oma
Synthesis of Benzothiadiazole-Based Liquid Crystalline Polyacrylates
for Polarized Light Emitting Diodes
Yung-Hsin Yao, Liang-Rern Kung and Chain-Shu Hsu*
Department of Applied Chemistry, National Chiao Tung University, 1001 Ta Hsueh Road, Hsinchu 300, Taiwan, Republic of China
(*Author for correspondence; Tel.: +886-3-5131523; Fax: +886-3-5131523; E-mail: [email protected])
Received 25 August 2005; accepted in revised form 6 December 2005
Key words: liquid crystalline polymer, polarized electroluminescence
Abstract
Two benzothiadiazole-based liquid crystalline polyacrylates were synthesized. These polymers revealed a nematic liquid crystal phase and exhibited photoluminescence as well as polarized electroluminescence when incorporated into light-emitting diode applications. The polymers showed dichroic ratios of about 8.3Y8.8 in UV-vis absorption and photoluminescence emission. The polymer with vinylene linkages (P2) showed better electroluminescence device performance than that with acetylene linkages (P1). The P2 device emitted red light at 604 nm with a turn-on voltage at 6 V, and a maximum polarized luminance of 235 cd/m2at 12 V, with an efficiency of 0.09 cd/A and a polarization ratio of 6.5.
Introduction
During the last few decades, liquid crystalline polymers have been extensively investigated, not least because of their unusual mechanical, optical and rheological proper-ties [1Y3], and the considerable possibility to utilize them as polarized emitters in organic light emitting diode struc-tures [4,5]. Such a polarized light source became of inter-est for a backlight in a liquid crystalline display, since it could replace a combination of a normal backlight and a polarizer. The expected benefit is that power efficiency can be enhanced because absorption by the polarizer can be avoided. With their unique self-organizing nature and fluidity, polymeric liquid crystalline materials generally have good processability, allowing simple fabrication by solution processes, and an intrinsically high durability [6,7].
The synthesis of several liquid crystalline polymers containing mesogenic conducting groups, either hole (ben-zothiadiazole) or electron (oxadiazole) transport moieties, has been reported [8Y11]. These materials showed charge transport and electroluminescence (EL) properties which resulted from high electron and/or hole drift mobilities owing to the aligned and stacked structures in the me-sophase [12]. Moreover, it was reported recently that elec-troluminescent devices using the aforementioned materials exhibited polarized emissions [13Y15]. Among various molecular alignment techniques, cooperative alignment of a pendant chromophore within a liquid crystalline material has been extensively studied [16]. The conventional
meth-od to achieve monmeth-odomain alignment of liquid crystals is to use an alignment layer. The alignment layer should permit hole injection and transport from the anode to the aligned emission layer. Common alignment materials include polyimide doped with hole-transporting materials [17], for example, poly(p-phenylene vinylene) [18Y20] and poly(3,4-ethylene-dioxythiophene) (PEDOT)-poly(styrene-sulfonic acid), which show good conducting, transparency and film-forming properties [21].
Recently, we reported polarized EL of a liquid crystal-line polyacrylate based on bistolane mesogen with PEDOT as an alignment layer [22]. This device emitted in the blue region with a dichroic ratio of 6.1 and a brightness of 3 cd/m2 at 14 V. In this work, the syntheses and comparative investigation of two benzothiadiazole-based liquid crystal-line polymers are described. The electronic properties as well as the polarized EL properties of these polymers have been evaluated.
Experimental
Materials and instrumentation
All manipulations involving air-sensitive reagents were performed under an atmosphere of dry argon. All reagents, unless otherwise specified, were obtained from Aldrich, Acros, and TCI Chemical Co. and were used without fur-ther purification. All solvents were carefully dried and pu-rified under a nitrogen flow. 1H- and 13C-NMR spectra
were recorded using a Varian 300 spectrometer and were referenced to tetramethylsilane. Gel permeation chroma-tography was performed with a VE-2001 gel permeation chromatograph in tetrahydrofuran (THF) using a calibra-tion curve of polystyrene standards. Polarized UV-vis spectra were measured with a Shimadzu UV-1601 spec-trophotometer with a polarizer placed between the sample and the detector. Spectra were measured with the polarizer aligned parallel and perpendicular to the rubbing direction. Polarization ratios were defined as the parallel to the perpendicular intensity. Polarized photoluminescence (PL) measurements were measured with a Shimadzu RF-5301 PC spectrofluorophotometer with a polarizer.
