The kinetics of transesterification in blends of liquid crystalline
copolyester and polycarbonate
Jia-Chong Ho, Kug-Hwa Wei*
Institute of Materirrl Science & Engineering, National Chiao Tung Universit;v, Hzinchu, Taivvan. 30049, Republic of China
Received 10 September 1997; revised 10 November 1997; accepted 17 December 1997
Abstract
The kinetics of transesterification in blends of thermotropic copoly(oxybenzoate-ethylene terephthalate) and polycarbonate at melt state were analysed quantitatively with 13C nuclear magnetic resonance, and were then modelled by a phenomenological equation. It was found that the activation energy for the transesterification reaction between the poly(oxybenzoate) segment in copoly(oxybenzoate-ethylene terephthalate) and polycarbonate was more than twice that for the transesterification reaction between the poly(ethylene terephthalate) segment and polycarbonate (i.e. 94.6 versus 41.8 kcal/mol). This is probably due to the stronger chemical bond in oxybenzoate-oxybenzoate that in ethylene-terephthalate. Additionally, owing to the rigid-rod nature of the poly(oxybenzoate) segment, the frequency factor in the transesterification reaction in poly(oxybenzoate) segment and polycarbonate is much higher than that in poly(ethylene terephthalate) segment and polycarbonate (i.e. lO36 versus lOL4 mini’). 0 1998 Elsevier Science Ltd. All rights reserved.
Keywords: Thermotropic copolymers blends; Kinetics; Transesterification
1. Introduction
It is well known that polyesters can have acidolysis by an acid-end group, alcoholysis by a hydroxy-end group, and midchain ester-ester interchange (transesterification) with itself or with other polymers at high temperatures [I] [2] (generally above 200°C). For high molecular weight poly- esters and their blends, the probability of transesterification occurring is much higher than that of acidolysis or alco- holysis occurring, because of relatively low end-group con- centrations. Therefore, transesterification usually dominates the reaction process. Transesterification has serious effects on the properties of polymers. For example, the molecular weight of polyester decreased with the increasing extent of transesterification [3]. On the other hand, the miscibility of polyester blends could be enhanced by transesterification due to the presence of resultant hybrid chemical structures [4] [.5]. The knowledge of the kinetics of transesterification in polyester blends is critically important to their physical properties.
Although thermotropic copolyesters and flexible-coil polymers are immiscible, transesterification in blends of thermotropic copoly(oxybenzoate-ethylene terephthalate)
* Corresponding author. Tel.: +00-886-3-5731871; fax: +OO-886-Y. 5724727
(POB-PET) at a molar ratio of 60140 and poly(butylene terephthalate) (PBT) [6], polycarbonate (PC) [7] [8] and poly(hexamethylene terephthalate) (PHMT) [9] have been found by differential scanning calorimetry analysis and were confirmed by nuclear magnetic resonance studies. From these studies, it was concluded that the miscibility in blends of PC/PET, PBT/POB-PET, PUPOB-PET and PC/poly(p-oxybenzoate-co-p-phenylene isophthalate) [lo] was found to increase with transesterification judging from the changes in the glass transition temperature of each component polymer. As transesterification in blends continues, new amorphous compatible blends will form.
For analysing transesterification in three components copolycondensates, Yamadera et aZ.[ 1 I] studied the average sequence length and the degree of randomness in blends of poly(ethylene terephthalate) (PET) and poly(ethylene seba- cate) (PES). For four-component copolycondensates, Devaux et al. [12] [13] [14] [15] investigated the chemical structures, the randomness, and the kinetics of transesterifica- tion in blends of PC/PBT and PC/PET. For the transesterifica- tion in five-component copolycondensates, we were the first group to study the mechanism of transesterification through a theoretical analysis on blends of random POB/PET (40/60) and PC with 13C nuclear magnetic resonance spectroscopy [16]. In the current study, we further analysed the kinetics of transesterification in blends of POB-PET and PC.
