, this study suggests that codon usage bias and protein functional conservation might have been more important than gene conversion for the decelerated evolution of WGD duplicate genes in yeasts. Note that gene conversion occurs only occasionally, whereas codon usage constraint and functional constraint of proteins are constant forces that slow down sequence evolution. Furthermore, the rate of gene conversion decreases as sequence divergence increases. For this reason gene conversion may not be an effective means for long-term maintenance of sequence similarity between duplicate genes in the absence of codon usage constraint or functional constraint. In contrast, both codon usage constraint and protein functional constraint can slow down sequence evolution in the absence of gene conversion. Of 14.
(26) course, the three factors can have synergistic effects in maintaining high sequence similarity between paralogues.. 15.
(27) Chapter 3 Protein complexity, gene duplicability and gene dispensability in the yeast genome. 16.
(28) Introduction. Previous studies have suggested that most genes (~80%) of the budding yeast (Saccharomyces cerevisiae) are nonessential under laboratory conditions (Winzeler et al., 1999; Glaever et al., 2002; Steinmetz et al., 2002). Two mechanisms have been proposed for explaining this phenomenon. The first is the existence of duplicate genes (e.g., Nowak et al., 1997; Gu et al., 2003; Conant and Wagner, 2004; Kafri, Bar-Even and Pilpel, 2005); that is, the loss of function in one copy can be compensated by the other copy or copies. The second mechanism stems from alternative metabolic pathways, regulatory networks, and so on (Wagner, 2000). Papp, Pal and Hurst (2004) used an in silico metabolic flux model of the yeast metabolic network to address the dispensability issue. They estimated that up to 68% of "dispensable" genes might actually be important, but under conditions yet to be examined in the laboratory, 15-28% of dispensable genes are compensated by a duplicate, while only 4-17% are buffered by flux reorganization of the metabolic network. In this study, I pursue the gene dispensability issue from the viewpoint of protein complexity. The number of domains in a polypeptide (He and Zhang, 2005) and the number of subunits in a protein complex (Yang, Lusk and Li, 2003) have been used to describe gene complexity and protein complexity, respectively. Here I define “domain complexity” as the number of domains in a polypeptide and “protein complexity” as the number of different subunit types in a protein complex. Although the number of protein interactions has been shown to correlate with protein deletion lethality (Jeong et al., 2001), there are four reasons to investigate protein complexity. First, the protein-protein interaction study was based on high-throughput data, which may have high false positive and false negative rates (von Mering et al., 2002). Second, subunits in a large complex without direct physical interactions to each other may not be detected by yeast two-hybrid analyses. Third, the number of protein interactions may reflect the number of functions or reactions that a polypeptide is 17.
(29) involved, while a large complex may have only one specific function. Fourth, I am also interested in comparing monomers and homo-multimers, which is not feasible from protein interaction data. Utilizing data on the fitness of heterozygotes for knockouts of essential genes in yeast, Papp, Pal and Hurst (2003b) found a greater decrease in heterozygote fitness if the gene is involved in a protein complex than if it is not, supporting the dosage balance hypothesis (Veitia, 2002; Veitia, 2003). However, homozygous gene deletion of a complex subunit may disrupt the protein function, which may be difficult to compensate by duplicated genes or alternative pathways if the function is cooperatively performed by multiple subunits. Further, Phadnis and Fry (2005) showed a negative correlation between homozygous effects and dominance of mutations (the ratio of heterozygous to homozygous effects) for all major categories of genes, which implies heterozygous and homozygous gene deletions may not have the same trend of fitness effect. It is therefore interesting to investigate whether the fitness effect of homozygous gene deletion increases with protein complexity and domain complexity. The second purpose of this study is to re-examine the issue of the effect of protein complexity on gene duplicability. Although Papp, Pal and Hurst (2003b) and Yang, Lusk and Li (2003) have shown that protein complexity is an important determinant of gene duplicability, the relationship between protein complexity and gene duplicability is still not very clear. This is particularly so with respect to the question of whether homo-complexes tend to have a higher gene duplicability than hetero-complexes; although Yang, Lusk and Li (2003) considered this question, their data was not sufficiently large to draw a clear conclusion. The third purpose is to investigate whether duplicate genes derived from the whole genome duplication event in the yeast (Wolfe and Shields, 1997; Dietrich et al.,. 18.