Polarized device fabrication and characterization
Polarized polymer light-emitting diodes were fabricated on indium tin oxide (ITO) coated glass substrates cleaned sequentially in ultrasonic baths of detergent, deionized water, 2-propanol, deionized water, and acetone. UV-ozone treatment was carried out for 3 min as the final step of cleaning to improve the contact angle just before film formation. Onto the ITO-coated glass a layer of PEDOT film was spin-coated from an aqueous dispersion. After baking, the PEDOT was rubbed by a rubbing machine and was used as the alignment layer. Solutions of the liquid crystalline polymers in toluene were prepared and spin-coated on top of the rubbed-PEDOT surface. After anneal-ing for 1 h, calcium (35 nm) and aluminum (100 nm) cathodes were deposited onto the aligned emitting films by thermal evaporation through a shadow mask. The po-larized device properties were determined with a Photo Research PR-650 Spectra Scan instrument. Spectra were measured with the polarizer aligned parallel or perpen-dicular to the rubbing direction.
Synthesis
6-{4-[2-(7-Bromo-2,1,3-benzothiadiazol-4-yl)-1-enthynyl]phenoxy}-1-hexanol (1)
Compound 1 was synthesized according to a similar method reported in the literature [23]. 6-[4-(1-Enthynyl)-phenoxy]-1-hexanol (0.92 g, 4.25 mmol), bis(triphenyl-phosphine) palladium(II) chloride (0.02 g, 0.03 mmol), copper(I) iodide (0.03 g, 0.14 mmol), triphenylphosphine (0.07 g, 0.26 mmol), and 4.7-dibromo-2,1,3-benzothiadia-zole (0.5 g, 1.7 mmol) were dissolved in 50 mL of triethylamine under an atmosphere of nitrogen. The mix-ture was heated at 80 -C for 24 h. The solvent was re-moved by a rotatory evaporator and ethyl acetate was added to the crude product. The ethyl acetate solution was extracted with ammonium chloride and then washed with water, saturated NaCl solution, and dried over anhydrous MgSO4. After the ethyl acetate solution had been removed,
the crude product was purified by column chromatography (silica gel, ethyl acetate-to-hexane ratio 1:1), with a yield
of 0.6 g (86%) of a yellow compound with a melting point of 102Y103 -C. 1 H-NMR (CDCl3, d, ppm): 1.40Y1.60 (m, 6H), 1.80Y 1.84 (m, 2H), 3.64Y3.69 (t, J=6.6 Hz, 2H), 3.97Y4.01 (t, J=6.6 Hz, 2H), 6.88Y6.91 (d, J=8.4 Hz, 2H), 7.61Y7.64 (d, J=7.5 Hz, 2H), 7.81Y7.84 (d, J=7.5 Hz, 2H). 13 C-NMR (CDCl3, d, ppm): 25.3, 26.2, 28.7, 32.1, 62.3, 67.8, 92.6, 92.9, 110.4, 114.1, 117.9, 122.0, 124.2, 136.0, 136.2, 151.0, 157.2, 159.6. 6-[4-(2-{7-[2-(4-Pentylphenyl)-1-enthynyl]2,1, 3-benzothiadiazol-4-yl}-1-ethynyl)phenoxy]-1-hexanol (2) 6-{4-[2-(7-Bromo-2,1,3-benzothiadiazol-4-yl)-1-enthynyl]-phenoxy}-1-hexanol (1.0 g, 2.25 mmol), bis(triphenylphos-phine) palladium(II) chloride (0.02 g, 0.02 mmol), copper(I) iodide (0.02 g, 0.1 mmol), triphenylphosphine (0.05 g, 0.18 mmol), and 1-ethynyl-5-pentylbenzene (0.6 g, 3.48 mmol) were dissolved in 50 mL of triethylamine under an atmosphere of nitrogen. The mixture was heated at 80 -C for 24 h. The solvent was removed by a rotatory evaporator and ethyl acetate was added to the crude product. The ethyl acetate solution was extracted with ammonium chloride and then washed with water, saturated NaCl solution, and dried over anhydrous MgSO4. After the
ethyl acetate solution had been removed, the crude prod-uct was purified by column chromatography (silica gel, ethyl acetate-to-hexane ratio 1:1), with a yield of 0.97 g (83%) of a yellow compound with a melting point of 87Y88 -C.
