Reactions of Ruthenium Acetylide and Vinylidene Complexes
Containing a 2-Pyridyl Group
Hsien-Hsin Chou, Ying-Chih Lin,* Shou-Ling Huang, Yi-Hong Liu, and Yu Wang
Department of Chemistry, National Taiwan UniVersity, Taipei, Taiwan 106, Republic of ChinaReceiVed March 18, 2008
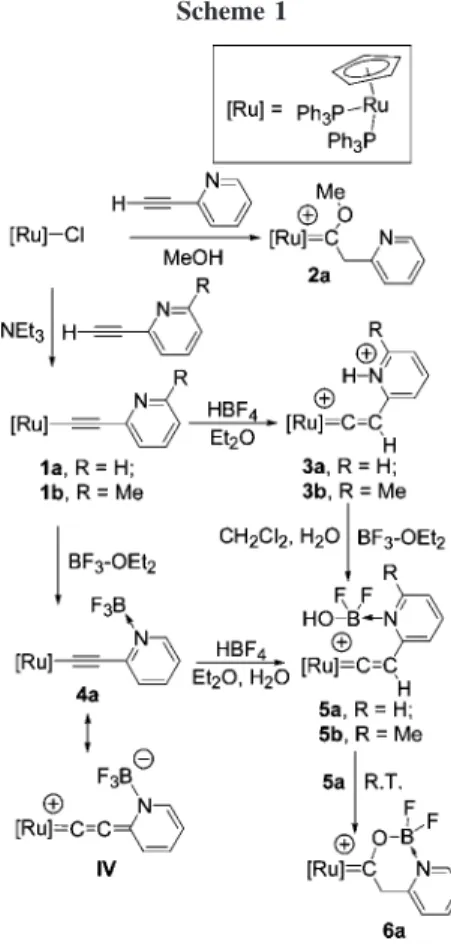
Two ruthenium acetylide complexes [Ru]CtC(C5H3RN) (1a, R ) H; 1b, R ) Me; [Ru] ) Cp(PPh3)2Ru) containing 2-pyridyl groups are prepared and their chemical reactivities are explored. Protonation of the ruthenium acetylide complex 1a with HBF4takes place at both the nitrogen atom and Cβ, giving the dicationic pyridiniumvinylidene complex {[Ru]dCdC(H)(C5H4NH)}(BF4)2(3a). Addition of BF3to 1a yields the Lewis acid/base adduct [Ru]CtC(C5H4NfBF3) (4a). In the presence of moisture both complexes 3a and 4a in solution transform into the cationic heterocyclic carbene complex {[Ru]dC(O)CH 2-(C5H4NfBF2)}BF4 (6a), for which the structure is confirmed by X-ray structure determination. The formation of 6a involves the intermediate {[Ru]dCdC(H)(C5H4NfBF2OH)}BF4(5a), characterized by spectroscopic methods. DFT calculations show that the Gibbs free energy change of the exothermic transformation of 5a to 6a is -20.59 kcal/mol. N-Alkylation reactions of 1b with two alkyl bromides BrCH2R′ (R′ ) CHdCHCO2Me and CO2Me) yield two pyridiniumacetylide complexes {[Ru]Ct C(C5H3MeNCH2R′)}Br (7b, R′ ) CHdCHCO2Me; 7c, R′ ) CO2Me, respectively). Complex 7c, characterized by X-ray structure determination, undergoes further protonation to give the pyridiniumvi-nylidene complex {[Ru]dCdC(H)(C5H4NCH2R′)2+(8c). Interestingly, the acetylide complex 7b undergoes a C-C coupling reaction of the acetylic Cβ with the CdC double bond to give the vinylidene complex
9b, characterized also by X-ray structure determination. Introduction
During the past decade, chemistry of transition metal complexes containing vinylidene ligands has attracted a great deal of attention because of their occurrence as key intermediates in many stoichiometric and catalytic transformations of organic molecules.1 Since the method for the preparation of cationic bisubstituted vinylidene complexes via electrophilic attack of metal acetylides was established, the diversity and applications of these metal vinylidene complexes have further expanded. Previously we have shown that ruthenium vinylidene complexes [Ru]dCdC(Ph)CH2R
+
([Ru] ) Cp(PPh3)2Ru) bearing an electron-withdrawing group R attached at Cγ undergo intramo-lecular cyclization under mild basic conditions.2On the basis of this approach, various mono- and multinuclear neutral metal cyclopropenyl or furyl complexes have been prepared. Recently, serendipitous results showed that the ruthenium vinylidene complex with a pendant terminal vinyl group exhibits excellent
metathesis reactivity of CdC double bonds so that skeletal rearrangement and cyclization are observed.3
Rich chemistry has been demonstrated for molecules contain-ing various pyridyl moieties. For example, ortho-substituted pyridyl groups usually act as building blocks in templating metal centers by means of coordination, which leads to supramol-ecules.4Very recently the metal-mediated C-H activation was reported to cause formation of pyridine/quinolidene ortho-carbene complexes.5ortho-Substitution of an ethynyl group on pyridine has been under investigation in the development of nonlinear optics.6 The coordination ability of the N atom of platinum acetylide complexes with 2-pyridyl functional groups has also been investigated.4d,eWe therefore set a goal to study * Corresponding author. E-mail: [email protected].
(1) (a) Bruce, M. I. Chem. ReV. 1991, 91, 197. (b) Werner, H. Angew.
Chem., Int. Ed. 1990, 29, 1077. (c) Trost, B. M.; Toste, F. D.; Pinkerton,
A. B. Chem. ReV. 2001, 101, 2067. (d) Valyaev, D. A.; Semeikin, O. V.; Ustynyuk, N. A. Coord. Chem. ReV. 2004, 248, 1679. (e) Bruneau, C.; Dixneuf, P. H. Angew. Chem., Int. Ed. 2006, 45, 2176. (f) Bruneau, C.; Dixneuf, P. H. Acc. Chem. Res. 1999, 32, 311. (g) Katayama, H.; Ozawa, F. Coord. Chem. ReV. 2004, 248, 1703. (h) Varela, J.; Saa´, C. Chem.-Eur.
J. 2006, 12, 6450.
(2) (a) Ting, P. C.; Lin, Y. C.; Lee, G. H.; Cheng, M. C.; Wang, Y.
J. Am. Chem. Soc. 1996, 118, 6433. (b) Lo, Y. H.; Lin, Y. C.; Lee, G. H.;
Wang, Y. Organometallics 1999, 18, 982. (c) Chang, C. W.; Lin, Y. C.; Lee, G. H.; Wang, W. Organometallics 2000, 19, 3211. (d) Chang, K. H.; Lin, Y. C.; Liu, Y. H.; Wang, Y. J. Chem. Soc., Dalton Trans. 2001, 3154. (e) Lin, Y. C. J. Organomet. Chem. 2001, 617-618, 141. (f) Lo, Y. H.; Hsu, S. C.; Huang, S. L.; Lin, Y. C.; Liu, Y. H.; Wang, Y. Organometallics 2004, 23, 5924. (g) Yen, Y. S.; Lin, Y. C. Organometallics 2007, 26, 1250. (h) Liu, C. W.; Lin, Y. C.; Huang, S. L.; Cheng, C. W.; Liu, Y. H.; Wang, Y. Organometallics 2007, 26, 3431.
(3) (a) Yen, Y. S.; Lin, Y. C.; Huang, S. L.; Liu, Y. H.; Sung, H. L.; Wang, Y. J. Am. Chem. Soc. 2005, 127, 18037. (b) Cheng, C. W.; Kuo, Y. C.; Chang, S. H.; Lin, Y. C.; Liu, Y. H.; Wang, Y. J. Am. Chem. Soc. 2007, 129, 14974.
(4) (a) Choi, M. Y.; Chan, M. C. W.; Peng, S. M.; Cheung, K. K.; Che, C. M. Chem. Commun. 2000, 1259. (b) Stang, S. L.; Lenz, D.; Paul, F.; Lapinte, C. J. Organomet. Chem. 1999, 572, 189. (c) Stang, S. L.; Lenz, D.; Paul, F.; Lapinte, C. Inorg. Chim. Acta 1999, 291, 403. (d) Berenguer, J. R.; Eguiza´bal, E.; Falvello, L. R.; Fornie´s, J.; Lalinde, E.; Martı´n, A.
Organometallics 1999, 18, 1653. (e) Ara, I.; Berenguer, J. R.; Eguiza´bal,
E.; Fornie´s, J.; Go´mez, J.; Lalinde, E.; Sa´ez-Rocher, J. M. Organometallics 2000, 19, 4385.
10.1021/om800253e CCC: $40.75 2008 American Chemical Society Publication on Web 09/26/2008
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
the chemistry of ruthenium acetylide and vinylidene complexes containing ortho-pyridyl groups. Herein we report the synthesis and reactivities of these acetylide and vinylidene complexes toward protic acids, Lewis acids, and electrophilic alkyl groups.