0032-3861/98/$ - see front matter 0 1998 Elsevier Science Ltd. All rights reserved
718 J.-C. Ho, K.-H. WdPolymer 40 (1999) 717-727
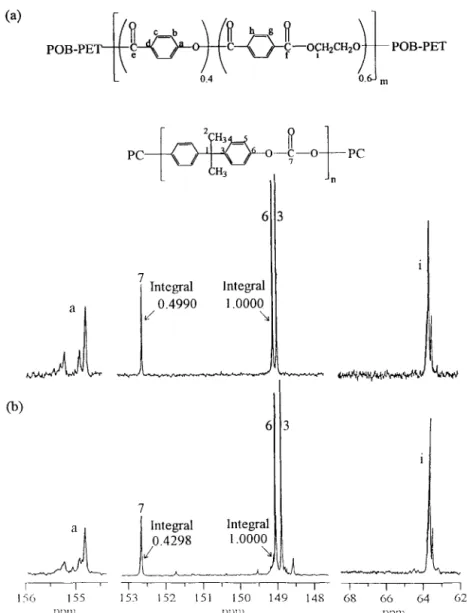
PC O-F-0 if pc
t n
Fig. I. The chemical structures of copoly(oxybenzoat+ethylene terephthalate) and polycarbonate
2. Experimental
POB-PET at molar ratio of 40/60 was synthesized in our laboratory following the method of Jackson [17] and was termed POB-PET in this study. Polycarbonate (PC) was purchased from General Electric Co. The trade name of PC is Lexan 121, and its molecular weight is M, = 158900. The intrinsic viscosity of POB-PET and PC were 0.53 and 1.15 dl/g, respectively, when they were pre- pared in mixed solvent of phenol and tetrachloroethane (50/ 50 by weight). The chemical structures of POB-PET and PC are shown in Fig. 1.
The solution blending of POB-PET and PC was carried out by dissolving POB-PET and PC at a weight ratio of 60/ 40 in 25 ml of a mixed solvent of phenol and tetra- chloroethane (50/50 by weight). The concentration of the solution containing POB-PET and PC is 2% by weight. The solution was maintained at 70°C. After the polymers were dissolved and became a one-phase solution for 30 min, the solution was precipitated in a lo-fold excess volume of methanol. The precipitated blends were washed five times, each time with 200 ml methanol. The blends were then dried in a vacuum oven at 100°C for 4 days. The thermal gravi- metric analysis of the dried blends showed no appreciable weight loss up to 350°C indicating a complete removal of the solvent. The blends were put into a high-temperature furnace at 240°C 250°C 260°C and were annealed for dif- ferent times under a purge of nitrogen at 100 cm3/min.
The thermal analysis of the blends was performed with a Du Pont 2910 differential scanning calorimetry (d.s.c.). The samples were heated from 25°C to 260°C with a heating rate of 20”C/min under nitrogen purge, and was annealed at 260°C for 1 min. Subsequently, the samples were quickly cooled down to 170°C and were allowed to crystallize at this temperature for 10 min. Then the samples were quenched to 25°C and were scanned again from 25°C to 260°C at the same scan rate. Without the isothermal crystallization, the T, of the blends were difficult to detect by d.s.c. because the d.s.c. curve cannot exhibit separate crystallization (by POB-PET) and glass transition (by PC) of the blends. Isothermal crystallization at 170°C for 10 min avoided the problem by allowing the samples to crystallize. The d.s.c.
curves of the samples were taken during the second heating. The midpoint of glass transition temperature was chosen as the glass transition temperature (T,).
POB-PET and PC were dissolved in deuterated trifluor- oacetic acid and deuterated chloroform (10/90, v/v) at a concentration of cu. 10%. Quantitative 13C n.m.r. analyses were carried out with a Bruker DMX-600 Spectrometer.
3. Results and discussion
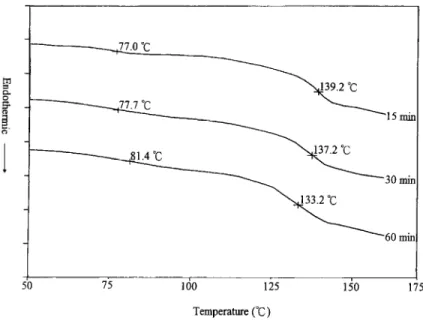
The d.s.c. curves of POB-PET and PC are presented in Fig. 2. In Fig. 2 the glass transition temperatures (Tss) of POB-PET and PC are 59.5”C and 148.O”C, respectively. The crystallization temperature (T,) for POB-PET IS 99.2”C. The broad endothermic peak is the melting transi- tion of PET-rich phase in POB-PET. For the 60/40 POB- PET/PC blends, the low T, represented the amorphous phase of POB-PET, and the high T, was due to PC. The partial and transient d.s.c. curves of the annealed 60/40 POB-PET/PC blends are shown in Fig. 3. The two T,s were about 62.2”C apart after the blend was annealed at 250°C for 15 min. The two Tgs converged on each other when the annealing time increased. After annealing the
I
0 50 100 150 200 250 :
Temperature (“C)
0
Fig. 2. The d.s.c. curves of copoly(oxybenzoate-ethylene terephthalate) and polycarbonate.