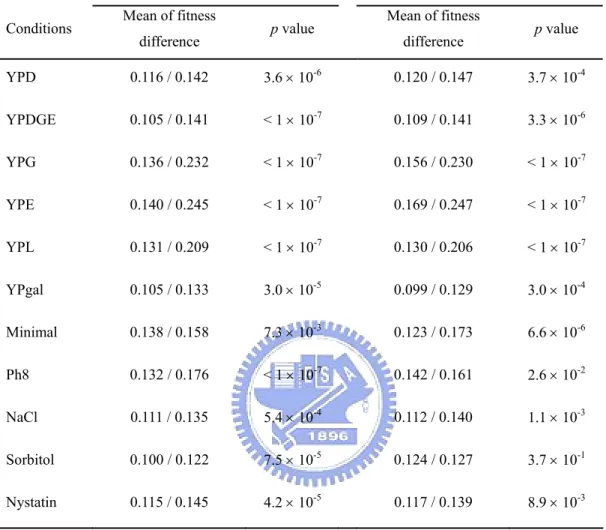
(30) 2004; Kellis, Birren and Lander, 2004) and non-WGD duplicate genes show similar relationships among protein complexity, duplicability, and dispensability. He and Zhang (2006) found that a less severe fitness consequence of deleting a duplicate gene than deleting a singleton gene is at least in part due to the reason that duplicate genes are intrinsically less important than singleton genes. I wish to obtain a better estimate of the contribution of duplicate genes to gene dispensability in the yeast genome because the estimate by Gu et al. (2003) did not subdivide duplicate genes into WGD and non-WGD genes and did not consider the possibility of different gene duplicabilities for homo- and hetero-complexes. In this study, for simplicity, I include monomers, which consist of a single polypeptide, in the class of homo-complexes because, as will be seen later, monomers and homo-complexes show small differences in both gene dispensability and gene duplicability. Materials and Methods Identification of duplicate genes and singletons. An all-against-all FASTA. (Pearson and Lipman, 1988) search was conducted for the whole set of S. cerevisiae protein sequences to obtain the list of singleton (single-copy) and duplicate genes as described in Gu et al. (2003). A whole genome duplication dataset was obtained from the genes listed in either Kellis, Birren and Lander (2004) or Dietrich et al. (2004). Although some gene pairs in the WGD dataset are quite diverged and may not satisfy the duplicate gene definition, they are still used in this analysis. The genes that did not satisfy the criteria for being singletons or duplicate genes were classified as twilight zone genes. The proportion of singleton families, P, was calculated as the number of singletons divided by the sum of the number of singletons and the number of duplicate gene families; 1 - P is used as a measure of gene duplicability. Data on fitness effect of gene deletion. The growth rates of each yeast. single-gene-deletion strain under various conditions were obtained from Steinmetz et al. (2002) (YDPM, http://www-deletion.stanford.edu/YDPM/YDPM_index.html) 19.
(31) with five growth media: YPD, YPDGE, YPG, YPE and YPL; and from Glaever et al. (2002) with six extra conditions: YPGal, Minimal, Ph8, NaCl, Sorbitol and Nystatin. Each strain contains the precise homozygous diploid deletion of one ORF in the yeast genome. Genes annotated as essential in MIPS (Mewes et al., 2002) (http://mips.gsf.de/) or in YDPM were removed from this growth rate dataset because there is a possibility that an essential strain could be detected due to cross hybridization of a tag from another non-essential strain. The remaining genes were used and I calculated the fitness values (f) as the extent of survival and reproduction of the deletion strain relative to the pool of all strains grown and measured collectively (Gu et al., 2003). Essential genes annotated in both MIPS and YDPM were sequentially included, and their fitness values were assumed to be 0. All genes were subdivided into four groups according to their f values: (1) the deletion has a weak or no fitness effect in all conditions studied if fmin ≥ 0.95, where fmin is the smallest f value among all 11 growth conditions; (2) the deletion has a moderate effect if 0.8 ≤ fmin < 0.95; (3) the deletion has a strong effect if 0 < fmin < 0.8; and (4) the deletion is lethal and f is set as 0. To avoid including pseudogenes and erroneously predicted genes, only ORFs with gene names in MIPS, YDPM or SGD (http://www.yeastgenome.org/) were kept for further analyses. Dispensable genes are defined as genes with a weak or no deletion fitness effect, i.e., fmin ≥ 0.95. Similar to Papp, Pal and Hurst (2003b), I only used the growth rates of heterozygous strains obtained on YPD substrate from Steinmetz et al. (2002) to estimate their haplosufficiency. Only genes with two measurements from repeat experiments were retained, and average growth rates were calculated. Relative heterozygous fitness was calculated as the relative growth rate to the pool of all strains. Collection of protein complexity data. Domain complexity data is obtained from. Deng et al. (2002). Protein complexity is defined here as the number of different polypeptide types in a protein complex, not as the number of polypeptide subunits as 20.