1
H-NMR (CDCl3, d, ppm): 0.87Y0.92 (t, J=7.2 Hz,
3H), 1.25Y1.62 (m, 12H), 1.80Y1.82 (m, 2H), 2.60Y2.66 (t, J=7.5 Hz, 2H), 3.66Y3.68 (t, J=5.4 Hz, 2H), 3.98Y 4.02 (t, J=6.3 Hz, 2H), 6.89Y6.91 (d, J=9.0 Hz, 2H), 7.20Y7.22 (d, J=8.1 Hz, 2H), 7.56Y7.60 (m, 4H), 7.74 (s, 2H). 13 C-NMR (CDCl3, d, ppm): 14.0, 22.5, 25.6, 26.3, 28.9, 31.0, 31.5, 32.1, 35.6, 62.3, 67.8, 92.5, 92.9, 114.1, 114.4, 117.9, 119.8, 133.4, 134.2, 136.0, 143.4, 157.4, 159.6. 6-[4-(2-{7-[2-(4-Pentylphenyl)-1-enthynyl]2,1, 3-benzothiadiazol-4-yl}-1-ethynyl)phenoxy]-1-hexyl acrylate (M1) 6-[4-(2-{7-[2-(4-Pentylphenyl)-1-enthynyl]2,1,3-ben-zothiadiazol-4-yl}-1-ethynyl)phenoxy]-1-hexanol (0.8 g, 1.53 mmol) was dissolved in a mixture of dried THF and triethylamine (1.1 mL, 7.65 mmol) under an atmosphere of nitrogen. After the solution had been cooled in an ice-water bath to 0 -C, acryloyl chloride (0.4 mL, 4.6 mmol) was added dropwise. The reaction mixture was allowed to warm slowly to room temperature and was stirred overnight. The solid formed was taken into ethyl acetate and washed with water. The organic layer was dried over anhydrous MgSO4. After filtration, the solvent was removed by rotary
evaporation and purified by column chromatography (silica gel, ethyl acetate-to-hexane ratio 1:4), with a yield of 0.5 g (86%) of a yellow compound with a melting point of 64Y65 -C.
1
H-NMR (CDCl3, d, ppm): 0.87Y0.92 (t, J=6.0 Hz, 3H),
1.25Y1.82 (m, 14H), 2.60Y2.66 (t, J=7.5 Hz, 2H), 3.98Y 4.02 (t, J=6.3 Hz, 2H), 4.16Y4.20 (t, J=6.6 Hz, 2H), 5.80Y5.84 (dd, J=10.5, 1.5 Hz, 1H), 6.08Y6.17 (dd, J=10.5, 17.4 Hz, 1H), 6.38Y6.44 (dd, J=17.1, 1.5 Hz, 1H), 6.89Y 6.91 (d, J=8.7 Hz, 2H), 7.20Y7.22 (d, J=8.1 Hz, 2H), 7.56Y7.60 (m, 4H), 7.74 (s, 2H). 13 C-NMR (CDCl3, d, ppm): 14.1, 22.5, 26.0, 26.2, 28.6, 28.9, 31.1, 31.5, 35.6, 62.7, 67.8, 92.6, 92.9, 114.1, 114.3, 118.0, 119.7, 128.7, 129.6, 133.4, 134.2, 136.0, 136.9, 143.5, 157.4, 159.6, 166.6. 6-{4-[(E)-2-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolane-2-yl)-1-ethenyl]phenoxy}-1-hexanol (3) 6-[4-(1-Enthynyl)phenoxy]-1-hexanol (3.0 g, 13.66 mmol) and pinacol borane (2.64 mL, 20.66 mmol) were dis-solved in 10 mL of toluene under an atmosphere of nit-rogen. The mixture was heated at 90 -C for 12 h. The product was isolated by pouring the solution into water and the solution was filtered. The crude product was puri-fied by column chromatography (silica gel, ethyl acetate-to-hexane ratio 1:4), with a yield of 4.5 g (90%) of a yellow liquid. 1 H-NMR (CDCl3, d, ppm): 1.20 (s, 12H), 1.40Y1.60 (m, 6H), 1.78Y1.82(m, 2H), 3.64Y3.69 (t, J=7.2 Hz, 2H), 3.97Y4.01 (t, J=6.3 Hz, 2H), 6.80Y6.92 (d, J=34.5 Hz, 2H), 6.94Y6.96 (d, J=6.0 Hz, 2H), 7.56Y7.60 (d, J=11.4 Hz, 2H). 13 C-NMR (CDCl3, d, ppm) : 24.1, 25.6, 26.2, 28.9, 32.1, 62.2, 67.8, 83.6, 114.1, 122.5, 128.3, 128.4, 150.5, 158.2. 6-{4-[(E)-2-(7-Bromo-2,1,3-benzothiadiazol-4-yl)-1-ethynyl]phenoxy}-1-hexanol (4)
Compound 4 was synthesized according to a similar method reported in the literature [24]. 6-{4-[(E)-2-(4, 4,5,5-Tetramethyl-1,3,2-dioxaborolane-2-yl)-1-ethenyl] phenoxy}-1-hexanol (0.77 g, 2.22 mmol), 4.7-dibromo-2,1, 3-benzothiadiazole (1.0 g, 3.4 mmol), K2CO3 (2.4 g,
16.8 mmol), Aliquat 336 (0.4 g, 1.0 mmol), and Pd(PPh3)4
(0.02 g, 0.02 mmol) were dissolved in a mixture of 10 mL of toluene and 2 mL of degassed water. The solution was refluxed with vigorous stirring for 48 h under an argon atmosphere. At the end of the reaction, the solid formed was taken into ethyl acetate and washed with water. The organic layer was dried over anhydrous MgSO4.