75 100 125 Temperature (“C)
150 1
Fig. 3. The partial d.s.c. curves of the 60/40 POB-PET/PC blend annealed at 250°C for different times.
blend at 250°C for 60 min, the difference in the two Tgs were about 51.8”C. The d.s.c. results of the POB-PET/PC blend at other annealing temperatures are listed in Table 1. In Table 1 the two T,s followed the same trend as that in the 250°C case. This result implied an improvement of the miscibility in the annealed 60/40 POB-PET/PC blend.
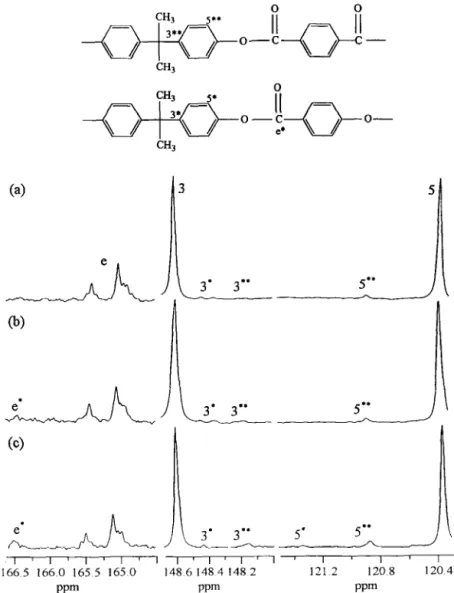
The partial 13C n.m.r. spectra of the 60/40 POB-PET/PC blends annealed at 250°C are shown in Fig. 4. These blends were completely dissolved in the n.m.r. solvent. After the blend was annealed at 250°C for 15 min, there were five new peaks 3**, 5**, 3*, 5*, and e* appearing at 148.15, 120.90, 148.40 and 166.50 ppm, respectively, as shown in Fig. 4. The new peaks 3”” and 5”” represented the new structure bisphenol A-terephthalate (A2B 1). The other new peaks 3*, 5* and e* were caused by the new structure bisphenol A- oxybenzoate (A2(AB)J. The model compound study of both structures has been carried out, and the peaks have been identified previously[l6]. In Fig. 4, the intensity of the new peaks increased gradually with annealing time at 250°C. The transient mole fraction of the dyad in the annealed 60/40 POB-PET/PC blend is determined from the product of the integral values under the resonance peak by its initial mole fraction, and they are listed in Table 3. In Table 3, the mole fraction of (A2B 1) and (Az(AB),) dyad increased with both the annealing time
and the annealing temperature. Specifically, the mole frac- tion of new dyad bisphenol A-terephthalate (AzB ,) became 0.009,0.017 and 0.038 after 60 min of annealing at 240,250 and 26O”C, respectively, whereas the mole fraction of the dyad bisphenol A-oxybenzoate (A2(AB) ,) became 0.001, 0.003 and 0.015 under the same thermal treatment. The loss of the mole fractions of the original dyads oxybenzoate-
oxybenzoate ((AB)I(AB)I), ethylene-oxybenzoate
(A,(AB) J, oxybenzoate-terephthalate((AB)lB J, ethylene- terephthalate (A,B ,), and bisphenol A-carbonate(A2B2) became the largest after 60 min of annealing at 260°C. This result can be used to interpret the effect of transesterification on the miscibility of the blend. In short, the disappearance of the dyad (AB) ,(AB) 1 (liquid crystalline) and the production of mixed dyads enhanced the miscibility between POB-PET and PC. Therefore, the miscibility of POB-PET/PC blends increased with the extent of transesterification.
Similar to the analysis in PC/PBT, we divided POB/PET and PC into five components, and used the notations (AB) ,, B, and A, standing for oxybenzoate, terephthalate, and ethylene in POB-PET, respectively. Bisphenol A and car- bonate are noted as AZ and Bz, as indicated in Table 2. The molecular weights of POB-PET and PC are high enough to neglect chain-end reaction in this analysis. These polymer melts have high viscosity, and therefore their molecular
Table 1
The evolution of the glass transition temperatures of the annealed 60/40 POB-PET blend at different temperatures Annealing temperature (“C)
Annealing time 240 250 260
(min) T,I T !A2 T &I T z2 T,I T 82
15 74.9 139.6 77.0 139.2 77.0 138.9
30 78.8 4 139.0 77.7 137.2 78.5 133.7
720 J.-C. Ho, K.-H. WdPolymer 40 (1999) 717-727
I’I’I’I’I 1’1’1’1 1 , 0 I ’ I
1665 1660 1655 1650 148614841482 121 2 120 8 1204
ppm ppm PPm
Fig. 4. The partial “C n.m.r. spectra of the 60/40 POB-PET/PC blend annealed at 250°C for (a) 15 min, (b) 30 min, (c) 60 min.