(32) defined in Yang, Lusk and Li (2003). The information of protein complexity was assembled from the complex or subunit descriptions in Swiss-Prot/TrEMBL (http://us.expasy.org/sprot/), MIPS, and SGD. A protein was regarded as a complex only when the descriptions of all components agreed with each other. A careful manual survey of published papers was made to verify these annotations. For example, in MIPS category 100, calcineurin B includes three entries; however, they do not form a hetero-trimer, but, instead, a regulatory subunit and two catalytic subunits form two kinds of hetero-dimers. I also used each gene name and several keywords to find literature on PubMed (http://www.ncbi.nlm.nih.gov/) and Google Scholar (http://scholar.google.com/) to increase the dataset. Homo-complexes (each composed of only one polypeptide type) were divided into monomers, homo-dimers, and homo-multimers, while hetero-complexes (each composed of more than one gene type) were classified according to the number of subunit polypeptide types. Polypeptides appearing in more than one complex were classified as multi-complex subunits, and the largest complex that a protein is involved was designated for the polypeptide. Cytoplasmic and mitochondrial ribosomal proteins were treated separately from other proteins. Fitness values and expression levels among complex subunits. Since a protein. complex could be a functional unit, its components should have similar deletion fitness effects. To test this hypothesis, hetero-complex genes, not including ribosomal and multi-complex proteins, were subdivided into dispensable (i.e., with a weak or no gene deletion effect) and indispensable, or lethal and nonlethal to examine if subunits of the same complex tend to have the same effect. I also wish to know, after excluding those dispensable and lethal genes, whether the fitness values of the subunits of a protein complex are still more similar than random gene pairs. For this purpose, I only keep genes with a strong or moderate deletion effect. The mean fitness difference between complex subunits is calculated and compared with the distribution of mean difference between randomly selected gene pairs. This random selection was repeated 21.
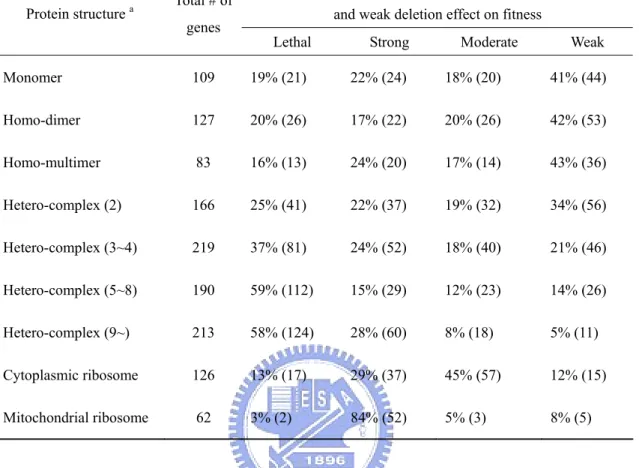
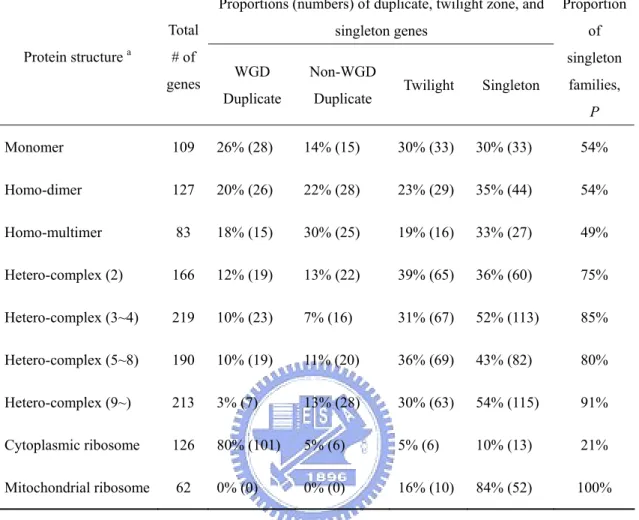
(33) 107 times. For comparison, the fitness difference between duplicate genes (Gu et al., 2003) is also examined using the present method. Similar procedures were applied to compare protein expression levels among complex subunits. TAP-tagged protein abundance data (Ghaemmaghami et al., 2003) were obtained from Yeast GFP Fusion Localization Database (http://yeastgfp.ucsf.edu/). Codon adaptation index (CAI) values, each of which indicates the strength of codon usage bias, were from MIPS. Results Protein complexity and gene dispensability. Previous studies used either only. complex/non-complex dataset (Ge et al., 2001; Papp, Pal and Hurst, 2003b; Poyatos and Hurst, 2004; Teichmann and Veitia, 2004; Phadnis and Fry, 2005) or used proteins of no recorded interaction as monomers (Yang, Lusk and Li, 2003). I collected a more extended and reliable protein complex dataset, so that an analysis of different protein complexities is feasible. Table 5 shows the fitness effects of gene deletions for subunits of homo-complexes and subunits of hetero-complexes. The proportions of genes with weak (or lethal) fitness effect of deletion for monomers, homo-dimers, and homo-multimers are not significantly different from one another (p > 0.1). Thus, the number of subunits in a homo-complex protein, including monomers, does not seem to affect gene dispensability significantly. In contrast, subunits of a hetero-complex tend to have a lower dispensability than subunits of a homo-complex, especially when the number of subunit types becomes larger than 2. This trend is also observed for the proportion of genes with lethal deletion effect (Table 5). Protein complexity and gene duplicability. I compared the proportions of singleton,. duplicate, and twilight zone genes for homo- and hetero-complex subunits (Table 6). The proportion of duplicate genes (including WGD and non-WGD duplicates) is consistently higher than 40% for all homo-complex proteins; the differences between 22.