Results and Discussion
Preparation and Reactions of Ruthenium Pyridylacetyl-ides. A convenient method was utilized to prepare a ruthenium
pyridylacetylide complex from ruthenium chloride. Thus the reaction of [Ru]-Cl ([Ru] ) Cp(PPh3)2Ru) with 2-ethynylpy-ridine in a mixed solvent (CHCl3/MeOH/NEt3) at room tem-perature exclusively gave the pyridylacetylide complex 1a as a yellow powder in good yield. Complex 1a shows a 31P resonance atδ 51.0 and CR resonance at δ 124.30 with JCP) 25.8 Hz in the31P and13C NMR spectra, respectively. These are comparable with those of analogous phenylacetylide derivatives.7a,bThe mass spectrum of 1a shows parent peaks at m/z ) 794.0 for M++ 1. Similarly the reaction of [Ru]-Cl with 6-methyl-2-ethynylpyridine yielded complex 1b, which has been characterized by single-crystal X-ray diffraction analysis. The molecular structure of complex 1b with selected bond lengths and bond angles is shown in Figure 1. The coordination sphere surrounding the ruthenium center of 1b adopts a three-legged piano-stool structure with a typical acetylide skeleton. The methyl and N atom in the pyridyl group face the same direction as the bonding of Ru toward the Cp ring centroid. The Ru-C(1) distance of 2.007(3) Å is a typical Ru-C single bond and is comparable to the corresponding one in [Ru]CtCPh (I, 2.017(5) Å)7a and [Ru]CtC(C
6H4-p-NO2) (II, 1.994(5) Å).7b The C(1)-C(2) distance of 1.216(4) Å, which is a CtC triple bond, is also comparable to that in I (1.214(7) Å) and the nickel
analogue Cp(PPh3)NiCtC(C5H3N-p-NO2) (III, 1.215(7) Å),7b but is slightly longer than that in II (1.202(8) Å).
On the other hand, when the reaction was carried out in a mixed solvent of CH2Cl2/MeOH at room temperature, the 2-picolylmethoxycarbene complex 2a was obtained (Scheme 1). This result is different from that in the ruthenium 4-pyridy-lacetylide complex reported by Lin et al.8aThe cationic complex
2a exhibits spectroscopic features different from those of 1a.
The13C NMR spectrum shows a characteristic carbenoic triplet resonance of CR atδ 306.0 with JCP) 12.8 Hz. The31P NMR spectrum of 2a shows a singlet atδ 46.6. Furthermore the31P NMR results indicate that during the reaction a singlet resonance atδ 42.0 appeared and diminished as 2a gradually formed. This intermediate is proposed to be the pyridylvinylidene complex {[Ru]dCdC(H)(C5H4N)}+. Because of its instability in the presence of alcohol and in the absence of NEt3, the intermediate complex will spontaneously undergo nucleophilic addition to yield the cationic methoxycarbene complex. The electron-withdrawing ortho-pyridine substituent seems to play a role in this reaction. Similar reactions involving various vinylidenes of the type Cp(PR3)2RudCdCRH can lead to alkoxycarbene in the presence of alcohol, albeit at elevated temperature.9This phenomenon prompted us to study the reactivities of the pyridine moiety implanted in the ruthenium acetylide backbone. The reactions of complex 1 toward protic acids, Lewis acids, and a (5) (a) Alvarez, E.; Conejero, S.; Paneque, M.; Petronilho, A.; Poveda,
M. L.; Serrano, O.; Carmona, E. J. Am. Chem. Soc. 2006, 128, 13060. (b) Esteruelas, M. A.; Ferna´ndez-Alvarez, F. J.; On˜ate, E. J. Am. Chem. Soc. 2006, 128, 13044. (c) Wiedemann, S. H.; Lewis, J. C.; Ellman, J. A.; Bergman, R. G. J. Am. Chem. Soc. 2006, 128, 2452. (d) Chen, D; Li, Y.; Wang, B.; Xu, S.; Song, H. Organometallics 2006, 25, 307.
(6) (a) Powell, C. E.; Humphrey, M. G. Coord. Chem. ReV. 2004, 248, 725. (b) Marder, T. B.; Lesley, G.; Yuan, Z.; Fyfe, H. B.; Chow, P.; Stringer, G.; Jobe, I. R.; Taylor, N. J.; Williams, I. D.; Kurtz, S. K. In Materials for
Nonnlinear Optics: Chemical PerspectiVes; Marder, S. R., John, S., Stucky,
G. D., Eds.; ACS Symp. Ser. A; 1991; Vol. 455, p 605. (c) Naulty, R. H.; Cifuentes, M. P.; Humphrey, M. G.; Houbrechts, S.; Boutton, C.; Persoons, A.; Heath, G. A.; Hockless, D. C. R.; Luther-Davies, B.; Samoc, M. J. Chem.
Soc., Dalton Trans. 1997, 4167.
(7) (a) Bruce, M. I.; Humphrey, M. G.; Snow, M. R.; Tiekink, E. R. T.
J. Organomet. Chem. 1986, 314, 213. (b) Whittall, I. R.; Humphrey, M. G.;
Hockless, D. C. R.; Skelton, B. W.; White, A. H. Organometallics 1995,
14, 3970.
(8) (a) Wu, I. Y.; Lin, J. T.; Luo, J.; Sun, S. S.; Li, C. S.; Lin, K. J.; Tsai, C.; Hsu, C.-C.; Lin, J.-L. Organometallics 1997, 16, 2038. (b) Bruce, M. I.; Ke, M.; Kelly, B. D.; Low, P. J.; Smith, M. E.; Skelton, B. W.; White, A. H. J. Organomet. Chem. 1999, 590, 184. (c) Szesni, N.; Drexler, M.; Maurer, J.; Winter, R. F.; de Montigny, F.; Lapinte, C.; Steffens, S.; Heck, J.; Weibert, B.; Fischer, H. Organometallics 2006, 25, 5774.
(9) (a) Takagi, Y.; Matsuzaka, H.; Ishii, Y.; Hidai, M. Organometallics 1997, 16, 4445. (b) Bruce, M. I.; Duffy, D. N.; Humphrey, M. G.; Swincer, A. G. J. Organomet. Chem. 1985, 282, 383. (c) Bruce, M. I.; Swincer, A. G. Aust. J. Chem. 1980, 33, 1471.
Figure 1. ORTEP drawing (30% thermal ellipsoid) of 1b with
phenyl groups on phosphine ligands (except ipso carbon) and hydrogen atoms eliminated for clarity. Selected bond distances (Å) and bond angles (deg): Ru1-C1, 2.007(3); C1-C2, 1.216(4); C2-C3, 1.422(4); Ru1-C1-C2, 174.7(2); C1-C2-C3, 172.5(3).
Scheme 1
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
few alkyl halides are thus investigated. Their further transforma-tions into vinylidene and heterocyclic carbene complexes are also studied.
Although many reports deal with the coordination ability of the N atom of the transition metal 2-pyridylacetylides,4d,e their interaction with protic acids and Lewis acids have not been thoroughly described. Furnished with acetylide and pyridyl groups both complexes 1a and 1b are expected to undergo electrophilic addition. Treatment of complex 1a with excess HBF4 in diethyl ether produces the bright orange pyridiniumvinylidene complex {[Ru]dCdC(H)(C5H4 NH)}-(BF4)2(3a) in high yield (Scheme 1). The31P NMR spectrum of 3a shows a singlet resonance at δ 37.3. The 1H NMR spectrum shows the characteristic broad proton resonance of NH atδ 12.7 and the relatively downfield singlet resonance of the vinylidene proton at Cβ at δ 5.90. This reveals that both the pyridyl group and Cβ are protonated; thus the basicity of the pyridine moiety is not diminished in forming a complex of this system.8 Addition of D
2O causes the exchange of the pyridinium and vinylidene protons of 3a as shown in the1H NMR spectrum. Surprisingly, this two-proton adduct is sufficiently acidic and can even protonate D2O to effectively restore 1a if excess D2O was added (observed in nearly 100% NMR yield). In a similar vein, protonation of complex 1b also gives the dicationic complex {[Ru]dCd C(H)(C5H3MeNH)}(BF4)2(3b).
On the other hand, a Lewis acid/base interaction was observed only at the pyridyl group, and the BF3 adduct of the pyridiniumacetylide complex [Ru]CtC(C5H4NfBF3) (4a) was obtained when complex 1a was treated with excess BF3-OEt2(Scheme 1). Binding of the BF3group is indicated by the singlet resonance at δ -152.2 in the 19F NMR spectrum, which is similar to that of the structurally similar BF4
-group atδ -152.0. The31P NMR spectrum of 4a shows a singlet resonance atδ 50.9, which is comparable to that of BF3-free 1a at δ 51.0. The 13C NMR triplet resonance of CR atδ 173.98 with JCP) 23.0 Hz is closer to the carbene region (generally at ca. δ 200) than that of complex 1a (δ 124.30 with JCP) 25.8 Hz), which is attributed to the partial contribution of the allenylidene structure (IV) in the ground-state geometry of complex 4a (Scheme 1).8c
Complex 4a is air and moisture sensitive and in chloroform can be cleanly transformed to 5a in the presence of moisture. In addition, treatment of 3a or 1a with an excess amount of BF3-OEt2in the presence of moisture also affords complex
5a (Scheme 1). The reaction of complex 4a with HBF4 in the presence of H2O at room temperature also gives 5a as a brown solid in good yield. Complex 5a is believed to be the cationic pyridiniumvinylidene complex {[Ru]dCdC(H)-(C5H4NfBF2OH)}BF4with a BF2OH group on the pyridine N atom. The 1H NMR spectrum of 5a shows a singlet resonance atδ 5.90 and a broad peak at δ 12.33 assigned to the vinylideneβ-proton and the OH group, respectively. The vinylidene backbone of 5a is reflected by the 13C NMR resonances of R- and β-carbons at δ 344.56 and 112.46, respectively. A 2D NMR 13C-1H HSQC experiment also confirms the vinylidene group by displaying a cross-peak between δHof 5.90 andδCof 112.46.