diffusions are slow. Although we have five reversible reac- where ki and ki (i = 1, 2, 3, 4, 5) are the forward and tions, these are independent of each other because of the the reverse reaction rate constant for each reaction, locality caused by the slow diffusion of dyads. These reac- respectively.
tions are given in the following. The POB-PET is very close to a random copolyester as
reaction I
P
- OCH$H,O - C -0 -
reaction II
kH3 @2B2> k2 k2 II wwIJ%) (G4BM32)reaction III
d&&O-!-O-
CH3 (Mb) O- GWWd+;&+&I- + - OCH,CH,O ---O-
CH3 (A2(mh) (AlB2)
reaction IV
CH3 642@3>1> (64w32)reaction V
ii - ii - OCH,CH,O - I C-@C- ks (A&h)722 J.-C. Ho, K.-H. WdPolymer 40 (1999) 717-727
z
POB-POB POB-PET 4 ---_~/~/ c? 9 0 3 18 I 1 I ’ I ’ 1 ’ I 7.55 7.50 1.45 7.40 7.35 7 30 mmFig. 5. (a) The full ‘H n.m.r. spectra of POB-PET, (b) the enlarged section of peak b in the full ‘H n.m.r. spectra of POB-PET identified from its ‘H n.m.r. spectrum[ 161. We will not con-
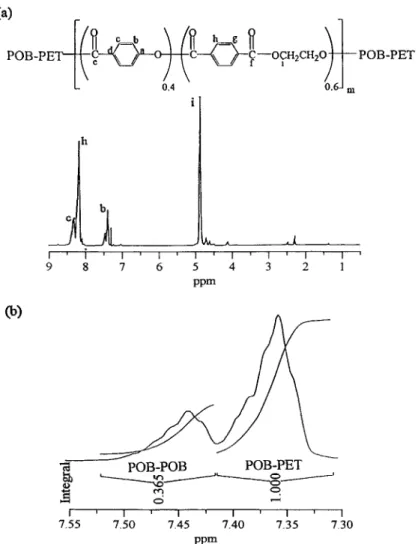
sider reaction V because reaction V has been in equilibrium during the synthesis of random POB-PET. Before the reac- tion started, we had AIBI, AI(A (AB)tBt, (AB), and AzB2. The initial mole fraction of these dyads have to be calculated first. The probability of a POB bonded to another POB and the probability of a POB bonded to a PET can be obtained from ‘H n.m.r. spectroscopy[ 181, as shown in Fig.
5a,b. From Fig. .5a,b, the integral under the peak of POB- POB and under the peak of POB-PET are 0.365 (Pt) and
1 .OOO (P2), respectively.
The initial dyad mole fractions are given in the following. _ mole fraction of POB-PET in
the blend X
mole fraction of POB in POB-PET X +
zz mole fraction of POB-PET in the blend X mole fraction of POB in POB-PET X & = mole fraction of POB-PET in
the blend X mole fraction of POB in POB-PET-
F~AB)I,ABU.O
FAIaI,a cannot be detected directly by n.m.r. spectroscopy. We can deduce it from the following method.
FAIBI,O = mole fraction of POB-PET in the blend X mole fraction of PET in POB-PET - mole fraction of PET bond to POB
The values of FAZBZ,O, FAZBZ,O, FAI(AB)I,O, F(AB)IBI,O and
FcAa)t(Aa)t,O were 0.300, 0.215, 0.205, 0.205 and 0.075, respectively.
When transesterification started we had new species A2Bl, A2(AB),, A,B? and (AB)tBz. The mole fraction of these new dyads based upon their individual reaction was defined as
x = FAZBI XI = (FAZBI), = (FAI,,),. XII = (FAX& = (F~ABMBz),,
Y = FAZ<M,I Y I = (FAZ(AB)I)III = (FAIB~)III~YII = (FAZ(AB,I)IV = (F~AB~IB~IV x=x,+x 11 Y = Y I + Y ,I
where the subscripts in x and y indicate the resulting reaction.