(34) monomers and homo-dimers or homo-multimers are not significant (p > 0.1). In contrast, subunits of hetero-complexes have a much lower proportion of duplicate genes; the proportion decreases from 25% to 16% as the number of complex subunit types increases from 2 to ≥ 9, though this weak trend is not statistically significant (p > 0.1). In terms of the proportion of singleton families (P), the P value increases from 75% to 91% as the number of subunit types in a hetero-complex increases from 2 to ≥ 9. Note that the differences in gene duplicability between subunits of homo-complexes (monomers, homo-dimers, or homo-multimers) and subunits of hetero-complexes (subdivided according to their subunit types) are all significant (p < 0.01). Yang, Lusk and Li (2003) showed that complex proteins are less duplicable than monomers. This study further indicates that in terms of gene duplicability the major distinction is between homo-complexes and hetero-complexes. It is likely that only duplication of a gene for a subunit in a hetero-complex may cause dosage imbalance. I then compared the proportion of haploinsufficient genes (heterozygous deletion fitness value obtained on YPD substrate < 0.99) among indispensable genes (homozygous deletion fitness value obtained on YPD substrate < 0.95) for homo-complex subunits. I found that homo-multimers are significantly more haploinsufficient (7/24) than homo-dimers + monomers (6/73, p < 0.05), which suggests that maintaining a sufficient protein dosage is more essential for homo-multimers (Kondrashov and Koonin, 2004). This result implies that many duplicates of genes for homo-multimer subunits were possibly retained due to protein dosage requirement. Compared to monomers, most duplicates of genes for homo-multimer subunits were from non-WGD events (p < 0.05, Table 6). This result supports the above observation because unlike WGD, which occurs rarely, non-WGD duplication can occur more frequently and duplicate genes can be retained if there is an increased requirement of protein dosage. I also found that the low duplicability of. 23.
(35) subunits of large hetero-complexes (composed of 9 or more subunit types) is largely due to their small number of WGD duplicates (p < 0.05, Table 6). Ribosomal proteins. Ribosomes are the largest protein complexes in the yeast. proteome. The WGD duplicates have been retained for most of the cytoplasmic ribosome proteins, but not for mitochondrial ones (Table 6). This phenomenon might be explained (1) by the dosage theory (Kondrashov and Koonin, 2004), i.e., after the WGD event a larger dosage would be required in the cytoplasm than in the mitochondria, and (2) by the dosage balance hypothesis (Veitia, 2002; Veitia, 2003), i.e., similar concentrations of subunits in the same protein complex are selectively preferred; otherwise, the imbalanced dosage of subunits may significantly reduce the final concentration of the protein complex. When a singleton cytoplasmic ribosomal subunit is deleted, its function cannot be compensated and the whole ribosome is not functional (10 out of 13 singleton genes have a lethal deletion effect), whereas deletion of a subunit with duplicates may only cause dosage deficiency and imbalance, but may not be lethal (91 out of 107 duplicate genes have strong or moderate deletion effects, but only 3 of them are lethal). Interestingly, most mitochondrial ribosome subunits are not essential (only with strong deletion effects), despite the fact that they are singleton genes. Sequence similarity and gene dispensability. The dataset was subdivided into WGD. and non-WGD sets, and also homo-complexes, hetero-complexes, and proteins without complex annotation (excluding ribosomal proteins). These genes were further subdivided according to the KA of each gene to its most similar paralogue in the genome. Their cumulative distributions of fitness effect of gene deletion were compared (Fig. 7). Surprisingly, the correlation between gene deletion fitness effect and KA is weak, especially for WGD genes (Kolmogorov-Smirnov test, p > 0.1), though this correlation was considered as strong evidence of functional compensation among duplicates (Gu et al., 2003). On the other hand, non-WGD hetero-complex 24.