Interestingly, in CDCl3solution, complex 5a further trans-forms into the new complex 6a in 4 days at room temperature in ca. 60% yield (Scheme 1). The cationic complex 6a contains a heterocyclic carbene ligand with a boron atom in the ring.10 The carbenoic triplet resonance of 6a atδC292.65 with JCP) 12.7 Hz shifts slightly upfield than the corresponding CR
resonance of the methoxy derivative 2a at δ 306.0. The IR spectrum of 6a reveals an absorption band atν ) 1350.5 cm-1, indicating the presence of a C-O single bond skeleton.10g Complex 6a has been characterized by X-ray structural deter-mination. Figure 2 displays an ORTEP drawing and selected bond lengths and angles of 6a. The Ru-C(1) bond distance of 1.944(3) Å is a typical RudC carbenoic double bond but is slightly longer than that of the methoxy derivative {[Ru]d C(OMe)Me}+(V, 1.931(9) Å),10ewhereas the C(1)-O(1) bond length of 1.308(4) Å is shorter than that in complex V (1.321(9) Å). The partial delocalization of the cationic charge around Ru-C(1)-O(1) bonds can be rationalized by the much upfield-shifted CR resonance of 6a in the13C spectrum. The O(1)-B(1) bond distance of 1.484(5) Å is slightly shorter than those in iron complexes10dand in rhodium complexes.10aFor compari-son, in a trinuclear Ru cluster the bond distances between B-O, O-C, and average C-Ru10bare 1.508(5), 1.262(4), and 2.004 Å, respectively.
In addition to complex 4a, the pyridiniumvinylidene complex
3a with two BF4
-counteranions in solution also yielded 6a in the presence of moisture. Upon NMR monitoring of a sealed tube containing a CDCl3solution of 3a under nitrogen, complex
6a, [Ru]-CO+, and triphenylphosphine oxide were observed in a 2:1:1 ratio based on31P NMR integration after 5 days at room temperature. The 31P NMR resonances of [Ru]-CO+
and OdPPh3appear atδ 42.5 and 30.0, respectively.11The1H and 13C signals of the Cp group atδ 5.00 and 91.4, respectively, confirm the formation of [Ru]-CO+. Perhaps trace moisture in the solution caused decomposition of the BF4-anion of complex
3a to form BF3,12which then reacted with 3a to result in the formation of 5a and finally 6a. The methyl derivative 1b sequentially gave complexes 3b and 5b. However, complex 5b
(10) (a) Yeston, J. S.; Bergman, R. G. Organometallics 2000, 19, 2947. (b) Chipperfield, A. K.; Housecroft, C. E.; Raithby, P. R. Organometallics 1990, 9, 479. (c) Lukehart, C. M.; Raja, M. Inorg. Chem. 1982, 21, 2100. (d) Butts, S. B.; Strauss, S. H.; Holt, E. M.; Stimson, R. E.; Alcock, N. W.; Shriver, D. F. J. Am. Chem. Soc. 1980, 102, 5093. (e) Bellachioma, G.; Cardaci, G.; Foresti, E.; Macchioni, A.; Sabatino, P.; Zuccaccia, C. Inorg.
Chim. Acta 2003, 353, 245. (f) Nakazawa, H.; Yamaguchi, Y.; Miyoshi,
K. Organometallics 1996, 15, 4661. (g) Butts, S. B.; Strauss, S. H.; Holt, E. M.; Stimson, R. E.; AIcock, N. W.; Shriver, D. F. J. Am. Chem. Soc. 1980, 102, 5093.
(11) Bruce, M. I.; Swincer, A. G.; Wallis, R. C. J. Organomet. Chem. 1979, 171, C5.
Figure 2. ORTEP drawing (30% thermal ellipsoid) of 6a with
phenyl groups on the phosphine ligands (except the ipso carbon) and hydrogen atoms eliminated for clarity. Selected bond distances (Å) and bond angles (deg): Ru1-C1, 1.944(3); C1-C2, 1.520(4); C2-C3, 1.484(5); C1-O1, 1.308(4); O1-B1, 1.484(5); B1-F1, 1.369; B1-F2, 1.352; Ru1-C1-C2, 123.4(2); C1-C2-C3, 113.7(3);Ru1-C1-O1,124.7(2);C2-C1-O1,111.8(3);C1-O1-B1, 125.4(3); O1-B1-N1, 105.9(3); F1-B1-F2, 112.8(3).
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
is relatively more stable toward cyclization in solution. Attempts to promote cyclization of 5b led to decomposition.
Given that the aforementioned transformations require the existence of moisture (either from the air or the solvent), a deuterium labeling experiment was carried out. To the complex
4a was added an equivalent amount of HBF4in the presence of excess D2O. The31P NMR spectrum of the product shows the resonance at δ 46.9 of 6a, while the disappearance of the methylene resonance atδ 5.15 in the1
H NMR spectrum reveals the doubly deuterated complex (6a-d2). As described below, because the protonation of a pyridiniumacetylide complex to give a pyridiniumvinylidene complex is easily achieved, we believe that the protonation of 4a here involves the formation of the pyridiniumvinylidene intermediate. Interestingly, no alkoxycarbene product from the addition at CR was observed during the reaction. This probably indicates that the pyridyl-BF3group is more reactive than the vinylidene group so that the substitution of F by OH proceeds faster than the nucleophilic attack at CR. When the 2-pyridyl group is bound to a less electron-deficient BH3 group, no substitution was observed. Namely, by treating the pyridylacetylide complex 1a with excess BH3-THF we prepared the pyridylacetylide complex [Ru]Ct C(C5H4NfBH3) (4c), which is sufficiently stable at ambient temperature even in solution for at least 1 week.13
The mechanism for the formation of 6a is thus suggested as followed. Treatment of 4a with HBF4immediately affords the vinylidene intermediate {[Ru]dCdC(H)(C5H4NfBF3)}
+ (VI). Substituting one fluoride of the pyridinium-BF3 group by a hydroxyl group subsequently gives the OH-substituted py-ridylvinylidene 5a with the formation of a B-OH bond.14Then two possible pathways were considered, namely, intramolecular nucleophilic addition of the OH group onto the vinylidene CR and 1,3-proton shift and direct nucleophilic addition of the O-H group onto the vinylidene CRdCβ bond, affording the final product 6a.15
According to this mechanism, it can be visualized that complex 6 can be prepared directly via a one-pot procedure from [Ru]-Cl. Actually, in chloroform solution, 10 molar equiv of BF3-OEt2and 2-ethynylpyridine were first mixed for 10 min followed by the addition of [Ru]-Cl. The resulting solution was stirred for 3 days at room temperature. Workup of the solution afforded complex 6a in high yield. However, without an excess amount of BF3, only [Ru]-Cl was obtained. Excess BF3probably induces dissociation of the chloride ligand and initiates the
reaction. The reaction of [Ru]-Cl with 2-ethynylpyridine in the presence of CH3I leads to [Ru]-I only.
Theoretical Calculations. DFT calculations at the B3LYP/
LanL2DZ16level by Gaussian 03 were performed to study the reaction of 5 to 6 in an effort to distinguish two pathways for the addition of OH to the CdC bond.17The phenyl groups of the PPh3 were replaced by hydrogen in our study. Figure 3 displays the optimized geometries and corresponding Gibbs free energy changes of the conversion of A to D, modeling the transformation of complex 5 to 6. The Ru-C bond of 1.95 Å, C-O bond of 1.35 Å, and O-B bond of 1.50 Å of the optimized structure reasonably match the experimental result obtained from
(12) (a) Cole-Hamilton, D. J. Science 2003, 299, 1702. (b) Ignat’ev, N. V.; Welz-Biermann, U.; Kucheryna, A.; Bissky, G.; Willner, H. J. Fluor.
Chem. 2005, 126, 1150. (c) Sellin, M. F.; Webb, P. B.; Cole-Hamilton,
D. J. Chem. Commun. 2001, 781. (d) Limmert, M.; Lorenz, I.-P.; Neubauer, J.; No¨th, H.; Habereder, T. Eur. J. Inorg. Chem. 2001, 1593.
(13) Lesley, M. J. G.; Woodward, A.; Taylor, N. J.; Marder, T. B.; Cazenobe, I.; Ledoux, I.; Zyss, J.; Thornton, A.; Bruce, D. W.; Kakkar, A. K. Chem. Mater. 1998, 10, 1355.
(14) The boron-water complex BF3-H2O has been studied for more than a decade. Decomposition of this complex into B(OH)3and BF4anion has also been reported; see for example: (a) Evans, D. G.; Yeo, G. A.; Ford, T. A Faraday Discuss. Chem. Soc. 1988, 86, 55. (b) Jacox, M. E.; Irikura, K. K.; Thompson, W. E. J. Chem. Phys. 2000, 113, 5705. (c) Ghaemy, M.; Khandani, M. H. Iran. Polym. J. 1997, 6, 5.