Table 2
The codes used in the dyads of 60/40 POB-PET/PC blend
POB-PET -OCH;?C&O - - Jk3 R / C- A1 Bl Gwl Polycarbonate
cases. The transient mole fraction of bisphenol A-ter- ephthalate (A2B1) is produced from both reactions I and II. We assumed that x and y are decoupled during the reac- tions i; order to simplify these equations. We defined y,, = dy.
s 0 dt -m reaction I and II, where t is the reaction time. The ytl is the mole fraction of bisphenol A-oxybenzo- ate (A2(AB),) at t = tl. Then we obtained Eq. (1) in the following form.
reaction I
+
k&m, -
x - yti
x, -
Fwq,,,,o XII - k_,xa2 reaction II= (FMBLO -x-Y,I) [J% (FA,B,,o -XI)
+~(F~AB~~,o-~II)~ - (k-d+kbx:,)
(1)
In the same way we defined xrl =
III and IV. s
*’ dx
- for x in reactions 0 dt
The x,, is the mole fraction of bisphenol A-terephthalate (A2B i) at t = t,. Thus we obtained Eq. (2) in the following form.
2 =
k(Lm.o
-
x,1-~)(F,u(,),,o
-
~1)
-
k-,x2
Y reaction III + k,(Lw -
X,1 - yx_
F(AB),(AB),,o
-+Ld
reaction IV= (FMBLO -x,1 -Y> [~~(FA,(AB)Lo -YI)
+~~(FAB~,(AB~,,o -yn)l- (ky: +LY:I) (2)
The dyad A ,BZ could not be detected because there were no new peaks split either from 13C in carbonate (peak 7) or from “C in ethylene (peak i). By the same reason, the dyad (AB) ,BZ could not be detected from 13C in POB (peak a) or from 13C in carbonate (peak 7), as shown in Fig. 6a,b where the partial n.m.r. spectra of the fresh blend and of the blend annealed for 60 min at 260°C are displayed. From another viewpoint, the amount of carbonate has been reduced after the reaction, since the ratio of the integral peak 6 (bisphenol A) to that of peak 7 (carbonate) in PC became smaller. This result can be used to explain the fact that there was decarboxylation in the reaction[l9]. Therefore, it is difficult to detect the new dyads A ,B 2 and (AB)iB2. Since only two new dyads (AZB, and AZ(AB),) can be detected by the most sensitive n.m.r. analysis, we will not be able to solve Eq. (1) and Eq. (2) in the present form, which contained four independent variables. Hence, we must make assumptions based upon the experimental results. We first deal with Eq. (1). Before the reaction started, the dyad mole fraction FAIBl,O is close to F~ABjIBl,~
(FAIBI,O = 0.215, FCAB)IBI,O = 0.205), and therefore we
assumed FAlsi,a G F~Aa)iai,a for simplifying the equations. Additionally, the dyad mole fractions FAiai,a and FcAB)iaI,a decreased at about the same rate as listed in Table 3. There- fore we assumed: X XI = x,, x1=x1,= - 2 kl = k2 = k, k_, =k_,=k_, Table 3
The transient mole fractions of various dyads in the annealed 60/40 POB-PET/PC blend
Annealing Annealing time F,,a,,oa,, FAI(ABU F,AB)IBI FAlBl F&WI FAZBl FAZBZ
temperature 0 min 0.075 0.205 0.205 0.215 0 0 0.300 240°C 15 min 0.075 0.205 0.203 0.212 0 0.005 0.295 30 min 0.075 0.205 0.202 0.212 0 0.006 0.294 60 min 0.075 0.204 0.201 0.210 0.001 0.009 0.290 250°C 15 min 0.75 0.204 0.202 0.211 0.001 0.007 0.292 30 min 0.75 0.204 0.200 0.210 0.002 0.010 0.288 60 min 0.074 0.203 0.197 0.206 0.003 0.017 0.280 260°C 15 min 0.074 0.204 0.200 0.209 0.002 0.011 0.287 30 min 0.073 0.200 0.193 0.20 0.007 0.025 0.268 60 min 0.07 1 1.194 0.187 0.195 0.015 0.038 0.247
724 J.-C. Ho, K.-H. WdPolymer 40 (1999) 717-727 ii PC o-q-0 -I- I
Jn
6 I 3 i PC i II I ’ I ’ I ’ I ’ I 1 I I * I 1 1 I 156 155 153 152 151 150 149 148 68 66 64 62 ppm ,‘~“,I pp11Fig. 6. The partial 13C n.m.r. spectra of (a) fresh 60/40 POB-PET/PC blend, (b) 60/40 POB-PET/PC blend annealed at 260°C for 60 min With these assumptions, Eq. (1) can be reduced to Eq. (3).