After filtration, the solvent was removed by rotary evapo-ration and purified by column chromatography (silica gel, ethyl acetate-to-hexane ratio 1:1), with a yield of 1.2 g (83%) of an orange compound with a melting point of 104Y105 -C.
1
H-NMR (CDCl3, d, ppm): 1.43Y1.62 (m, 6H),
1.78Y1.82 (m, 2H), 3.63Y3.67 (t, J=7.5 Hz, 2H), 3.96Y4.00 (t, J=6.3 Hz, 2H), 6.89Y6.92 (d, J=8.7 Hz, 2H), 7.39Y7.56 (m, 4H), 7.78Y7.91 (m, 2H). 13 C-NMR (CDCl3, d, ppm): 25.6, 26.2, 28.9, 32.1, 62.3, 67.8, 110.3, 113.8, 115.5, 119.1, 129.6, 131.6, 134.2, 141.5, 142.0, 157.9, 158.3. 4,4,5,5-Tetramethyl-2-[(E)-2-(4-pentylphenyl)-1-ethynyl]-1,3,2-dioxaborolane (5)
1-Ethynyl-5-pentylbenzene (4.8 g, 31.66 mmol) and pina-col borane (6.4 mL, 47.46 mmol) were dissolved in 10 mL of toluene under an atmosphere of nitrogen. The mixture was heated at 90 -C for 12 h. The product was isolated by pouring the solution into water and the solu-tion was filtered. The crude product was purified by col-umn chromatography (silica gel, ethyl acetate-to-hexane ratio 1:4), with a yield of 8.4 g (91%) of a yellow liquid. 1 H-NMR (CDCl3, d, ppm): 0.92Y0.97 (t, J=7.2 Hz, 3H), 1.20 (s, 12H), 1.40Y1.60 (m, 6H), 2.61Y2.67 (t, J=6.3 Hz, 2H), 6.80Y6.92 (d, J=32.7 Hz, 2H), 6.93Y6.96 (d, J=8.7 Hz, 2H), 7.55Y7.60 (d, J=7.2 Hz, 2H). 13 C-NMR (CDCl3, d, ppm) : 14.0, 22.5, 24.0, 31.5, 35.5, 83.6, 122.5, 124.9, 127.7, 133.3, 144.1, 150.4. 6-[4-((E)-2-{7-[(E)-2-(4-Pentylphenyl)-1-ethenyl]-2,1, 3-benzothiadiazol-4-yl}-1-ethynyl) phenoxy]-1-hexanol (6) 6-{4-[(E)-2-(7-Bromo-2,1,3-benzothiadiazol-4-yl)-1-ethynyl]phenoxy}-1-hexanol (0.6 g, 1.38 mmol), 4,4,5,5-tetramethyl-2-[(E)-2-(4-pentylphenyl)-1-ethynyl]-1,3, 2-dioxaborolane (0.59 g, 2.07 mmol), K2CO3(1.22 g, 10.9
mmol), Aliquat 336 (0.2 g, 0.5 mmol), and Pd(PPh3)4(0.01
g, 0.01 mmol) were dissolved in a mixture of 10 mL of toluene and 2 mL of degassed water. The solution was refluxed with vigorous stirring for 48 h under an argon atmosphere. At the end of the reaction, the solid formed was taken into ethyl acetate and washed with water. The organic layer was dried over anhydrous MgSO4. After
filtration, the solvent was removed by rotary evapora-tion and purified by column chromatography (silica gel, ethyl acetate-to-hexane ratio 1:1), with a yield of 0.52 g (83%) of an orange compound with a melting point of 82Y83 -C.