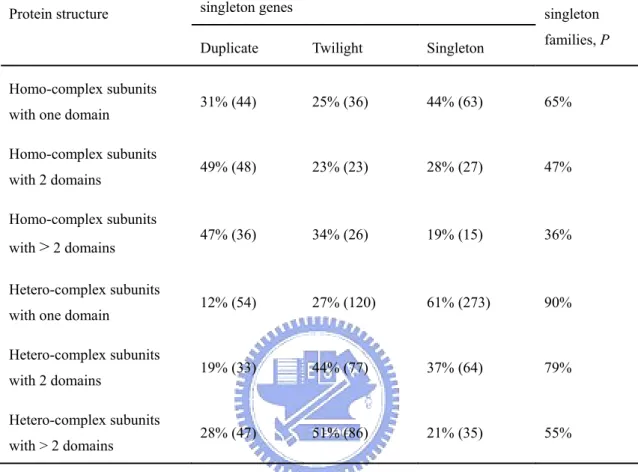
(36) subunits with KA < 0.4 are more dispensable than subunits with KA > 0.4 (Fig. 7B, p < 0.001); however, subunits with KA > 0.4 are less dispensable than twilight zone and singleton genes (p < 0.001). For non-WGD homo-complex subunits, genes with KA > 0.4 have similar fitness distributions (Fig. 7D), while genes with KA < 0.4 are more dispensable (p < 0.05). Similar results are found for non-WGD genes without protein complex annotations (Fig. 7F). In this case, gene dispensability is increased when KA is < 0.6 (p < 0.05). Because protein complexity is an important determinant of gene duplicability (Papp, Pal and Hurst, 2003b; Yang, Lusk and Li, 2003), one may suspect that the higher dispensability of subunits of a homo-complex protein is mainly due to a higher proportion of duplicate genes for subunits of homo-complex proteins in the genome. Figure 7 indicates that when the distance of each gene to its most similar paralogue is controlled, homo-complex subunits still are much more dispensable than hetero-complex subunits, especially for non-WGD genes. This result suggests that the higher dispensability for homo-complex subunits is not due to their abundance of duplicate genes. I further analyzed gene dispensability with protein complexity for hetero-complex subunits. When I removed duplicate genes to regenerate the relationships between fitness effect of gene deletion and protein complexity (Fig. 8), the observation that gene dispensability decreases as the number of subunit types in a protein complex increases (Table 5) still holds, except that the dispensability of homo-complex subunits is slightly decreased. Therefore, I suggest that the higher dispensability of genes coding for subunits of small hetero-complexes (or homo-complexes) cannot be attributed to functional compensation of duplicated genes. On the other hand, protein complexity may serve as a better indicator of gene dispensability than does gene duplication, as will be discussed later. Domain complexity and protein complexity. Since a protein domain may be the. functional unit, one may expect multi-domain polypeptides to have lower 25.
(37) dispensability. Indeed, Figure 9 shows that multi-domain polypeptides (with ≥2 domains) are significantly less dispensable than single-domain polypeptides. This difference is more significant when polypeptides for which no domain information is available are included in single-domain polypeptides. I found that 43% of hetero-complex subunits and 55% of homo-complex subunits are multi-domain polypeptides (p < 0.001). This result suggests that the proportion of multi-domain polypeptides cannot explain the low dispensability of hetero-complex subunits. On the other hand, one may suspect that the larger number of domains in the homo-complex avoids the need for a hetero-complex, implying that it might be the total number of domains of all the subunits of a protein complex that is a determinant of gene duplicability. To test this hypothesis, I only consider subunits of homo-complex or hetero-complex for which the summation of domain numbers in a complex is 2~4. Duplicate genes are excluded. The result indicates that hetero-complex subunits are still less dispensable than homo-complex ones (51 genes out of 192 genes with weak deletion fitness effects for hetero-complex subunits; 30 genes out of 70 genes for homo-complex subunits; p < 0.05). Among these genes, hetero-complex subunits should have fewer domains than homo-complex subunits. Therefore, I suggest protein complexity should be a more important determinant of gene dispensability than domain complexity. Previous studies showed that domain complexity (He and Zhang, 2005) and protein complexity (Papp, Pal and Hurst, 2003b; Yang, Lusk and Li, 2003) both are important determinants of gene duplicability. Therefore, it is interesting to investigate whether these two factors correlate with each other since homo-complex subunits have a higher proportion of multi-domain polypeptides. Table 7 reveals that when the domain number in a polypeptide increases from one to > 2, its gene duplicability (1 P) also increases from 35% to 64% for homo-complex subunits (p < 10-2), and from 10% to 45% for hetero-complex ones (p < 10-9). Moreover, homo-complex subunits are more duplicable than hetero-complex subunits when the number of domains is 26.