dx
z=k,[(F,zsz,o-~t~)
-x]
[(F,,,,,o+F(,n,,,,,,)
-x]
- ;k_.n’We followed the method which Devaux[lS] used to inves- tigate the dyad concentration in PC/PBT blends. The initial mole fraction of participating dyads should be normalized, so we obtained Eq. (4).
and then X is obtained in the following form.
x= X
(Fm,o -Y,,) + (PAIBI,O + P~~n)m,,a) (5)
Substituting Eq. (4) and Eq. (5) into Eq. (3), we converted Eq. (3) into the following form:
dX
[email protected]?n~.o-~t,) + (FA,~,,~+F~AB)~B,,o)](A-X)
X (B -X) - +-a [(km,” -
yt,)
+ (%,~,,a+F~.m~m,,o)lX2
=I,(A-X)(B -x)- kLax2
(6)
Where k, and k_, are defined in Eq. (7)
k =k,[(P,,,,,, -YI,) + (P/,mt,o+P~,,m,,,)]
k~n=k-.[(FA2n2,O-yrl)+ (FAIBI,o+F~AB)~B~,“)] (7)
Since the copolyester is random at equilibrium ($$ = 0), and we put X, = AB and A + B = 1 into Eq. (6), we get Bq. (8).
(8)
Putting Eq. (8) into Eq. (6), we derive Eq. (9).
Integrating Eq. (9), and defining the transesterification ratio r, = X/A for reaction I and II, we obtain a kinetic expression for the dyad A2B ,.
In ~
[
(B
_”
r.J
1
=
kt
(10)
By the same method, in Eq. (2), the initial dyad mole frac- tion F AI(Au~t.a was approximately three times the initial mole fractions F(AB)I(AB)I,O, FAI(AB),I,O~ and F(AB)I(AB)I,O decreased at
the same rate. Therefore we roughly assumed:
F At(Au)t,a F(AB)I(AB)I.D I s?L 4’11
Z 3 y, GG ;y yrr &Z ;y
k,k4=kb k_3=kp4=k_b
With these assumptions, Eq. (2) is rewritten as Eq. (11). dy ,=k,[(FA*B*,o-~t,)-Y] [(FA~(A~)~.O+F~AB)~(AB)~,O)-Y] 5 _- k by2 8 -
(11)
The initial mole fraction of participating dyads were normalized in the following.
c =
(F,,B,,o - %,)(F AZBLO - 4 t + ( FA1(AB)I,O + F(~~),(~~)~,~ ) D = (F~,~~~)~,~ +~(AEwAB),,o)
CF.42B2,0 -X,1) + (FA,(ABJ,,o+FcAB)I(ABJ,,o)
(12)
Y can then be obtained in the following form.
Y= Y
(FAZBLO --Q> + (FA,(AB),,o +FcAB),(AB),,o) (13) By substituting Eqs. (12) and (13) into Eq. (1 l), we obtained Eq. (14).
dY
~‘k,[(FA2sz,0 -%I) + (FA,(ABJ, +FcAB),(ABJ,)]
x CC-Y)(D-Y)- $‘[(&mz,o-&,) + (FA,(AB), +F(AB),(AB)I)]~~
=&(C-Y)(D-Y)- &Y2 (14)
where I$, and ,& _ b are defined as