1
H-NMR (CDCl3, d, ppm):0.92Y0.97 (t, J=7.5 Hz,
3H), 1.25Y1.65 (m, 12H), 1.80Y1.85 (m, 2H), 2.61Y2.66 (t, J=7.8 Hz, 2H), 3.65Y3.70 (t, J=6.6 Hz, 2H), 3.99Y4.02 (t, J=6.6 Hz, 2H), 6.91Y6.94 (d, J=8.7 Hz, 2H), 7.20Y 7.23 (d, J=8.1 Hz, 2H), 7.55Y7.66 (m, 8H), 7.90Y7.98 (m, 2H). 13 C-NMR (CDCl3, d, ppm): 14.0, 22.5, 25.6, 26.2, 28.8, 31.0, 31.5, 32.1, 35.6, 62.2, 67.8, 114.3, 115.4, 126.3, 129.0, 129.4, 130.8, 132.3, 133.7, 135.5, 144.0, 149.1, 158.2. 6-[4-((E)-2-{7-[(E)-2-(4-Pentylphenyl)-1-ethenyl]-2,1, 3-benzothiadiazol-4-yl}-1-ethynyl) phenoxy]hexyl acrylate (M2)
6-[4-((E)-2-{7-[(E)-2-(4-Pentylphenyl)-1-ethenyl]-2, 1,3-benzothiadiazol-4-yl}-1-ethynyl)phenoxy]-1-hexanol (0.4 g, 0.78 mmol) was dissolved in a mixture of dried THF and triethylamine (0.56 mL, 3.9 mmol) under an atmosphere of nitrogen. After the solution had been cooled in an ice-water bath to 0 -C, acryloyl chloride (0.18 mL, 1.95 mmol) was added dropwise. The reaction
mixture was allowed to warm slowly to room temper-ature and was stirred overnight. The solid formed was taken into ethyl acetate and washed with water. The or-ganic layer was dried over anhydrous MgSO4. After
fil-tration, the solvent was removed by rotary evaporation and purified by column chromatography (silica gel, ethyl acetate-to-hexane ratio 1:4),with a yield of 0.4 g (90%) of an orange compound with a melting point of 71Y 72-C. 1 H-NMR (CDCl3, d, ppm): 0.92Y0.97 (t, J=7.2 Hz, 3H), 1.25Y1.82 (m, 14H), 2.61Y2.64 (t, J=7.5 Hz, 2H), 3.98Y4.03 (t, J=6.3 Hz, 2H), 4.16Y4.20 (t, J=6.6 Hz, 2H), 5.80Y5.84 (dd, J=10.5, 1.5 Hz, 1H), 6.08Y 6.17 (dd, J=10.2, 17.4 Hz, 1H), 6.38Y6.44 (dd, J= 17.1, 1.5 Hz, 1H), 6.89Y6.94 (d, J=8.7 Hz, 2H), 7.20Y7.22 (d, J=8.4 Hz, 2H), 7.55Y7.63 (m, 8H), 7.90Y7.92 (m, 2H). 13 C-NMR (CDCl3, d, ppm):13.9, 22.5, 26.1, 26.3, 28.6, 28.9, 31.1, 31.5, 35.6, 62.7, 67.8, 114.3, 115.4, 115.6, 126.3, 129.0, 129.4, 130.8, 132.1, 133.8, 135.5, 144.0, 149.2, 158.2, 166.5. Poly{6-[4-(2-{7[2-(4-pentylphenyl)-1-enthynyl]2,1, 3-benzothiadiazol-4-yl}-1-ethynyl) phenoxy]-1-hexyl acrylate} (P1)
Monomer M1 (0.5 g) and initiator 2,20 -azobisisobutyroni-trile (AIBN) (1 wt%) were dissolved in THF (1.0 g/mL). The mixture was stirred at 60-C for 24 h under a nitrogen atmosphere. The product was isolated by pouring the re-action mixture into methanol and was purified by rep-recipitation in methanol three times, giving a yield of 0.38 g (76%) of a yellow compound.