(15) (a) Li, W. T.; Pan, M. H.; Wu, Y. R.; Wang, S. L.; Liao, F. L.; Liu, R. S. J. Org. Chem. 2000, 65, 3761. (b) Chen, M. J.; Chang, S. T.; Liu, R. S. Tetrahedron 2000, 56, 5029. (c) Chen, M. J.; Lo, C. Y.; Chin, C. C.; Liu, R. S. J. Org. Chem. 2000, 65, 6362.
(16) (a) Vosko, S. H.; Wilk, L.; Nusair, M. Can. J. Phys 1980, 58, 1200. (b) Becke, A. D. Phys. ReV. A 1988, 38, 3098. (c) Lee, C.; Yang, W.; Parr, R. G. Phys. ReV. B 1988, 37, 785. (d) Hay, P. J.; Wadt, W. R. J. Chem.
Phys. 1985, 82, 299.
(17) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y. ; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S. ; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Revision D.02; Gaussian, Inc.: Wallingford, CT, 2004.
Figure 3. Optimized geometries and relative Gibbs free energies (kcal/mol) for the conversion of {Cp(PH3)2RudCdC(H)(C5H4NfBF2OH)} +
(A) to heterocyclic carbene {Cp(PH3)2RudCCH2(C5H4Nf BF2O)} +
(D). Values in parentheses are relative electronic energies.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
the crystal structure of complex 6a (1.944(3), 1.308(4), and 1.484(5) Å, respectively). In addition, the Ru-C-O bond angle of 121.4° and C-O-B bond angle of 124.6°also resemble those of 6a (123.4(2)° and 125.4(3)°, respectively). NBO analysis18of D, the modeling complex of 6a, reveals that the Ru-C and C-O bond orders are 1.46 and 1.95, respectively, indicating partial delocalization of π-electrons and positive charge at the carbene moiety. For compound A, the modeling complex of 5a, the LUMO and LUMO+1 orbitals are largely localized at CR, showing that an electrophilic attack might take place there (see Figure S1 in the Supporting Information). Consequently, the approach of the hydroxyl group of BF2OH to CR in A caused the formation of the intermediate B, which is lower in Gibbs free energy than A by -1.66 kcal/mol. B is formally an intramolecular cyclization product of A, where the oxygen atom is bound to boron, CR, and a proton. The transition state (TSAB) is 5.87 kcal/mol higher in free energy than A.
Following the reaction coordinate, the conversion of B to D first requires the rotation of the Ru-C bond to become the rotamer C, which is higher in free energy than B by 1.66 kcal/ mol. This can be rationalized by the metal-carbonπ-bonding resulted from different preferred orbital overlap of metal dπ
-orbital with the carbene and vinylidene p--orbital.19The conver-sion of C to D is exothermic by -20.59 kcal/mol. However, this conversion via the transition state TSCDhas an activation
free energy of 42.07 kcal/mol. Since experimentally we have observed that complex 5 spontaneously undergoes isomerization to give 6 at room temperature in solution, the pathway C f
TSCDfD is thus unfavorable. That is, the direct O-H addition is less likely. Alternatively, another pathway via a stepwise deprotonation-protonation process through the intermediate E is suggested. Although we cannot accurately calculate the relative energy of E in this system, this C f E f D pathway is much preferred due to the fact that the spontaneous transformation of 5 to 6 often occurs in acidic conditions, i.e., in the presence of an excess amount of HBF4 during the synthesis of 6. The overall conversion of A to D is exothermic by a free energy of -20.59 kcal/mol, which reasonably matches our experimental observation.
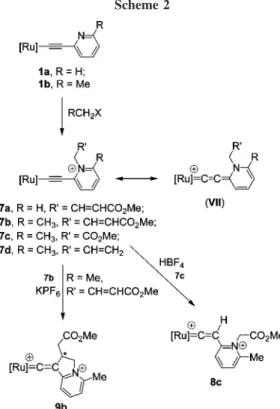
Alkylation Reactions. Reactions of 1 with various alkyl
halides are also studied. At room temperature, treatment of 1a with 4-bromomethylcrotonate produces the air-stable golden-yellow product 7a in high yield. The alkylation takes place at the nitrogen atom. Similarly, facile N-alkylation is observed in the case of 1b. Treatment of 1b with functionalized alkyl halides also generates 7b, 7c, and 7d in high yield (Scheme 2). Complexes 7a-7d were characterized by1H,31P, and13C NMR and mass spectra. 2D NMR HMBC and HSQC techniques were also applied to confirm the connectivity between the pyridiyl and the alkylated group. For example, the13C NMR spectrum of 7c shows a triplet resonance assigned to the Ru-CR carbon atδ 181.8 with JCP) 22.3 Hz. The1H NMR spectrum of 7c displays a singlet downfield shift resonance of the methylene protons of the ester group atδ 5.96. Of the most important is the 1H-13C HMBC spectrum, which shows long-range cou-plings between the methylene protons at δ 5.96 of the ester group to both Cβ at δ 114.9 and one of the pyridyl carbons at δ 153.7, but no cross-peak is observed with the CR resonance at δ 181.8. This demonstrates that the methylene protons of the ester group are not in the vicinity of CR.
The molecular structure of complex 7c established by single-crystal X-ray diffraction analysis is shown in Figure 4 with selected bond lengths and bond angles. Bonding of the pyri-dinium N-C bond faces the direction parallel to that of the Ru center to the Cp centroid. The Ru-C(1) single bond distance (1.977(4) Å) is slightly shorter than that in complex 1b (2.007(3) Å), whereas the C(1)-C(2) triple bond distance (1.219(5) Å) is slightly longer than that in 1b (1.216(4) Å) but is shorter that in the chromium allenylidene complex.8cThe Ru-C1-C2 bond angle is approximately the same as that in 1b (174.4(7)°). This fact might indicate partial contribution of the allenylidene resonance structure VII shown in Scheme 2 in the solid state. The13C resonance of CR of 7d atδ 181.8 compared to that of
1b atδ 117.6 provides further support.
The fact that complexes 1a and 1b underwent addition exclusively at the N atom rather than at Cβ might stem from the intrinsic reactivity of the pyridyl group. A similar event was observed in the alkylation reaction of tungsten and chromium pyridylacetylide and indolylacetylide complexes to give
alle-(18) Martin, F.; Zipse, H. J. Comput. Chem. 2005, 26, 97.
(19) (a) Schilling, B. E. R.; Hoffmann, R.; Lichtenberger, D. L. J. Am.
Chem. Soc. 1979, 101, 585. (b) Kostic´, N. M.; Fenske, R. F. Organometallics
1982, 1, 974.
Scheme 2
Figure 4. ORTEP drawing (30% thermal ellipsoid) of 7c with
phenyl groups on the phosphine ligands (except the ipso carbon), hydrogen atoms and the anion eliminated for clarity. Selected bond distances (Å) and bond angles (deg): Ru1-C1 1.977(4), C1-C2 1.219(5), C2-C3 1.400(5), N1-C9 1.458(5), Ru1-C1-C2 174.7(2), C1-C2-C3 174.1(3).
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
nylidene complexes.8c Also in ruthenium 4-pyridylacetylide complexes, the dangling pyridine was protonated, methylated, or ligated to tungsten carbonyl fragments to give various pyridiniumacetylide complexes.8aInterestingly, when the alky-lation reaction of 1b was carried out in methanol at 60°C, no reaction was observed. On the other hand, protonation took place when strong acid-like HBF4was used. Treatment of 7a, 7b,
7c, and 7d with excess HBF4 in CH2Cl2 at 0 °C produced various dicationic pyridiniumvinylidene complexes 8. However, among these spectroscopically observed pyridiniumvinylidene complexes only complex 8c, with the CH2CO2Me group, was isolated. The 31P resonance of phosphorus ligands and 1H resonance of the Cp group of complex 8c resemble that of complex 3. Other protonated products could not be isolated, since theβ-hydrogen of the product is relatively acidic, and these revert rapidly to pyridiniumacetylides in the presence of a weak base such as trace amounts of water in the solvent.20 Surprisingly, addition of KPF6to an acidic solution of 7b at room temperature resulted in the formation of the brown dicationic ruthenium vinylidene complex 9b (Scheme 2). A cyclization by the formation of a C-C bond takes place between Cβ of the vinylidene ligand and one of the olefinic carbons, leading to a heterocyclic fused-ring ligand. The 31P NMR spectrum of 9b displays a set of doublets of doublets atδ 41.3 and 39.6 with JPP) 24.6 Hz due to the presence of a stereogenic center. The1H NMR spectrum shows this methine resonance atδ 4.72, while the 13C NMR spectrum displays a resonance of this carbon atδ 32.62. The two methylene groups both split into multiplets in the1H NMR spectrum (δ 2.14 and 2.42 for NCH2; δ 2.38 and 2.42 for the other CH2) because of the adjacent stereogenic center. Finally, the characteristic R-carbon resonance at δC of 343.42 and the downfield-shifted Cp resonance atδHof 5.77 andδCof 97.55 indicate the presence of a vinylidene structure. 2D NMR COSY, HSQC, and HMBC techniques are also applied in the structure determination of
9b. The HSQC spectrum clearly displays C-H cross-peaks of
the two methylene groups, and cross-peaks in the HMBC between the methine and two methylene groups clearly indicate their connectivity. The structure of 9b is confirmed by X-ray diffraction analysis. Figure 5 shows an ORTEP drawing and selected bond lengths and angles of 9b. The Ru(1)-C(1) bond length of 1.820(6) Å and the C(1)-C(2) bond length of 1.330(9)
Å reveal a typical vinylidene skeleton. The Ru(1)-C(1)-C(2) bond angle of 166.0(5)°is bent from linear arrangement, which is possibly due to the steric effect between the CO2Me group and two PPh3ligands.2hAttempts to promote a similar coupling reaction for complex 7d failed to yield the desired product, indicating the requirement of the activating ester group in this C-C bond forming reaction.