(38) controlled (p < 10-7 for single-domain polypeptides; p < 10-3 for polypeptides with 2 domains; the difference is not significant for polypeptides with > 2 domains due to the small sample size). This result suggests domain complexity and protein complexity are largely independent with respect to gene duplicability. Similar dispensabilities and expression levels for the subunits of a complex.. Complex subunits were subdivided into dispensable (i.e. with a weak or no gene deletion effect) and indispensable, or into lethal and nonlethal genes (Table 8). The proportions of each combination of subunit pairs found in the same complex and those of randomly selected gene pairs were compared. The observed number of subunit pairs with the same fitness effect category was found to be much higher than expected. Therefore, complex subunits tend to display similar fitness effects of gene deletion. It has been reported that proteins in the same interaction module also have similar dispensability (Poyatos and Hurst, 2004). Because most genes are distributed at the two extreme ends of fitness effect of gene deletion, it is interesting to ask whether the above conclusion still holds if only genes with strong or moderate deletion effects are considered. The answer is yes, no matter which growth condition is considered (Table 9). Although duplicated genes may have a chance to compensate each other’s function (Gu et al., 2003), I found that under most conditions duplicate gene pairs are not as similar to each other in gene deletion effect on fitness as the subunits of a complex. The reason might be that many duplicated genes have already functionally diverged, whereas the subunits of a complex usually play the same functional role. Under the dosage balance hypothesis, complex subunits should have similar protein expression levels. Using the same method described above, i.e., comparing with randomly selected gene pairs, I find that similarity indeed exists for protein expression levels of complex subunits. The mean logarithm difference of TAP-tagged protein abundance values between hetero-complex subunits is significantly less than 27.
(39) the mean difference between random gene pairs (p << 10-7). For proteins that do not have abundance data (~one third of the genes), the codon adaptation index (CAI) was used to infer the expression level. I found that the mean difference in CAI values between subunits of a protein complex is only half of the mean difference between random gene pairs (p << 10-7). This result is comparable to Ge et al.’s (2001) finding that genes encode interacting proteins tend to have similar expression profiles. Discussion Different trends of dispensability and duplicability for hetero-complexes. It was. noted above that although subunits of hetero-complexes composed of 2 subunit types are less dispensable compared with subunits of homo-complexes (Fig. 8), the difference is not significant (p > 0.1). In contrast, the dispensability of hetero-complexes composed of 3~4 subunit types is significantly lower (p < 0.01). In other words, when the number of subunit types increases, gene dispensability decreases gradually, instead of a sharp difference between homo- and hetero-complexes. On the other hand, although gene duplicability (1 - P) correlates with protein complexity, the difference in gene duplicabilities between subunits of hetero-complexes composed of 2 and > 9 subunit types is not significant. Although the insignificance could be due to a small sample size, both duplicabilities are significantly less than the duplicability of homo-complex subunits. The duplicability dramatically decreases from 46% ~ 51% for homo-complex subunits to 9% ~ 25% for hetero-complexes subunits (Table 6). The reason might be only duplication of a gene for a hetero-complex subunit may cause serious dosage imbalance. This observation suggests that the dosage balance hypothesis can explain gene duplicability of complex proteins well (Papp, Pal and Hurst, 2003b; Yang, Lusk and Li, 2003), but cannot completely explain the difference in dispensabilities between hetero-complex subunits.. 28.
(40) It is likely that knocking out a gene coding for a hetero-complex subunit would disrupt the function of the whole complex. This viewpoint is supported by the above result that subunits in the same complex tend to have similar deletion fitness effects. If the function of a protein complex is determined by most or all of its subunits, to compensate its lost function may need another complex either from duplicated genes or from alternative pathways. This effect may be more harmful than a complex concentration reduction derived from subunit duplication or heterozygous deletion (dosage imbalance). On the other hand, the formation of a large protein complex may take a long time in evolution. Therefore, losing the function of a large complex may be more severe than losing the function of a small one. This might explain why the dispensability of hetero-complexes decreases with protein complexity. Functional compensation by duplicate genes. Non-WGD genes were subdivided. into hetero-complexes, homo-complexes, and genes without complex annotations (Fig. 7). The contribution of duplicate genes to genetic robustness was estimated using Gu et al.’s method (2003). The result indicates that the dispensability of 1, 10, and 106 genes out of 104, 93, and 1086 dispensable genes might be attributed to gene duplication for these three categories, respectively. The proportion of the contribution of duplicate genes to genetic robustness is thus estimated to be 9% (117/1283) for non-WGD genes. He and Zhang (2006) found that less important genes tend to have a higher gene duplicability than important genes and suggested that this difference can partly account for a less severe fitness effect of deleting a duplicate gene than deleting a singleton gene. In the case studied here, the high dispensability of non-WGD genes with KA < 0.4 may partly be due to recent duplications of less important genes, rather than all from functional compensation by duplicates. Therefore, the contribution of duplicate genes to dispensability may not be as high as previously estimated (23%, Gu et al., 2003).. 29.