Poly{6-[4-((E)-2-{7-[(E)-2-(4-pentylphenyl)-1-ethenyl]-2, 1,3-benzothiadiazol-4-yl}-1-ethynyl)phenoxy]hexyl acrylate} (P2)
Monomer M2 (0.5 g) and initiator AIBN (1 wt%) were dissolved in THF (1.0 g/mL). The mixture was stirred at 60 -C for 24 h under a nitrogen atmosphere. The product was isolated by pouring the reaction mixture into meth-anol and was purified by reprecipitation in methmeth-anol
Scheme 1. Synthesis of 6-[4-(2-{7-[2-(4-Pentylphenyl)-1-enthynyl]2,1,3-benzothiadiazol-4-yl}-1-ethynyl)phenoxy]-1-hexyl acrylate (M1) and poly{6-[4-(2-{7[2-(4-pentylphenyl)-1-enthynyl]2,1,3-benzothiadiazol-4-yl}-1-ethynyl) phenoxy]-1-hexyl acrylate} (P1).
Scheme 2. Synthesis of 6-[4-((E)-2-{7-[(E)-2-(4-Pentylphenyl)-1-ethenyl]-2,1,3-benzothiadiazol-4-yl}-1-ethynyl) phenoxy]hexyl acrylate (M2) and poly{6-[4-((E)-2-{7-[(E)-2-(4-pentylphenyl)-1-ethenyl]-2,1, 3-benzothiadiazol-4-yl}-1-ethynyl)phenoxy]hexyl acrylate} (P2).
three times, giving a yield of 0.4 g (80%) of an orange compound.
Results and discussion Synthesis and characterization
The benzothiadiazole-based liquid crystalline polyacrylates were prepared according to the synthetic routes depict-ed in Schemes 1and2. The acrylate monomer M1 was obtained by coupling of dibromobenzothiadizole with 6- [4-(1-ethynyl)-phenoxy]-1-hexanol and 1-ethynyl-5-pentyl ben-zene followed by esterification with acryloyl chloride, whereas M2 was obtained from coupling of dibro-mobenzothiadizole with ethenyldioxaborolane, 3 and 5, then esterification with acryloyl chloride. The polymer-izations of M1 and M2 were performed as shown in Scheme1 using AIBN as the initiator in THF.
Table1lists the molecular weights and phase-transition temperatures of P1 and P2.
Liquid crystalline phase behavior
Liquid crystalline behavior and phase transitions of P1 and P2 were examined with a polarizing optical microscope equipped with a hot stage. The results are summarized in Table 1. Figure 1 depicts the representative differential scanning calorimetry (DSC) thermograms of P1. The heat-ing scan shows a crystalline-to-nematic phase transition at 92.4-C and a nematic-to-isotropic phase transition at 169.7 -C. On the cooling scan, the isotropic-to-nematic and crystallization endotherms at 155.5 and 65.0 -C are ob-served. Figure 2 exhibits the typical nematic texture for polymer P1. As can be seen from Table1, polymer P2 also reveals the nematic liquid crystalline behavior. No glass transition was observed for both polymers in the DSC curves.
Optical properties
Aligned samples for further experiments were prepared by annealing the polymers on PEDOT-coated glass plates. The PEDOT surface was mechanically rubbed prior to spin-coating of the polymers.
The optical properties of aligned P1 and P2 were characterized by UV-vis absorption along with PL emis-sion, as shown in Table 2. To estimate the degree of alignment achieved, the polarized spectra were recorded
Figure 1. Differential scanning calorimetry thermogram of poly{6-[4-(2-{7[2-(4-pentylphenyl)-1-enthynyl]2,1,3-benzothiadiazol-4-yl}-1-ethynyl) phenoxy]-1-hexyl acrylate} (P1): A second heating scan, B first cooling scan.
Table 1. Polymerization results and phase-transition temperatures of polymers poly{6-[4-(2-{7[2-(4-pentylphenyl)-1-enthynyl]2,1,3-benzothiadiazol-4-yl}-1-ethynyl) phenoxy]-1-hexyl acrylate} (P1) and poly{6-[4-((E)-2-{7-[(E)-2-(4-pen-tylphenyl)-1-ethenyl]-2,1,3-benzothiadiazol-4-yl}-1-ethynyl)phenoxy]hexyl acrylate} (P2). Polymer Yield (%) Mn Mw Mw/Mn Phase-transition temperatures P1 76.3 8,600 15,600 1.81 G 92-C N 170 -C I I 156-C N 65 -C G P2 80.4 9,600 18,400 1.92 G 79-C N 126 -C I I 124-C N 59 -C G G glassy state, N nematic phase, I isotropic.