On the basis of the aforementioned protonation reactions of pyridylacetylides 1 and pyridiniumacetylides 7, it is reasonable to assume that formation of 9b from 7b proceeds via an addition of the activated olefin to Cβ, leading to C-C bond formation generating the RudCdC vinylidene moiety. Addition of a proton possibly from acid or moisture at the other olefinic carbon atom then gave the final product. This C-C coupling reaction occurs between the internal carbon of the allylic moiety and the acetylide Cβ without cleavage of the C-N bond. Reactions involving coupling of the allylic terminal carbon with transition metal acetylide complexes have been reported in the literature.21 Preparations of transition metal vinylidene complexes with monosubstituted Cβ are well known in the literature. Yet preparation of vinylidenes with quarternary Cβ directly via highly substituted alkyl halides5a,bwere only scarcely demon-strated.22The established alkylating protocol for metal acetylides provided by Bruce11,23in reactions with alkyl halides is limited to primary ones. To our knowledge, these highly branched ruthenium pyridiniumvinylidenes are unprecedented. Our ob-servation in the formation of 9b unequivocally serves as an alternative approach to prepare Cβ,Cβ-disubstituted vinylidenes with a stereogenic center simultaneously generated at Cγ.
Conclusion
Protonation of the two ruthenium pyridylacetylide complexes
1a and 1b gave pyridiniumvinylidene complexes 3a and 3b,
respectively. Additions of BF3to 1a take place at the N atom of the pyridyl group, yielding 4a. This reaction is rationalized by the Lewis acid/base interaction. Subsequent protonation of
4a in the presence of moisture caused substitution of one F in
the BF3bonded to the 2-pyridyl group of 4a by an OH group, giving the vinylidene complex 5a. This is followed by a spontaneous cyclization reaction, resulting in the formation of the cationic carbene complex 6a. This spontaneous transforma-tion of 5a to 6a is further confirmed by DFT calculatransforma-tions using model complexes. Analogous vinylidene complex 5b could be obtained from 1b. However, the cyclization process is not observed in the case of complex 5b. Alkylation of 1a and 1b also took place preferentially at the N atom of the pyridyl group, yielding the pyridiniumacetylide complexes 7, which could be protonated to give pyridiniumvinylidene complexes 8 detected
(20) Bustelo, E.; Jime´nez-Tenorio, M.; Mereiter, K.; Puerta, M. C.; Valerga, P. Organometallics 2002, 21, 1903.
(21) (a) George, D. S. A.; Hilts, R. W.; McDonald, R.; Cowie, M.
Organometallics 1999, 18, 5330. (b) Winter, R. F.; Klinkhammer, K.-W;
Za´lisˇ, S. Organometallics 2001, 20, 1317.
(22) (a) Chang, C. W.; Lin, Y. C.; Lee, G. H.; Wang, Y. J. Chem. Soc.,
Dalton Trans. 1999, 4223. (b) Chang, C. W.; Lin, Y. C.; Lee, G. H.; Huang,
S. L.; Wang, Y. Organometallics 1998, 17, 2534. (c) Ipaktschi, J.; Mu¨ller, B. G.; Glaum, R. Organometallics 1994, 13, 1044. (d) Birdwhistell, K. R.; Templeton, J. L. Organometallics 1985, 4, 2062. (e) Bruce, M. I.; Koutsantonis, G. A.; Liddell, M. J.; Tiekink, E. R. T. J. Organomet. Chem. 1991, 420, 253. (f) Bruce, M. I.; Cifuentes, M. P.; Snow, M. R.; Tiekink, E. R. T. J. Organomet. Chem. 1989, 359, 379. (g) Fischer, H.; Scheck, P. A. Chem. Commun. 1999, 1031. (h) Lugan, N.; Kelley, C.; Terry, M. R.; Geoffroy, G. L.; Rheingold, A. L. J. Am. Chem. Soc. 1990, 112, 3220. (i) Werner, H.; Schneider, D.; Schulz, M. J. Organomet. Chem. 1993, 451, 175.
(23) (a) Davison, A.; Selegue, J. P. J. Am. Chem. Soc. 1978, 100, 7763. (b) Abbott, S.; Davies, S. G.; Warner, P. J. Organomet. Chem. 1983, 246, C65. (c) Bianchini, C.; Peruzzini, M.; Vacca, A.; Zanobini, F.
Organome-tallics 1991, 10, 3697. Figure 5. ORTEP drawing (30% thermal ellipsoid) of 9b with
phenyl groups of PPh3ligands (except ipso carbon atoms), hydrogen
atoms and the anion are eliminated for clarity. Selected bond distances (Å) and bond angles (deg): Ru1-C1, 1.820(6); C1-C2, 1.330(9); C2-C3, 1.447(9); C3-N1, 1.333(8); N1-C9, 1.473(9); Ru1-P1, 2.3715(16); Ru1-P2, 2.3560(16); Ru1-C1-C2, 166.0(5); C1-C2-C3, 126.3(6); C3-C2-C10, 108.3(5); P1-Ru1-P2, 99.93(5); C1-Ru1-P1, 97.94(17); C1-Ru1-P2, 94.01(19).
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
spectroscopically. For the pyridiniumacetylide complex 7d, containing an unsaturated functional group CH2CHdCHCO2Me on the pyridinium moiety, the C-C coupling of the acetylide Cβ with the CdC double bond yielded 9b.
Experimental Section
General Procedures. All manipulations were performed under
nitrogen using vacuum-line, glovebox, and standard Schlenk techniques unless mentioned otherwise. Hexanes and CH2Cl2were
distilled from CaH2, diethyl ether and THF from sodium
benzophe-none ketyl, and methanol from Mg/I2. All other solvents were of
reagent grade and were used as received. NMR spectra were recorded on Bruker Avance-400 and DMX-500 FT-NMR spec-trometers at room temperature (unless stated otherwise) and are reported in units ofδ with residual protons in the solvents as a
standard (CDCl3, δ 7.24; d6-acetone,δ 2.04). FAB mass spectra
were recorded using a JEOL SX-102A spectrometer using 3-ni-trobenzyl alcohol (NBA) as the matrix. Infrared spectra were recorded on a Nicolet-MAGNA-550 spectrometer. X-ray diffraction studies were carried out at the Regional Center of Analytical Instrument at the National Taiwan University. All reagents were obtained from commercial suppliers. RuCl3· xH2O was purchased
from Strem Chemicals. Cp(PPh3)2RuCl24was prepared following
the method reported in the literature.
Synthesis of [Ru]CtC(C5H3RN) (R ) H, 1a; R ) CH3, 1b).
In a Schlenk flask containing a mixed solvent of CHCl3/MeOH/
NEt3(15:15:1 mL) were added at ambient temperature [Ru]Cl (1.03
g, 1.42 mmol) and 2-ethynyl-6-methylpyridine (406 mg, 3.1 mmol), and the resulting solution was stirred for 18 h. After that the volatiles were removed to obtain an oily product, which was redissolved in 10 mL of CH2Cl2followed by filtration via a small pack of Celite
into 60 mL of methanol. The precipitate thus formed was collected by filtration and dried under vacuum to afford 1b as a yellow powder (824 mg, 72.0% yield). The rest of the methanol solution was evaporated to dryness, and the residue was recrystallized from CH2Cl2/cold pentane to bring about more desired product 1b as a
brown microcrystal, 34.09 mg (yield ca. 18.3%). Anal. (%) Calcd for C49H41NP2Ru: C, 72.94; H, 5.12; N, 1.74. Found: C, 72.78; H,
5.10; N, 1.82. FAB mass (m/z): 807.3 (M+), 546.1 (M+- PPh3), 468.1, 429.1 ([Cp(PPh3)Ru] + ). IR (KBr, cm-1):ν 2064.6 (CtC). 1H NMR (CDCl 3):δ 7.46-7.04 (Ph and Py), 6.70 (d,3JHH) 7.7 Hz, 1H, Py), 6.43 (d,3J HH) 7.7 Hz, 1H, Py), 4.35 (s, 5H, Cp), 2.50 (s, 3H, CH3).13C NMR (CDCl3):δ 132.86, 131.98, 131.22,
128.98 (Py), 138.66-127.35 (Ph), 123.38 (Py), 117.57 (RuCC), 115.38 (t,2J
CP) 5.2 Hz, RuC), 86.02 (Cp), 23.39 (CH3).31P NMR
(CDCl3):δ 51.2 (s).