kb = kb[(FAzm,o --Q) + (FA,(AB), +F~AB),(AB),)] 5, = k-b[(FAm,o --%> + (FANABII +FcABJ,(AB),)]
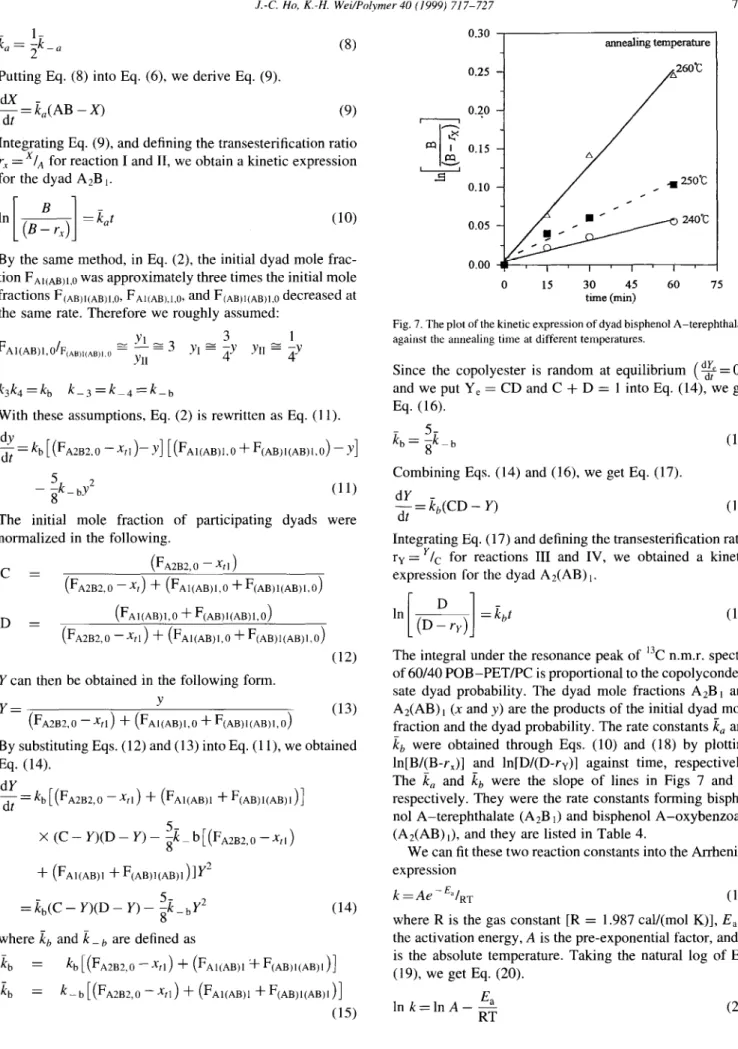
(15) 0.30 0.25 annealing temperature 0 15 30 45 60 75 time (min)
Fig. 7. The plot of the kinetic expression of dyad bisphenol A-terephthalate against the annealing time at different temperatures.
Since the copolyester is random at equilibrium (% = 0)) and we put Y, = CD and C + D = 1 into Eq. (14), we get Eq. (16).
Combining Eqs. (14) and (16), we get Eq. (17). dY -
dt = kb(CD - Y) (17)
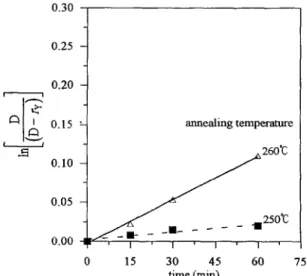
Integrating Eq. (17) and defining the transesterification ratio ry = ‘Ic for reactions III and IV, we obtained a kinetic expression for the dyad Az(AB) 1.
(18)
The integral under the resonance peak of ‘jC n.m.r. spectra of 60/40 POB-PET/PC is proportional to the copolyconden- sate dyad probability. The dyad mole fractions A2B1 and A2(AB), (x and y) are the products of the initial dyad mole fraction and the dyad probability. The rate constants I, and kb were obtained through Eqs. (10) and (18) by plotting ln[Bl(B-r,)] and ln[D/(D-r,)] against time, respectively. The k, and kb were the slope of lines in Figs 7 and 8, respectively. They were the rate constants forming bisphe- no1 A-terephthalate (A2B J and bisphenol A-oxybenzoate (A2(AB) i), and they are listed in Table 4.
We can fit these two reaction constants into the Arrhenius expression
k = Ae - EdlRT (19)
where R is the gas constant [R = 1.987 cal/(mol K)], E, is the activation energy, A is the pre-exponential factor, and T is the absolute temperature. Taking the natural log of Eq. (19), we get Eq. (20).
726 J.-C. Ho, K.-H. WdPolymer 40 (I 999) 717-727 0.25 1 0.20 v _ . 1 cl I-” 0.15 . ra v, .5 0.10 1 / 26OC 0 15 30 45 60 75 the (min)
Fig. 8. The plot of the kinetic expression of dyad bisphenol A-oxybenzoate against the annealing time at different temperatures.
In k, and lnkb were plotted against l/T in Fig. 9. In Fig. 9, the slopes of these lines were - E,IR. We than obtained the following equation.