(41) It is worth noting that, except for ribosomal proteins, most of the ~400 WGD gene pairs that have been retained (Dietrich et al., 2004; Kellis, Birren and Lander, 2004) are dispensable (Fig. 7). For genes with the same protein complexity and with the same range of KA to their most similar paralogues in the genome, WGD genes are consistently more dispensable than non-WGD duplicate genes. The difference is statistically significant for hetero-complexes, for homo-complexes with KA > 0.4, and for genes without complex annotation and with KA > 0.6 (KS test, p < 0.05). This result implies that in the majority of cases the dispensability of WGD genes may not be due to functional compensation from their duplicates, because functional compensation should have similar effects for WGD and non-WGD duplicates. An alternative explanation is that dispensable genes might have a higher chance to be retained than indispensable genes following the WGD event. This result echoes the previous observation that dispensable (less important) genes have a higher duplicability (He and Zhang, 2006). The reason Gu et al. (2003) overestimated the contribution of duplicate genes to dispensability is likely because their singleton dataset includes many hetero-complex subunits, while duplicate gene dataset includes many homo-complex subunits and WGD genes, which are dispensable intrinsically. Another set of WGD genes are cytoplasmic ribosomal proteins, which, as mentioned earlier, tend to be indispensable. While deletion of a singleton cytoplasmic ribosomal subunit usually has lethal effect, deletion of a ribosomal subunit with duplicates may only cause dosage deficiency and imbalance (strong or moderate effect), but may not be lethal (Table 5). This fact suggests functional compensation exist for these WGD ribosomal proteins. However, although deletion of a ribosomal subunit with duplicates may not be lethal, such deletion is still evolutionarily deleterious. It is likely that their duplicates are retained mainly due to dosage requirement, but not due to functional compensation.. 30.
(42) Chapter 4 Protein structure and evolutionary rate. 31.
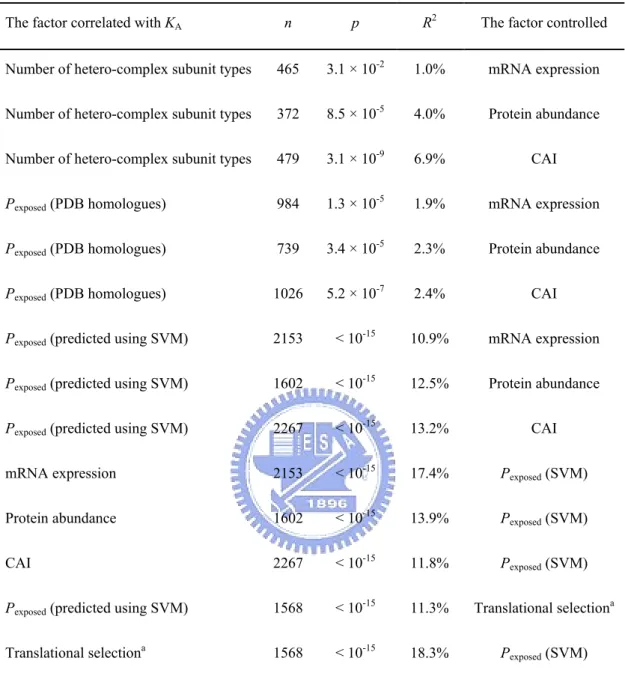
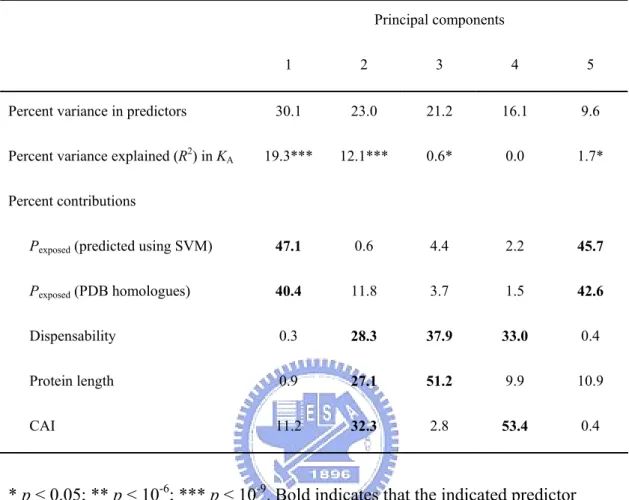
(43) Introduction. The issue of what factors determine protein evolutionary rate has drawn much attention in recent years. Two major theories have been proposed to explain the variance of protein evolutionary rates. One is that functionally less important proteins evolve faster than more important ones (Ohta, 1973; Kimura and Ohta, 1974; Wilson, Carlson and White, 1977), which was supported by the weak but significant correlation between gene dispensability and protein evolutionary rate (Hirsh and Fraser, 2001; Yang, Gu and Li, 2003; Wall et al., 2005; Zhang and He, 2005). The other is that the rate is primarily determined by the proportion of residues involved in specific functions (functional density, Zuckerkandl, 1976). Fraser et al. (2002) suggested that proteins with more interactions evolve more slowly because they have higher functional density. Other studies reported that a protein evolves slowly if (1) the protein is highly expressed (Pal, Papp and Hurst, 2001; Rocha and Danchin, 2004; Wall et al., 2005), (2) the protein is involved in stable complexes (Teichmann, 2002), (3) the protein is involved in interaction hubs situated in single modules (Fraser, 2005), or (4) the protein occupies a central position in networks (Hahn and Kern, 2005). However, some of the above results were questioned because the levels of gene expression were not controlled (Bloom and Adami, 2003; Pal, Papp and Hurst, 2003). Recently, Drummond, Raval, and Wilke (2006) proposed that translational selection is the single dominant determinant, and provided an explanation why gene or protein expression level governs the evolutionary rate (Drummond et al., 2005). Most of these studies only considered characters of a whole protein, but did not look into differences in evolutionary constraints among residues. It is therefore interesting to investigate whether there are other dominant determinants. Materials and Methods Yeast genomic data. I obtained nonsynonymous rates (KA) from Wall et al. (2005),. protein interaction modules from Han et al. (2004), mRNA expression level from 32.