Complex 1a was prepared in 73% yield from 2-ethynylpyridine following the same procedure as that of 1b. Spectroscopic data for
1a: Anal. (%) Calcd for C48H39NP2Ru: C, 72.71; H, 4.96; N, 1.77.
Found: C, 71.88; H, 4.91; N, 1.63. ESI mass (m/z): 794.0 (M++ 1).1H NMR (CDCl 3):δ 8.39 (d,3JHH) 7.6 Hz, Py), 7.30 (t,3JHH ) 7.6 Hz, Py), 7.43-7.05 (Ph), 6.83 (t,3J HH) 7.6 Hz, Py), 6.64 (d,3J HH) 7.6 Hz, Py), 4.36 (s, 5H, Cp).13C NMR (CDCl3): δ 149.03, 147.81 (Py), 139.11-127.01 (Ph), 125.51 (Py), 124.30 (t, 2J
CP) 25.8 Hz, RuC), 117.73 (Py), 116.52 (RuCC), 85.65 (Cp). 31P NMR (CDCl
3):δ 51.0.
Synthesis of {[Ru]dC(OMe)CH2(C5H4N)}PF6 (2a). To a
Schlenk flask charged with [Ru]Cl (254.2 mg, 0.35 mmol) and NaPF6(56.0 mg, 0.47 mmol) were added 2-ethynylpyridine (0.5
mL, 0.5 mmol) and 35 mL of mixed solvent (CH2Cl2/methanol,
3:4, v/v) under nitrogen. The resulting solution was stirred at room temperature for 13 h. After that, volatiles were removed and the solid residue was extracted with 5 mL of CH2Cl2 followed by
reprecipitation with 60 mL of diethyl ether. The precipitate thus formed was collected in a glass frit, washed with diethyl ether, and dried under vacuum. The final product can be obtained as a brown powder and was identified as complex 2a (255.7 mg, 75% yield). Anal. (%) Calcd for C49H44F6NOP3Ru: C, 60.62; H, 4.57;
N, 1.44. Found: C, 60.71; H, 4.71; N, 1.32. FAB mass (m/z): 826.3 (M+), 719.2, 564.1 (M+- PPh3), 429.1 ([Cp(PPh3)Ru] + ). IR (KBr, cm-1):ν 1588.9 (RudC), 1263.7 (br, C-O).1H NMR (CDCl 3):δ 8.26 (d,3J HH) 6.8 Hz,1H, Py), 7,90 (t,3JHH) 6.8 Hz, 1H, Py), 7.65 (t,3J
HH) 6.8 Hz, 1H, Py), 7.96-6.38 (Ph and Py), 4.99 (s,
2H, CH2), 4.78 (s, 5H, Cp), 3.38 (s, 3H, OCH3).13C NMR (CDCl3): δ 306.0 (t,2J
CP) 12.8 Hz, RuC), 154.77, 148.91, 137.82 (Py)
136.16-127.62 (Ph), 124.49, 122.30 (Py), 91.84 (s, Cp), 64.05 (s,
CH2), 62.23 (s, OCH3).31P NMR (CDCl3):δ 46.6.
Protonation of 1a and 1b. Complex 1a (182.2 mg, 0.23 mmol)
was dissolved in 80 mL of diethyl ether. The resulting solution was cooled to 0 °C followed by addition of HBF4(54 wt % in
diethyl ether, 0.5 mL) via a syringe, and the solution was allowed to warm to room temperature and stirred for 1 day. After that, the precipitate was collected by a glass frit, washed with diethyl ether, and dried under vacuum to afford 3a as an orange powder (211.7 mg, 95% yield). Anal. (%) Calcd for C48H41B2F8NP2Ru: C, 59.53;
H, 4.27; N, 1.45. Found: C, 59.83; H, 4.10; N, 1.52. 1H NMR (CDCl3):δ 12.7 (br, 1H, NH), 8.29 (1H, Py), 8.14 (1H, Py), 7.93 (1H, Py), 7.59-6.89 (Ph), 5.90 (s, 1H, dCdCH), 5.51 (s, 5H, Cp). 13C NMR (CDCl 3):δ 346.41 (t, 2JCP) 14.6 Hz, RuC), 145.96, 139.47 (Py), 133.22-128.00 (Ph), 125.86, 122.35 (Py), 112.40 (RuCC), 96.58 (Cp).31P NMR (CDCl 3):δ 37.3 (s).
Complex 3b was similarly obtained from 1b in 91% yield. Spectroscopic data for3b: Anal. (%) Calcd for C49H43B2F8NP2Ru:
C 59.90, H 4.41, N 1.43. Found: C, 59.78; H, 4.50; N, 1.52.1H
NMR (CDCl3):δ 12.28 (br, 1H, NH), 8.13 (t,3JHH) 7.8 Hz, 1H,
Py), 7.59 (d,3J
HH) 7.8 Hz, 1H, Py), 7.60-7.03 (Ph and Py), 6.10
(s, 1H, RuCCH), 5.50 (s, 5H, Cp), 2.65 (s, 3H, CH3).31P NMR
(CDCl3):δ 37.8 (s).
Synthesis of [Ru]CtC(C5H4NfBY3) (Y ) F, 4a; Y ) H, 4c). At room temperature complex 1a (90.3 mg, 0.11 mmol) was
weighted into a Schlenk flask under nitrogen. Diethyl ether (80 mL) was added into the flask via a cannula followed by addition of BF3-OEt2(ca. 48%, 0.02 mL) under nitrogen. The resulting
orange cloudy solution was then stirred for 8 h. After that, the mixture was filtered through a glass frit under nitrogen, and the solid was washed with diethyl ether and dried under vacuum to afford 4a as a golden powder (85.9 mg, 88% yield). Anal. (%) Calcd for C48H39BF3NP2Ru: C, 66.99; H, 4.57; N, 1.63. Found: C,
66.79; H, 4.60; N, 1.52.1H NMR (CDCl 3):δ 8.29 (d,3JHH) 8.3 Hz, 1H, Py), 7.68 (t,3J HH ) 8.3 Hz, 1H, Py), 7.11-7.39 (Ph), 7.04 (t,3J HH) 8.3 Hz, 1H, Py), 6.70 (d,3JHH) 8.3 Hz, 1H, Py), 4.54 (s, 5H, Cp).13C NMR (CDCl 3):δ 173.98 (t,2JCP) 23.0 Hz, RuC), 142.53, 139.58, 136.41 (Py), 137.78-127.45 (Ph), 117.15 (Py), 113.08 (RuCC), 87.44 (Cp).31P NMR (CDCl 3):δ 50.9.19F NMR (CDCl3):δ -152.2.
Complex 4c was similarly synthesized from 1a and BH3-THF
in 61% yield. Spectroscopic data for 4c: Anal. (%) Calcd for C48H42BNP2Ru: C, 71.47; H, 5.25; N, 1.74. Found: C, 71.68; H,
5.10; N, 1.62.1H NMR (CDCl
3):δ 8.51 (d,3JHH) 8.0 Hz, 1H,
Py), 7.07-7.44 (Ph and Py), 6.82 (d,3J
HH) 8.0 Hz, 1H, Py), 4.51 (s, 5H, Cp), 2.5-3.5 (br, 3H, BH3).13C NMR (CDCl3):δ 153.47 (t,2J CP ) 23.5 Hz, RuC), 147.37, 143.20, 136.55, 128.23 (Py), 138.71-127.39 (Ph), 117.03 (Py), 115.39 (RuCC), 86.58 (Cp).31P NMR (CDCl3):δ 51.5.
Synthesis of {[Ru]dCdC(H)(C5H3RNfBF2OH)}BF4(5a, R
) H; 5b; R ) Me). To a Schlenk flask containing a CH2Cl2
solution (25 mL) of complex 1a (139.8 mg, 0.18 mmol) was added BF3-OEt2(1.25 mL) at room temperature, and the resulting solution
was stirred for 12 h, while the color of the solution turned from yellow to orange-red. Then volatiles of the solution were removed
(24) Bruce, M. I.; Hameister, C.; Swincer, A. G.; Wallis, R. C. Inorg.
Synth. 1990, 28, 270.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
under vacuum, and 60 mL of diethyl ether was added into the flask. The resulting mixtures were stirred vigorously under nitrogen for 1.5 h. Precipitates thus formed were collected by a glass frit, washed with diethyl ether, and dried in vacuo to give complex 5a as a pale orange powder (120.9 mg, 72%). Anal. (%) Calcd for C48H41B2F6NOP2Ru: C, 60.91; H, 4.37; N, 1.48. Found: C, 60.74;
H, 4.10; N, 1.48.1H NMR (CDCl 3):δ 12.33 (br, 1H, OH), 8.14 (t, 3J HH) 7.2 Hz, Py), 7.71 (d,3JHH) 7.2 Hz, Py), 7.43-6.95 (Ph and Py), 5.90 (s, 1H, dCdCH), 5.49 (s, 5H, Cp). 13C NMR (CDCl3):δ 344.56 (t,2JCP) 15.7 Hz, RuC), 146.02, 145.94, 139.46
(Py), 133.48-126.50 (Ph), 125.93, 122.42 (Py), 112.46 (RuCC), 96.62 (Cp).31P NMR (CDCl
3):δ 37.6 (s).19F NMR (CDCl3):δ
-149.6 (br, BF2), -150.7 (BF4).