Figure 2. Polarizing optical micrograph of P1: nematic droplet texture obtained at 150-C.
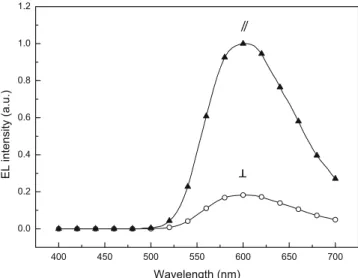
with the polarizer placed parallel and perpendicular to the rubbing direction. Figure 3 shows polarized UV and PL spectra of P1. The results indicate that absorbance parallel to the rubbing direction was much higher than that perpendicular to the rubbing direction. This means that the mesogens of polyacrylate are aligned parallel to the rubbing direction. The dichroic ratios calculated from the polarized UV and PL spectra are in the range from 8.3 to 8.8 (Table2).
Polymer light-emitting diodes
To examine the potential application of these polymers, light-emitting devices were constructed with P1 or P2 as the active layer between the ITO anode and the Ca/Al cathode. Luminance-voltage and current density-voltage characteristics for the EL devices are illustrated in Figures 4and5. P1 and P2 devices yielded maximum emissions at 580 and 604 nm with turn-on voltages of 4 and 6 V, respectively. The intensity of the emitted light recorded parallel to the rubbing direction was approximately 1 order of magnitude greater than that from the perpendicular direction at the same applied voltage. This indicates that the chromophores were aligned predominantly along the rubbing direction. Table 3 summarizes all the EL data for two devices. Compared with other EL polymers with mesogen pendants [25], the single-layer EL devices consisting of P1 or P2 showed lower turn-on voltages. The luminescence intensity increases as the applied volt-age increases and achieves a maximum luminance of 85 and 235 cd/m2 for P1 and P2, respectively. These lumi-nances are significantly better than those in our previous
Figure 3. Polarized UV-vis absorption and polarized photoluminescence emission spectra of P1. (// polarizer parallel to rubbing direction, ± polarizer perpendicular to rubbing direction).
Figure 4. Polarized electroluminescence (EL) spectra of poly{6-[4-((E)-2- {7-[(E)-2-(4-pentylphenyl)-1-ethenyl]-2,1,3-benzothiadiazol-4-yl}-1-ethy-nyl)phenoxy]hexyl acrylate} (P2) in the indium tin oxide (ITO)/aligned poly(3,4-ethylene-dioxythiophene)-poly(styrenesulfonic acid) (PEDOT)/ P2/Ca/Al device.
Table 3. Polarized device properties of polymers P1 and P2. Polymer ELmax(nm) at Vturn on Vturn on (V) Luminance (max) (cd/m2) Efficiency (max) (cd/A) Polarization ratio (EL///EL±) P1 580 5 85 0.03 4.0 P2 604 6 235 0.09 6.5
Table 2. The UV-vis absorption, photoluminescence (PL) emission, and polarization ratios of polymers P1 and P2.
Polymer UV-vis absorption (nm) PL emission (nm) Polarization ratio UV-vis (UV///UV±) PL (PL///PL±) P1 492 583 8.5 8.3 P2 517 603 8.8 8.6
Figure 5. J-V and L-V curves of P2 in the ITO/aligned PEDOT/P2/Ca/Al device.
report for a similar pendant liquid crystalline polymer [22]. Compared with our previous report, the middle phe-nyl ring of the chromophore was replaced with a ben-zothiadiazole group. According to the literature [8,9], the mesogen with a benzothiadiazole group has better hole-transport properties, which not only increases the current density but also increases the efficiency of the recombina-tion of electrons and holes. This is the reason why P1 and P2 show much higher EL efficiencies than previous polymers.
Conclusion
The results presented here illustrate that, in principle, benzothiadiazole-based bistolane or bisstilbene incorporat-ed onto polyacrylate exhibits liquid crystalline behavior and gives polarized EL. The observation of polarized EL from aligned P1 and P2 illustrates the potential use of mesogenic materials for polarized emitter applications. Further sys-tematic studies are now necessary to improve the dichroic ratio and to increase the efficiency of these systems.