(44) Holstege et al. (1998), protein abundance from Ghaemmaghami et al. (2003), and codon adaptation index (CAI) values from Drummond, Raval and Wilke (2006); CAI indicates the strength of codon usage bias (Sharp and Li, 1987). Protein complexity and gene dispensability data are obtained as described in chapter 2. Genes without gene names were excluded. Principal component regression was performed using R with the package ‘pls’ (Ihaka and Gentleman, 1996). Protein abundance and mRNA expression level was log transformed. Solvent accessibility predicted using homology model. PDB homologues for each. open reading frame (ORF) were obtained from the Saccharomyces Genome Database (SGD, http://www.yeastgenome.org/). The solvent accessible surface areas (ACC) for each residue of the PDB homologue with the highest p value were obtained from DSSP (http://swift.cmbi.ru.nl/gv/dssp/) (Kabsch and Sander, 1983). Relative solvent accessibility (RelACC) was the ACC for each residue subdivided by the maximum value of ACC for the certain amino acid (represented using percentage), which is estimated from a Gly-X-Gly extended tripeptide conformation. Here I define residues with RelACC higher than 25% as exposed residues, and the others as buried. The proportion of exposed residues (Pexposed) for each PDB homologue was thus calculated. Solvent accessibility predicted using supporting vector machine (SVM). SVM. prediction was performed as described in Hsu (2005) using 480 proteins from Kim and Park (2004) as training dataset, and position-specific scoring matrices (PSSM), secondary structure profiles, and hydropathy indexes as feature factors. A 7-fold cross validation test yields 78% accuracy. Results and Discussion. I first studied the relationship between the number of hetero-complex subunit types (excluding ribosomal proteins) and nonsynonymous substitution rates (KA) for 33.
數據



+7
相關文件
The four e/g-teaching profiles identified in this study are outlined as follows: parsimony (low e-teaching and medium, below- average g-teaching), conservation (low e-teaching and
The case where all the ρ s are equal to identity shows that this is not true in general (in this case the irreducible representations are lines, and we have an infinity of ways
In addressing the questions of its changing religious identities and institutional affiliations, the paper shows that both local and global factors are involved, namely, Puhua
For the data sets used in this thesis we find that F-score performs well when the number of features is large, and for small data the two methods using the gradient of the
In the work of Qian and Sejnowski a window of 13 secondary structure predictions is used as input to a fully connected structure-structure network with 40 hidden units.. Thus,
assembly of the genome of that species will be far better if read lengths are longer than N... Accurate but
Conserved complexes are connected sub- graphs within the bacteria-yeast alignment graph, whose nodes represent orthologous protein pairs and edges represent conserved
are the largest manufacturers, but the operating performance that the Asia Cement Co’s average of four indicators are leading the same industry and named in the third