Complex 5b was prepared using the same procedure as that of
5a in 79% yield. Spectroscopic data for 5b:1H NMR (CDCl 3):δ
12.14 (br, 1H, OH), 7.99-7.67 (m, Py), 7.54-6.99 (Ph and Py), 6.09 (s, 1H, dCH), 5.46 (s, 5H, Cp), 2.58 (s, 3H, CH3).13C NMR
(CDCl3):δ 345.82 (t,2JCP) 16.2 Hz, RuC), 151.96, 145.77, 145.46
(Py), 134.58-128.55 (Ph), 123.05, 122.59 (Py), 112.48 (RuCC), 96.44 (Cp), 19.47 (CH3).31P NMR (CDCl3):δ 37.7 (s).19F NMR
(CDCl3):δ -150.4 (BF4).
Synthesis of {[Ru]dC(O)CH2(C5H4NBF2)}BF4(6a). To a 10
mL chloroform solution of 2-ethynylpyridine (0.4 mL, 3.96 mmol) at room temperature was added an aliquot of BF3-OEt2(ca. 48%
in ether, 2 mL, 7.57 mmol). The solution was stirred for 10 min under nitrogen. Subsequently the solution was transferred to a 25 mL chloroform solution containing [Ru]Cl (514.3 mg, 0.71 mmol) under nitrogen. After stirring for 2 days the solvent of the resulting solution was removed and the crude product was extracted with CH2Cl2. The solution was concentrated and added into diethyl ether,
and the precipitate thus formed was collected by a glass frit, washed with diethyl ether and hexane, and dried under vacuum to afford the yellow product 6a (485.2 mg; 85% yield). Anal. (%) Calcd for C49H43B2F6NOP2Ru: C, 61.27; H, 4.51; N, 1.46. Found: C, 61.51;
H, 4.60; N, 1.42. FAB mass (m/z): 860.4 (M+). IR (KBr, cm-1): ν 1350.5 (νCO), 1081.9 (m, BF), 1030.2 (m, BF). 1H NMR (CDCl3):δ 8.46 (d,3JHH) 7.2 Hz, 1H, Py), 8.30 (t,3JHH) 7.2 Hz, 1H, Py), 8.03 (d,3J HH) 7.2 Hz, 1H, Py), 7.51 (t,3JHH) 7.2 Hz, 1H, Py), 7.31-6.95 (Ph), 5.15 (s, 2H, CH2), 4.94 (s, 5H, Cp). 13C NMR (CDCl 3):δ 292.65 (t, 2JCP) 12.7 Hz, RuC), 148.54, 145.31, 140.21 (Py), 135.96-128.04 (Ph), 126.60, 124.16 (Py), 92.18 (Cp), 63.11 (CH2).31P NMR (CDCl3):δ 46.9 (s).19F NMR (CDCl3):δ -155.8 (br, 2F, BF2), -152.4 (4F, BF4).
Synthesis of Complexes {[Ru]CtC(C5H3RNCH2R′)}X (R ) H, R′) CHdCHCO2CH3, 7a; R ) Me, R′) CHdCHCO2CH3, 7b; R ) Me, R′) CO2CH3, 7c; R ) Me, R′) CHdCH2, 7d).
Two synthetic methods are used. Method A for 7d: an aliquot of allyl iodide (1 mL) was added into a 20 mL of CH2Cl2solution of
1b (507 mg, 0.63 mmol) under nitrogen, and the solution was stirred
at 0°C overnight. The volume of the resulting solution was reduced
to 5 mL and was added into 50 mL of diethyl ether. A bright yellow precipitate thus formed was filtered through a glass frit, washed three times with diethyl ether and hexane, and dried under vacuum to obtain complex 7d (432 mg, 81% yield). Method B: to a Schlenk flask containing 50 mL of a diethyl ether solution of 1b (47.5 mg, 0.06 mmol) was added 0.1 mL of allyl iodide and the resulting solution stirred overnight. The golden precipitate thus formed was collected, washed, and dried under vacuum to afford 7d. The yield is comparable. Anal. (%) Calcd for C52H46INP2Ru: C, 64.07; H,
4.76; N, 1.44. Found: C, 64.18; H, 4.60; N, 1.52. FAB mass (m/z): 848 (M+).1H NMR (CDCl 3):δ 7.68 (t,3JHH) 7.8 Hz, 1H, Py), 7.41-7.05 (Ph and Py), 6.57 (d,3J HH) 7.8 Hz, 1H, Py), 5.89 (s, 2H, CH2), 5.03 (s, 1H, dCH), 4.45 (s, 5H, Cp), 3.71 (s, 2H, dCH2), 2.82 (s, 3H, CH3).13C NMR (CDCl3):δ 181.33 (t, 2JCP) 22.5 Hz, RuC), 166.86 (dCH2), 152.97, 141.26, 139.26 (Py), 137.40-127.75 (Ph), 127.38, 120.53 (Py), 114.32 (RuCC), 90.82 (dCH), 87.05 (Cp), 55.01 (CH2), 53.18 (CH2), 22.46 (CH3).31P NMR (CDCl3): δ 49.8.
Complexes 7a, 7b, and 7c were prepared following the same procedure as that in 7d. Spectroscopic data for 7a (method B, in 85% yield): Anal. (%) Calcd for C53H46BrNO2P2Ru: C, 65.50; H,
4.77; N, 1.44. Found: C, 65.74; H, 4.81; N, 1.32.1H NMR (CDCl 3): δ 9.30 (d,3J
HH) 8.1 Hz, 1H, Py), 7.72 (t,3JHH) 8.1 Hz, 1H,
Py), 7.40-6.89 (Ph and Py), 6.75 (d,3J
HH) 8.1 Hz, 1H, Py), 6.17 (d,3J HH) 15.6 Hz, 1H, dCH), 5.89 (s, 2H, NCH2), 4.45 (s, 5H, Cp), 3.67 (s, 3H, OCH3).13C NMR (CDCl3):δ 181.15 (t,2JCP) 22.6 Hz, RuC), 165.56, 141.39, 140.05 (Py), 137.41-127.23 (Ph), 124.56, 118.82 (Py), 113.39 (RuCC), 86.85 (Cp), 57.01 (NCH2), 51.60 (OCH3).31P NMR (CDCl3):δ 49.5.
Spectroscopic data for 7b (method B, in 98% yield): Anal. (%) Calcd for C54H48BrNO2P2Ru: C 65.79, H 4.91, N 1.42. Found: C,
65.78; H, 5.04; N, 1.41. FAB mass (m/z): 906.3 (M+), 719.3, 645.3, 429.0.1H NMR (CDCl
3):δ 7.62-6.98 (Ph, dCH and Py), 6.66
(d,3J
HH) 8.2 Hz, 1H, Py), 6.03 (d,3JHH) 2.7 Hz, 2H, CH2),
5.77 (m, 1H, dCHCO2Me), 4.47 (s, 5H, Cp), 3.67 (s, 3H, OCH3),
2.91 (s, 3H, CH3).13C NMR (CDCl3):δ 180.27 (t, 2JCP) 22.5
Hz, RuC), 165.35 (CdO), 152.48, 140.86, 140.15 (Py), 137.40-127.24 (Ph), 122.81, 120.69 (Py), 114.18 (RuCC), 97.50 (dCH), 90.78 (dCH), 86.93 (Cp), 54.62 (CH2), 51.69 (OCH3), 22.08 (CH3).31P
NMR (CDCl3):δ 49.6.