I;, = 5.39 X lOI exp( -41814/,,)
&l 1.17 X 1O36 exp( -94645/,T)
(21) =
From Eq. (21), the activation energy of forming bisphenol A-oxybenzoate, A;?(AB),, is more than twice of that of forming bisphenol A-terephthalate, A2BI (94.6 versus 41.8 kcal/mol). These activation energies for the transester- ification in POB-PET/PC are much larger than the activa- tion energy for the transesterification in PET/PC (3 1.1 kcal/ mol) [4]. The difference in activation energy can be explained by the fact that due to the conjugation effect, the chemical bond between oxybenzoate and oxybenzoate is much stronger than that between ethylene and terephtha- late. Therefore, the PET segment is more susceptible to transesterification with PC than POB segment with PC. The pre-exponential factor for forming bisphenol A-oxy- benzoate was much larger than that for forming bisphenol A-terephthalate were ( 1O36 versus 1014 min-‘). This is due to the rigid-rod structure of POB segment. The more rigid POB segment had a larger radius of gyration compared to that of the flexible-coil PET segment. Since in the trans- esterification reaction the collision frequency is proportional to the square of the size of the reacting segment, this
Table 4
The rate constants &, and &,, as a function of temperature Anealing temperature (“C) r;, x 10’ K, x 101 (min-‘) (min-‘) 240 0.88 * 250 I .66 0.33 260 4.09 1.86
*Absence of the bisphenol A-oxybenzoate dyad.
J -5.0 -:_: * v.lope=- a, R \
\
-8.0 m -9.0 I-I
1.86 1.88 1.90 1.92 1.94 1.96+x
lo3(
K-‘)Fig. 9. The change in the rate constants I;, and & of transesterification in the 60/40 POB-PET/PC blend with the annealing temperature.
accounts for the high pre-exponential factor in producing new dyad bisphenol A-oxybenzoate.
From the above analysis in pre-exponential factor and in activation energy, we knew that the production of dyad bisphenol A-terephthalate dominated the transesterification process in the blend of POB-PET and PC at low tempera- tures such as at 240°C. As the temperature increased to 260°C the production of dyad bisphenol A-oxybenzoate increased very quickly.
4. Conclusions
We have carried out a study of the kinetics of transester- ification in POB-PET/PC blend. In this study, we found that the activation energy of forming dyad bisphenol A-oxy- benzoate resulted from the transesterification between the POB segment in POB-PET and PC is more than twice that of forming dyad bisphenol A-terephthalate resulting from the transesterification between PET segment in POB-PET and PC (94.6 versus 41.8 kcal/mol). This is probably due to the more stable chemical bond in oxybenzoate and oxy- benzoate than that in ethylene and terephthalate. On the other hand, owing to the rigid-rod nature of POB segment, the frequency factor in the transesterification reaction in POB segment and PC is much higher than that in PET seg- ment and PC (i.e. 10xh versus 1014 min-‘).
Acknowledgements
The authors thank the financial support provided by the National Science Council through project NSC 86-2216-E- 009-006.
References
[I] Kotilar AM. J Polym Sci, Macromol Rev 1981;16:367.
[2] Pilati F, Marianucci E, Berti Cl. .I Appl Polym Sci 1985;30:1267. [3] Kimura M, Porter RS. J Polym Sci Polym Phys Ed 1983;21:367. [4] Godard P, Dekonick JM, Devlesaver V, Devaux J. J Polym Sci, Part
A: Polym Chem 1986;24:3315.
[5] Henrichs PM, Tribone J, Mass DJ, Hewitt JM. Macromolecules 1988;21:1282.
[6] Kimura M, Porter RS. J Polym Sci Polym Phys Ed 1983;22: 1697. [7] Su KF, Weig KH. J Appl Polym Sci 1995;56:79.
[E] Wei KH, Hwang WJ, Tyan HL. Polymer 1996;37:2087. [9] Laivins GV. Macromolecules 1989;22:3974.
[IO] Wei KH, Jang CJ, Ho JC. Polymer 1997;38:3521.
[I l] Yamadera R, Murano MJ. J Polym Sci Polym Chem Ed 1967;5:2259. [12] Devaux P, Godard P, Mercier JP. J Polym Sci Polym Phys Ed
1982;20: 1875.
[13] Devaux P, Godard P, Mercier JP. J Polym Sci Polym Phys Ed 1982;20:1881.
[14] Devaux P, Godard P, Mercier JP. J Polym Sci Polym Phys Ed 1982;20: 1895.
1151 Devaux P, Godard P, Mercier JP. J Polym Sci Polym Phys Ed 1982;20: 1901.
[ 161 Wei KH, Ho JC. Macromolecules 1997;30: 1592.
1171 Jackson JR, Kuhfuss HF. J Polym Sci Polym Chem Ed 1976; 14:2043. [ 181 Nicely VA, Dougherty JT, Renfro LW. Macromolecules 1987;20:573. [ 191 Schilling FC, Ringo WM. Jr, Sloane NJ, Bovey FA. Macromolecules