Acknowledgments
The authors are grateful to the National Science Council of the Republic of China for financial support of this work (NSC-92-2216-E009-015).
References
1. S. Poser, H. Fischer and M. Arnold, Prog. Polym. Sci., 23, 1337 (1998).
2. N. A. Plate, in: Advances in Polymer Science, Vol. 59Y61, M. Gordon, Ed., Springer, Berlin Heidelberg New York, 1984, p 140.
3. R. A. Weiss and C. K. Ober, Liquid-Crystalline Polymers. American Chemical Society, Washington, DC, 1990, 241.
4. M. Grell, D. D. C. Bradley, E. P. Woo and M. Inbasekaran, Adv. Mater., 9, 798 (1997).
5. M. Grell, M. Redecker, K. S. Whitehead, D. D. C. Bradley, M. Inbasekaran, E. P. Woo and W. W. Wu, Liq. Cryst., 26, 1403 (1999).
6. J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 347, 539 (1990).
7. G. Gustafsson, Y. Gao, G. M. Treacy, F. Klavetter, N. Colaneri and A. J. Heeger, Nature, 357, 477 (1992).
8. M. Funahashi and J. Hana, Jpn. J. Appl. Phys., 35, L703 (1996). 9. M. Funahashi and J. Hana, Phys. Rev. Lett., 78, 2184 (1997). 10. H. Tokuhisa, M. Era and T. Tsutsui, Adv. Mater., 10, 404 (1998). 11. H. Mochizuki, T. Hasui, M. Kawamoto, T. Shiono, T. Ikeda, C. Adachi,
Y. Taniguchi and Y. Shirota, Chem. Commun., 19, 1923 (2000). 12. A. J. Campbell, D. D. C. Bradley and H. Antoniadis, Appl. Phys.
Lett., 79, 2133 (2001).
13. H. Tokuhisa, M. Era and T. Tsutsui, Appl. Phys. Lett., 72, 2639 (1998). 14. K. U. W. Clauswitz, F. Geffarth, A. Greiner, G. Lu¨ssem and J. H.
Wendorff, Synth. Met., 111Y112, 169 (2000).
15. T. Miteva, A. Meisel, M. Grell, H. G. Nothofer, D. Lupo, A. Yasuda, W. Knoll, L. Klopperburg, U. H. F. Bunz, U. Scherf and D. Nehr, Synth. Met., 111Y112, 173 (2000).
16. M. Grell and D. D. C. Bradley, Adv. Mater., 11, 895 (1999). 17. M. Grell, W. Knoll, D. Lupo, A. Meisel, T. Mikeva, D. Neher,
H. G. Nothofer, U. Scherf and A. Yasuda, Adv. Mater., 11, 671 (1999).
18. K. S. Whitehead, M. Grell, D. D. C. Bradley, M. Jandke and P. Strohriegl, Appl. Phys. Lett., 76, 2946 (2000).
19. M. Jandke, P. Strohriegl, J. Gmeiner, W. Bu¨tting and M. Schwoerer, Synth. Met., 111Y112, 117 (2000).
20. K. S. Whitehead, M. Grell, D. D. C. Bradley, M. Inbasekaran and E. P. Woo, Synth. Met., 111Y112, 181 (2000).
21. L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 12, 481 (2000).
22. S. W. Chang, A. K. Li, C. W. Liao and C. S. Hsu, Jpn. J. Appl. Phys., 41, (2002) 1374.
23. C. S. Hsu, K. F. Shyu and Y. Y. Chiang, Liq. Cryst., 27, 283 (2000). 24. N. Miyaura and A. Suzuki, Chem. Rev., 95, 2457 (1995). 25. M. Kawamoto, H. Mochizuki, A. Shishido, O. Tsutsumi, T. Ikeda,
![Figure 1. Differential scanning calorimetry thermogram of poly{6-[4-(2- poly{6-[4-(2-{7[2-(4-pentylphenyl)-1-enthynyl]2,1,3-benzothiadiazol-4-yl}-1-ethynyl) phenoxy]-1-hexyl acrylate} (P1): A second heating scan, B first cooling scan.](https://thumb-ap.123doks.com/thumbv2/9libinfo/7939658.157497/5.892.79.437.807.1086/differential-scanning-calorimetry-thermogram-pentylphenyl-enthynyl-benzothiadiazol-acrylate.webp)