Spectroscopic data for 7c (method A, in 86% yield): Anal. (%) calcd for C52H46BrNO2P2Ru: C, 65.07; H, 4.83; N, 1.46. Found:
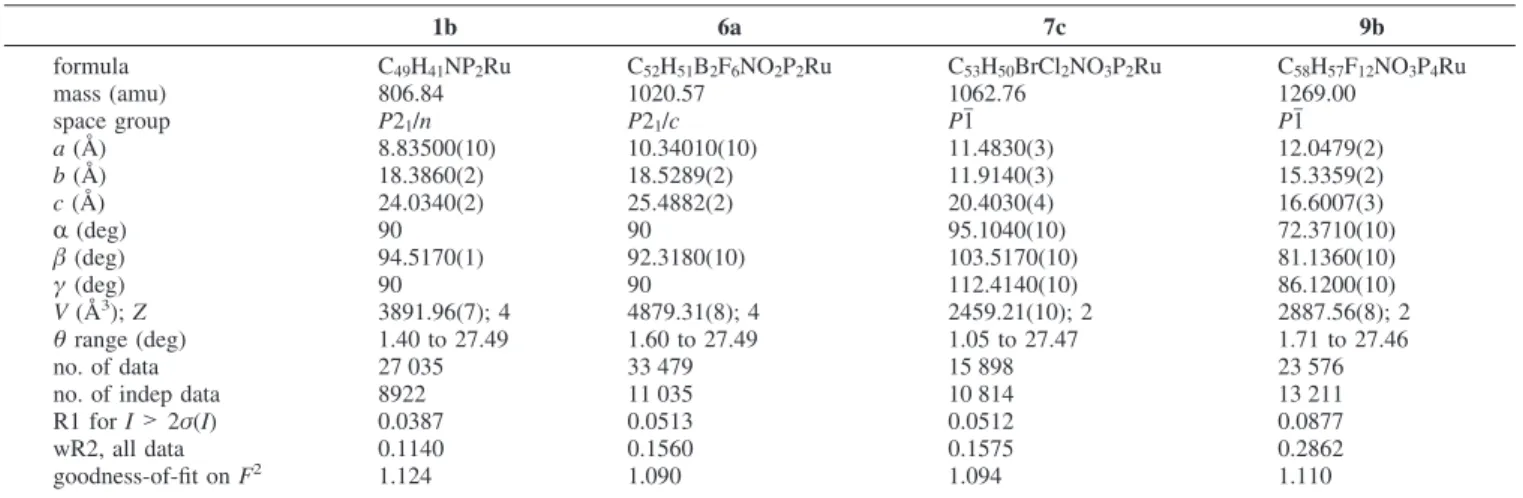
C, 65.17; H, 4.91; N, 1.48. FAB mass (m/z): 880.1 (M++ 1).1H NMR (CDCl3):δ 7.70 (d,3JHH) 8.1 Hz, 1H, Py), 7.26-7.12 (Ph and Py), 6.56 (d,3J HH) 8.1 Hz, 1H, Py), 5.96 (s, 2H, CH2), 4.46 (s, 5H, Cp), 3.72 (s, 3H, OCH3), 2.87 (s, 3H, CH3).13C NMR (CDCl3):δ 181.76 (t,2JCP) 22.3 Hz, RuC), 167.54 (CdO), 153.74, 141.90, 139.95 (Py), 138.06-128.37 (Ph), 128.00, 121.18 (Py), 114.95 (RuCC), 87.68 (Cp), 55.76 (CH2), 53.78 (OCH3), 23.15 (CH3).31P NMR (CDCl3):δ 49.8. Table 1. Crystal Data and Refinement Parameters for Complexes 1b, 6a, 7c, and 9b
1b 6a 7c 9b formula C49H41NP2Ru C52H51B2F6NO2P2Ru C53H50BrCl2NO3P2Ru C58H57F12NO3P4Ru mass (amu) 806.84 1020.57 1062.76 1269.00 space group P21/n P21/c P1j P1j a (Å) 8.83500(10) 10.34010(10) 11.4830(3) 12.0479(2) b (Å) 18.3860(2) 18.5289(2) 11.9140(3) 15.3359(2) c (Å) 24.0340(2) 25.4882(2) 20.4030(4) 16.6007(3) R (deg) 90 90 95.1040(10) 72.3710(10) β (deg) 94.5170(1) 92.3180(10) 103.5170(10) 81.1360(10) γ (deg) 90 90 112.4140(10) 86.1200(10) V (Å3); Z 3891.96(7); 4 4879.31(8); 4 2459.21(10); 2 2887.56(8); 2 θ range (deg) 1.40 to 27.49 1.60 to 27.49 1.05 to 27.47 1.71 to 27.46 no. of data 27 035 33 479 15 898 23 576
no. of indep data 8922 11 035 10 814 13 211
R1 for I > 2σ(I) 0.0387 0.0513 0.0512 0.0877
wR2, all data 0.1140 0.1560 0.1575 0.2862
goodness-of-fit on F2 1.124 1.090 1.094 1.110
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
Synthesis of {[Ru]dCdC(H)(C5H3MeNCH2CO2CH3)}(BF4)2 (8c). To a Schlenk flask containing a solution of 7c (45.5 mg, 0.05
mmol) in 20 mL of CH2Cl2at 0°C was added HBF4(54 wt %
diethyl ether solution, 0.1 mL) via a syringe. The resulting solution was allowed to warm to room temperature in 10 min, and the color of the solution turned from yellow to brown. Volatiles were removed under vacuum, and the residue was washed with diethyl ether. A brown solid thus formed was collected and dried in vacuo to afford 8c (38.3 mg, 77% yield).1H NMR (CDCl 3):δ 8.20 (t, 3J HH) 8.2 Hz, Py), 7.72-6.98 (Ph, Py), 5.85 (d,3JHH) 8.2 Hz, 1H, Py), 5.58 (s, 5H, Cp), 5.01 (s, 2H, NCH2), 4.27 (m, 1H, dCdCH), 3.77 (s, 3H, OCH 3), 2.65 (s, 3H, CH3). 13C NMR (CDCl3): δ 341.77 (t, 3JCP ) 14.6 Hz, RuC), 189.28 (CdO), 153.58-129.03 (Ph, Py), 101.38 (RuCC), 97.82 (Cp), 54.03 (OCH3),19.85 (CH3).31P NMR (CDCl3):δ 38.9.
Transformation of Complex 7b to 9b. A CHCl3solution (4
mL) containing complex 7b (80.4 mg, 0.08 mmol) and KPF6(33.9
mg, 0.18 mmol) was stirred at ambient atmosphere for 3 days. Volatiles were removed and the residue was extracted with CH2Cl2
(10 mL), which was added into diethyl ether to form a precipitate, which was collected, washed with diethyl ether and cold THF, and dried under vacuum to afford 9b as a brown powder (48.4 mg, 50% yield). Anal. (%) Calcd for C54H49F12NO2P4Ru: C, 54.19; H,
4.13; N, 1.17. Found: C, 54.07; H, 4.10; N, 1.12. FAB mass (m/z): 906.4 (M++ 1), 726.1, 644.2 (M2+- PPh 3), 429 ([Cp(PPh3)Ru] + ). 1H NMR (CDCl 3): δ 7.40-6.90 (Ph and Py), 5.77 (s, 5H, Cp), 5.59 (m, 1H, NCH), 4.72 (br, 1H, CH), 4.42 (d,3J HH) 11.3 Hz, 1H, NCH), 3.53 (s, 3H, OCH3), 2.74 (s, 3H, CH3), 2.42 (m, 2H) and 2.38 (dd, JHH) 7.0 Hz, 1H, CH), 2.14 (d,3JHH) 16.1 Hz, 1H, CH).13C NMR (CDCl 3):δ 343.42 (t,3JCP) 13.4 Hz, RuC), 171.38 (CO), 152.21, 149.21, 143.44 (Py), 133.87-128.43 (Ph), 124.75, 120.70 (Py), 97.55 (Cp), 61.03 (NCH2), 52.01 (OCH3), 39.59 (CH2), 32.62 (CH), 20.79 (CH3).31P NMR (CDCl3):δ 41.3, 39.6 (2 d,2J PP) 24.6 Hz).
X-ray Structure Determinations. Details of the structure
analyses carried out on complexes 1b, 6a, 7c, and 9b are given in Table 1. A single crystal of 1b suitable for an X-ray diffraction study was glued to a glass fiber and mounted on a Nonius Kappa CCD diffractometer. The diffraction data were collected using 3 kW sealed-tube molybdenum KR radiation (T ) 295 K). Exposure time was 5 s per frame. Multiscan absorption correction was applied, and decay was negligible. Data were processed, and the structures were solved and refined by the SHELXTL program.25The structure
was solved using direct methods and confirmed by Patterson methods, and refining on intensities of all data gave R1 and wR2 for unique observed reflections (I > 2σ(I)). Hydrogen atoms were
placed geometrically using the riding model with thermal parameters set to 1.2 times that for the atoms to which the hydrogen is attached and 1.5 times that for the methyl hydrogens. Structures of complexes
6a, 7c, and 9b were similarly determined. Table 1 lists the crystal
data and refinement parameters for complexes 1b, 6a, 7c, and 9b.
Computational Methods. Theoretical calculations have been
carried out using the Gaussian 03 program17at the DFT level by
means of the Becke 3 and Lee-Yang-Parr (B3LYP) composite exchange-correlation functional.16The LanL2DZ basis sets was
used throughout the calculation, where for the Ru atom the innermost electrons are replaced by a relativistic ECP and the 18 valence electrons are explicitly treated by a double-ξ basis set. Full
geometry optimization without symmetry restrictions was performed for all structure. Before performing optimization of ground-state geometries at the B3LYP/LanL2DZ level of theory, the molecular structures were initially optimized at the semiempirical PM326level
of calculation. Harmonic frequencies were calculated at the optimization level using the same basis sets, and the nature of the stationary points was determined in each case according to the right number of negative eigenvalues of the Hessian matrix. For each of the stationary points, the presence of one imaginary frequency indicates a transition state, while no imaginary frequency indicates a local minimum. The intrinsic reaction coordinate (IRC) path-ways27from the transition structures have been followed using a
second-order integration method28to verify the expected
connec-tions of the first-order saddle points with the correct local minima found on the potential energy surface.
Acknowledgment. This research is supported by the National Science Council and National Center for High-Performance Computing of Taiwan, Republic of China.
Supporting Information Available: Complete crystallographic
data for 1b, 6a, 7c, and 9b (CIF) and detailed computational results. This material is available free of charge via the Internet at http://pubs.acs.org.
OM800253E
(25) SHELXTL: Structure Analysis Program, version 5.04; Siemens Industrial Automation Inc.: Madison, WI, 1995.
(26) (a) Stewart, J. J. P. J. Comput. Chem. 1989, 10, 207. (b) Stewart, J. J. P. J. Comput. Chem. 1989, 10, 221.
(27) Fukui, K. Acc. Chem. Res. 1981, 14, 363.
(28) Gonzalez, C.; Schlegel, H. B. J. Phys. Chem. 1990, 94, 5523.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009